

Abstract
Misexpression and intracellular retention of peripheral myelin protein 22 (PMP22) is associated with hereditary neuropathies in humans, including Charcot-Marie-Tooth disease type 1A (CMT1A). Mice expressing extra copies of the human PMP22, termed C22, display morphologic and behavioral characteristics of CMT1A. In neuropathic Schwann cells, the turnover of the newly-synthesized PMP22 is decreased, leading to the formation of cytosolic protein aggregates. To aid the processing of PMP22 and alleviate the associated myelin defects, we pharmacologically stimulated the expression of protein chaperones by synthetic small-molecule inhibitors of heat shock protein 90 (HSP90). The exposure of Schwann cells to these compounds enhanced the levels of cytosolic chaperones in a time- and dose-dependent manner, with minimal cytotoxicity. Treatment of dorsal root ganglion (DRG) explants from neuropathic mice improved myelin formation and the processing of PMP22. These results warrant further studies with HSP90 inhibitors as potential therapeutic candidates for hereditary demyelinating neuropathies.
Keywords: CMT1A neuropathy, demyelination, Schwann cells, protein misfolding, heat shock response, heat shock proteins, HSP90 inhibitors, myelination, myelin, peripheral myelin protein 22
Introduction
Hereditary peripheral neuropathies comprise a heterogeneous group of disorders, among which Charcot–Marie–Tooth disease type 1A (CMT1A) is the most prevalent form (Shy et al., 2001). CMT1A is a demyelinating neuropathy mainly associated with a 1.5-megabase duplication on human chromosome 17 that includes the peripheral myelin protein 22 (PMP22) locus (Lupski et al., 1991). Transgenic rodent models based on the overexpression of the wild type (Wt) PMP22 reproduce features of the human condition and provide experimental models to study disease pathogenesis (Huxley et al., 1996; Magyar et al., 1996; Sereda et al., 1996; Perea et al., 2001; Robertson et al., 2002). One of these transgenic lines termed C22, express approximately 1.7-fold higher levels of the PMP22 mRNA and display slowed nerve conduction velocities and a reduction in the percentage of myelinated fibers (Huxley et al., 1996; Huxley et al., 1998; Robertson et al., 1999; Robertson et al., 2002).
Eukaryotic cells maintain protein homeostasis by using a collection of quality control pathways known as the unfolded protein response (UPR). Induction of UPR leads to the attenuated protein translation in the ER, ER-assisted folding and ER-associated degradation (ERAD) via the proteasome (Kincaid and Cooper, 2007). PMP22 folds with only a modest efficiency even under normal conditions (Sanders et al., 2001), as approximately eighty percent of the newly-synthesized protein is degraded by the proteasome (Pareek et al., 1997; Notterpek et al., 1999). In response to PMP22 overexpression, the quality control system appears to be overwhelmed and protein aggregates form. In the C22 mouse model, a reduced turnover of the newly-synthesized PMP22 is associated with the presence of cytosolic protein aggregates within Schwann cells (SCs) and impaired proteasome activity (Fortun et al., 2006). In myelinating dorsal root ganglion (DRG) explant cultures, the retention of PMP22 within the SC cytosol decreases the amount of protein at the plasma membrane (Fortun et al., 2006), which could contribute to the observed myelin defects in affected mice.
A promising therapeutic approach for protein misfolding disorders, such as PMP22-associated neuropathies, involves the enhancement of chaperone expression (Muchowski and Wacker, 2005). Inhibitors of heat shock protein 90 (HSP90), including geldanamycin (GA) and its pharmacologically improved derivatives, 17-DMAG and 17-AAG, have been shown to suppress aggregation of mutant huntingtin and α-synuclein in cultured cells (Nixon et al., 1994; Sittler et al., 2001; McLean et al., 2004; Herbst and Wanker, 2007). A family of small molecule HSP90 inhibitors significantly reduce tau protein levels and selectively clear specific phospho-tau aggregates in association with an increase in the levels of HSP27, HSP40 and HSP70 (Dickey et al., 2005; Dickey et al., 2006). These studies underscore the importance of HSPs in the elimination of misfolded proteins in neurodegenerative diseases; however their potential application for hereditary peripheral neuropathies has not been tested. Here we show that out of fifteen small molecule inhibitors of HSP90, EC137 effectively enhances chaperone levels and improves myelination, along with the trafficking of PMP22, in dorsal root ganglion (DRG) explant cultures from neuropathic mice.
Materials and methods
Mouse colonies
The PMP22 overexpressor (C22) (Huxley et al., 1996) mouse breeding colony is housed under SPF conditions at the University of Florida, McKnight Brain Institute animal facility. The use of animals for these studies has been approved by an Institutional Animal Care and Use Committee (IACUC). Genomic DNA was isolated from tail biopsies of mouse pups (less than 10 days old) and litters were genotyped by PCR (Huxley et al., 1996).
Non-myelinating SC cultures
Primary SC cultures from genotyped postnatal day 6 (P6) Wt and C22 mouse pups, or neonatal rat pups, were prepared and maintained as described (Ryan et al., 2002). Cells were grown to ~80% confluency in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum (Hyclone, Logan, UT, USA), 2.5 (mouse) or 5 μM (rat) forskolin (Calbiochem, La Jolla, CA, USA) and 10 μg/mL bovine pituitary extract (Biomedical Technologies Inc, Stoughton, MA, USA).
Cellular toxicity assay
Non-myelinating rat SC cultures were treated with small-molecule, synthetic HSP90 inhibitors at 50 nM and 500 nM (Dickey et al., 2005; Dickey et al., 2006; Dickey et al., 2007) concentrations for 16 h and then incubated in the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) solution (0.5 mg/ml) for 5 h at 37 °C, producing the formazan product as a result of the cleavage of the tetrazolium salt MTT by the mitochondrial enzyme succinate-dehydrogenase (Mosmann, 1983). The amount of blue formazan product is directly proportional to the number of viable cells present. The optical density (OD) of each well was measured using an automated plate reader (550 nm) (Heaton et al., 2004). The toxicity of each test compound with respect to DMSO treated control cells from three independent experiments was determined and graphed as the percentage of cell death. Statistical significance was determined by using Student’s t-test.
Dorsal root ganglion (DRG) explant cultures and compound treatment paradigms
Mouse DRG explant cultures were established as described (Cosgaya et al., 2002). Pregnant Wt and heterozygous C22 mice were sacrificed according to guidelines of University of Florida Institutional Animal Care and Use Committee. DRGs were collected from embryonic day 12–14 mice, digested with 0.25 % trypsin (Gibco, Rockville, MD), dissociated and plated on rat tail collagen-coated (Biomedical Technologies, Inc.) glass coverslips. DNA was isolated from each embryo for genotyping by PCR, as described above. Explants were maintained in minimum essential medium (MEM; Gibco) supplemented with 10% fetal calf serum (Hyclone, Logan, UT), 0.3 % glucose (Sigma-Aldrich, St. Louis, MO), 10 mM HEPES (Cellgro; Mediatech, Inc., Herndon, VA), and 100 ng/ml nerve growth factor (Harlan Bioproducts for Science, Madison, WI) for 7 days. In the case of the GA paradigm, myelination was initiated for 10 days by the addition of ascorbic acid (50 μg/ml; Sigma-Aldrich) which was followed by GA exposure for 72 h. For exposure with EC137, cultures were maintained under myelination-promoting conditions for 14 days and treated with compound for two 48 h periods, with a 48 h washout, in between. After the second 48 h treatment and a 16 h washout, the cultures were processed for immunostaining and Western blot analyses (Fig, 5A). For SC depleted neuronal cultures (Einheber et al., 1993), the explants were treated for 24 or 48 h with HSP90 inhibitors.
Figure 5. Myelin production by neuropathic explant cultures is enhanced by EC137.
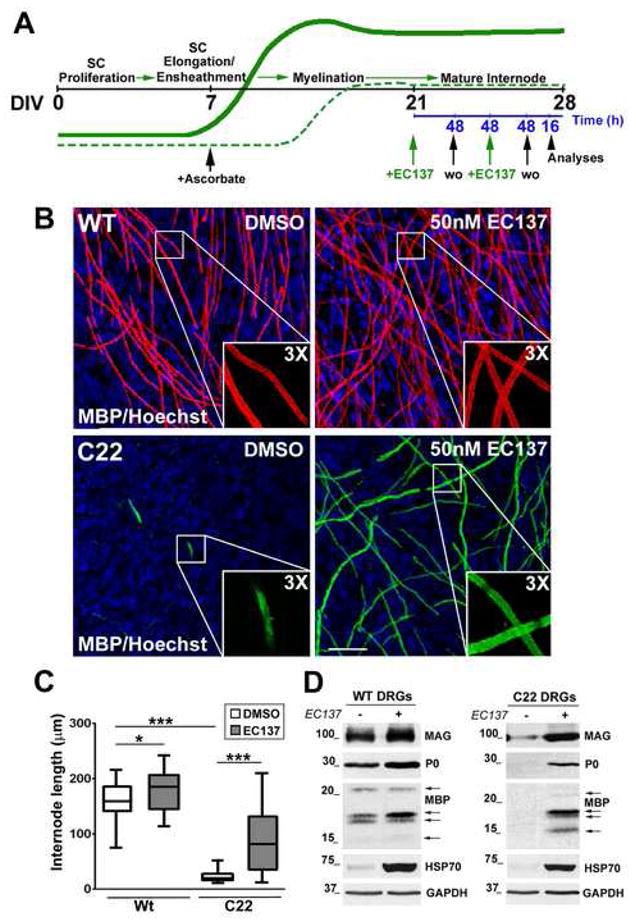
(A) Schematic of the treatment paradigm for DRGs from Wt and C22 mice with EC137. The black line indicates the time-scale for days in vitro (DIV). The lines in green, bold and dashed, represent the expression profiles of myelin proteins in DRGs from Wt and C22 mice respectively. The blue line indicates the time-scale (h) for EC137 treatment. Starting on DIV21, a pulse treatment of EC137 (50 nM) (green arrows) was added for 48 h, followed by 48 h washout (wo, black arrows). This sequence was repeated and a second wash out (16 h) was followed by analyses of the samples (arrow head). (B) DRG explant cultures from Wt (top panel) and C22 neuropathic (bottom panel) mice, under myelinating conditions, were treated with DMSO (control) or EC137 (50 nM) for a total of 96 h as described (A), and stained with an anti-MBP antibody. Insets show the outlined regions at 3X magnification. Hoechst dye was used to stain the nuclei. Scale bar, 40 μm. (C) The lengths of the myelin internodes (n is at least 100 for each condition) were measured in explant cultures from Wt and C22 mice treated with DMSO or EC137 (50nM), using Spot Advanced software. *p<0.05, ***p< 0.001. Error bars show SEM. (D) DRG explants from Wt and C22 mice were treated as described (A) and whole protein lysates (40 μg/lane) were analyzed for the levels of myelin proteins MAG, P0 and MBP, and of HSP70 from at least three independent experiments. Arrows on the MBP blots indicate the 21.5, 18.5, 17 and 14 kDa isoforms. GAPDH serves as a loading control. Molecular mass in kDa.
Primary Antibodies
Antibodies for protein chaperones included anti-HSP70, -HSP40, -αB-crystallin and -calnexin (all polyclonal rabbit antibodies, from Stressgen, Victoria British Columbia, Canada), rat anti-HSP90 (Stressgen) and goat anti-HSP27 (Santa Cruz, CA). To monitor the heat shock transcriptional response, a polyclonal rabbit anti-heat shock factor 1 (HSF1) antibody (Stressgen) was employed. Antibodies for myelin proteins included monoclonal mouse-anti myelin associated glycoprotein (MAG), rat anti-myelin basic protein (MBP) (both from Chemicon, Temencula, CA, USA), mouse anti-P0 (Archelos et al., 1993). To detect PMP22, a 1:1 mixture of two rabbit polyclonal antibodies, developed against a peptide corresponding to the second extracellular loop of the human or the rat PMP22, was used (Pareek et al., 1997; Fortun et al., 2006). Monoclonal anti-actin, -tubulin (both from Sigma), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (clone 1D4, EnCor Biotechnology Inc., Alachua, FL, USA) served as protein loading controls. Polyclonal rabbit anti-ubiquitin (Dako, Carpinteria, CA) was purchased from the indicated supplier.
Immunolabeling studies
Non-myelinating SCs and myelinating DRG explant cultures on glass coverslips were fixed with 4% paraformaldehyde for 10 min and permeabilized with 100 % methanol for 5 min at −20 °C. After blocking with 10 % normal goat serum, the samples were incubated with the indicated primary antibodies overnight at 4°C, followed by the appropriate secondary antibodies, including Alexa Fluor 594 goat anti-rabbit IgG, Alexa Fluor 488 goat anti-rat IgG and Alexa Fluor 488 goat anti-mouse IgG (all from Molecular Probes, Eugene, OR). Hoechst dye (Molecular Probes) was included in the secondary antibody solution at 10 μg/ml to visualize nuclei. Coverslips were mounted using the ProLong Antifade kit (Molecular Probes). Samples were imaged with a Spot camera attached to a Nikon Eclipse E800 microscope, or a Leica TCS SP2 AOBS Spectral confocal microscope and were formatted for printing by using Adobe Photoshop 5.5.
Biochemical studies
Untreated control and compound treated cultures were lysed in sodium dodecyl sulfate (SDS) gel sample buffer (62.5 nM Tris, pH 6.8, 10 % glycerol, 3 % SDS) and protein concentrations were determined using BCA assay (Pierce, Rockford, IL, USA). Samples were analyzed on polyacrylamide gels under reducing conditions (except for the determination of MAG), and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were blocked in 5 % non-fat milk in PBS and incubated overnight with primary antibodies. After washing, anti-mouse, anti-rabbit or anti-rat HRP-linked secondary antibodies were added for 2 h. Bound antibodies were visualized using an enhanced chemiluminescence detection kit (PerkinElmer Life Sciences, Boston, MA, USA). Films were digitally imaged using a GS-710 densitometer (Bio-Rad Laboratories) and were formatted for printing by using Adobe Photoshop 5.5. Densitometric analysis of Western blots was performed using Scion image software.
Quantification of myelin internode lengths
DRG explant cultures were subjected to the treatment paradigms described above and immunostained with an anti-MBP antibody to label internodal myelin segments (Amici et al., 2007). Internode lengths from Wt and C22 cultures from three independent experiments were measured with Spot RT software (Diagnostic Instruments, Inc., Sterling Heights, MI). Measurements were collected from three coverslips per genotype per treatment paradigm. Statistical significance was determined by using Student’s t-test using GraphPad Prism software.
Results
Myelin production in C22 neuropathic samples is enhanced by geldanamycin
In the DRG explant model, sensory neurons and SCs from normal mouse embryos produce many myelinated segments, while samples from neuropathic mice only form a few shortened segments (Fortun et al., 2006; Amici et al., 2007). SCs cultured with DRG neurons from C22 neuropathic mice accumulate PMP22 within their cytosol (Fig. 1A, upper left, arrows) (Fortun et al., 2006). To test, whether enhancement of protein chaperone expression might be beneficial for myelination in these samples, DRG explants from C22 mice were treated with GA for 72 h and processed for immunolabeling and Western blots (Fig. 1). GA is a naturally occurring ansamycin antibiotic which inhibits HSP90, thereby activating HSF1 and the expression of chaperones, including HSP70, HSP40 and HSP27 (McDonough and Patterson, 2003). Exposure of explant cultures from neuropathic mice to 50 nM GA for 72 h reduced the presence of cytosolic PMP22 aggregates (Fig. 1A, upper right), as compared to DMSO-treated control samples (Fig. 1A, upper left, arrows). Treatment with GA also increased myelin production, as judged from immunolabeling with an anti-MBP antibody (Fig. 1A, lower right). As shown in previous studies with GA (Nixon et al., 1994; Kim et al., 1999; Petrucelli et al., 2004), in total protein lysates we detected a prominent induction of HSP70 and HSP27, as compared to DMSO-treated controls (Fig. 1B). In agreement with the enhanced myelination (Fig. 1A), we also detected an increase in the steady-state level of PMP22 (Fig. 1B), which is known to correlate with compact myelin formation (Notterpek et al., 1999). While additional studies of GA-treated cultures indicate a consistent improvement of myelination in explants from neuropathic mice, prolonged exposure to this compound is known to be toxic (Miyata, 2005). Therefore, we decided to test a class of synthetic HSP90 inhibitors, which cause less toxicity in cultured neuroglioma cells and have the potential for in vivo application (Dickey et al., 2005; Dickey et al., 2007).
Figure 1. Myelin production is stimulated by geldanamycin.
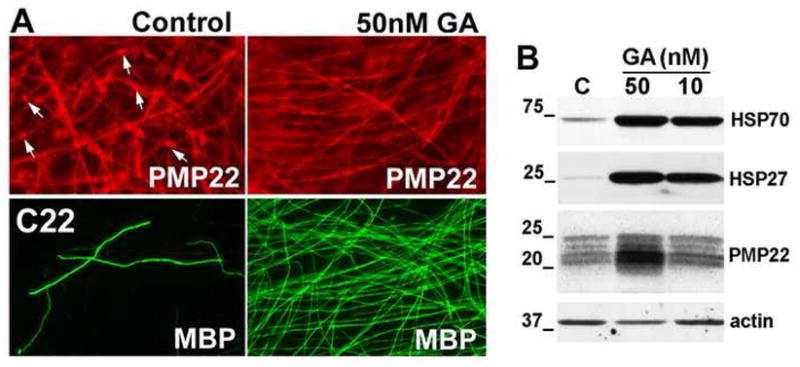
(A) DRG explant cultures from C22 mice under myelinating conditions, were treated with DMSO or GA (50 nM) for 72 h, and stained with polyclonal anti-PMP22 (red) or anti-MBP (green) antibodies. Enhanced PMP22-like immunoreactivity is associated with the SC bodies (arrows) in the DMSO-treated samples. Magnification, X40. (B) The steady-state levels of HSP70, HSP27 and PMP22 were determined in total lysates of DRG explant cultures (20 μg/lane) from C22 neuropathic mice after 72 h of GA-treatment (10 and 50 nM), as compared to DMSO control (C). Actin serves as a protein loading control. Molecular mass in kDa.
Small-molecule HSP90 inhibitors enhance chaperone expression in SCs
To begin our studies with the synthetic HSP90 inhibitors (EC compounds), non-myelinating primary rat SC cultures were treated for 16 h at 50 and 500 nM concentrations, followed by MTT cellular toxicity assay (Fig. 2A). We used rat SCs for the initial screening of the compounds due to the relative ease of obtaining a large number of homogeneous cell populations from neonatal rat nerves, as compared to mouse. In comparison to GA, which led to a significant 20–25 % glial cell death (p<0.001), several EC compounds were less toxic to SCs. For example, EC137 at 50 nM has significantly less cellular toxicity as compared to GA at 50 nM (p<0.001) (Fig. 2A). In parallel with the cellular toxicity studies, we have tested the ability of these compounds to induce HSP70 expression (Fig. 2B). Eight out of fifteen tested compounds induced HSP70 at 50 nM, while thirteen out of fifteen were effective at 500 nM (Fig. 2B). Furthermore, two negative controls, EC116 and EC117 that are structurally-related inactive HSP90 inhibitors, failed to induce HSP70. The levels of calnexin, which is an ER chaperone rather than a HSP, are unaltered by exposure of the cells to the HSP90 inhibitors (Fig. 2B). From these results, we chose EC137, EC119, EC127 and EC139 for further studies at 50 nM concentrations. While all four of these compounds at 50 nM concentrations enhanced HSP70 expression in rat SCs, pilot studies with neuropathic samples identified EC137 as the most effective compound in reducing the levels of poly-ubiquitinated proteins (Fig. 3A). In agreement with our previous studies (Ryan et al., 2002; Fortun et al., 2003), the basal levels of HSP70 are elevated in SCs from neuropathic mice, which is further enhanced upon treatment with HSP90 inhibitors.
Figure 2. Cellular toxicity and chaperone expression for the HSP90 inhibitors.
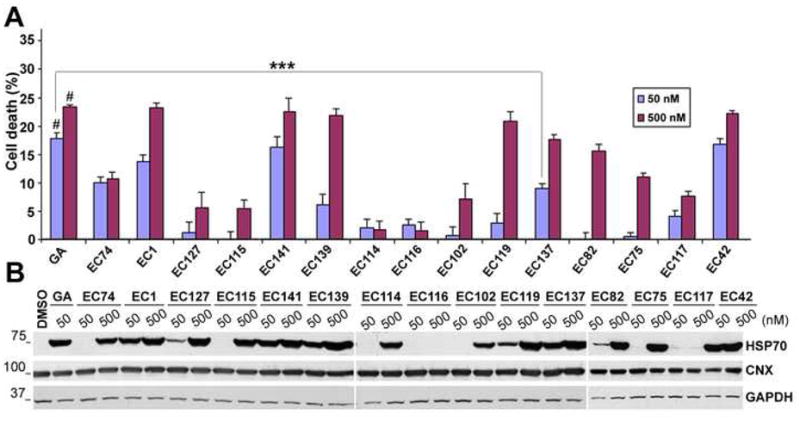
(A) Non-myelinating rat SCs were treated with DMSO (control) or HSP90 inhibitors at 50 and 500 nM concentrations for 16 h and then incubated in MTT (0.5 mg/ml) for 5 h at 37 °C. The toxicity of the test compounds was determined from three independent experiments with respect to DMSO treated control cells, which was set at 0 % cell death (***p<0.001 and #p<0.001 with respect to DMSO control). Error bars indicate SEM. (B) The levels of HSP70 and calnexin (CNX) were determined by Western blot analyses in total cell lysates (20 μg/lane) after 16 h treatment. GAPDH is shown as a protein loading control. Molecular mass in kDa.
Figure 3. EC137 stimulates chaperone production in non-myelinating SCs in dose and time-dependent manner.
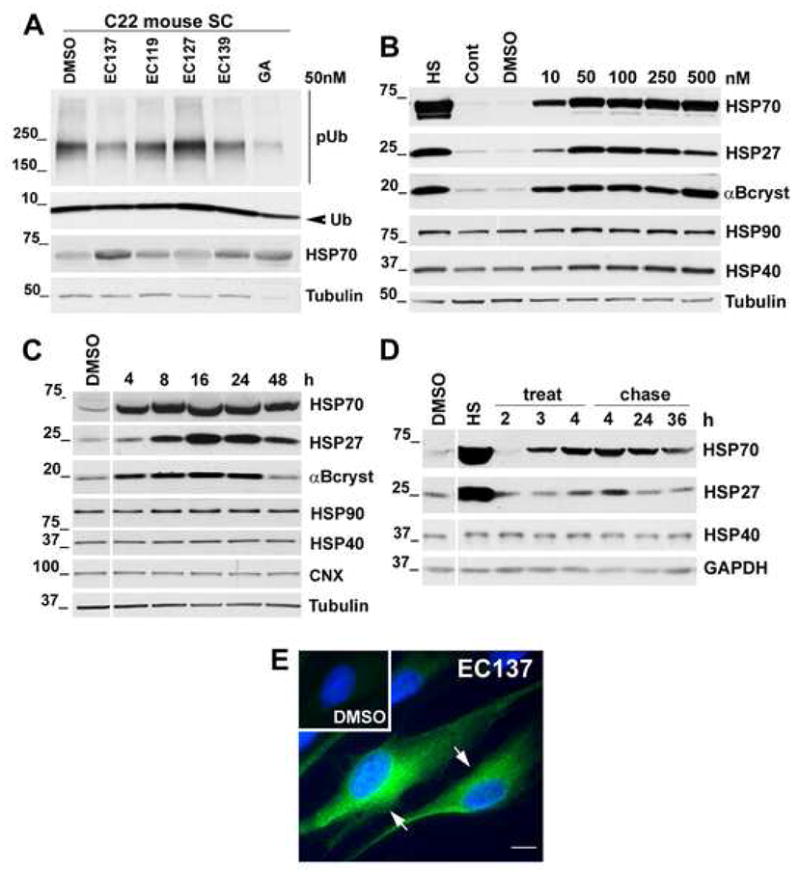
(A) SCs from C22 neuropathic mice were treated with low toxicity HSP90 inhibitors (EC137, EC119, EC127, EC139; all at 50 nM) and GA (50 nM) for 16 h and the levels of poly-ubiquitinated (pUb) substrates and HSP70 were analyzed by Western blots (20 μg/lane). Arrowhead indicates mono-ubiquitin (Ub). (B) For the dosage response, the levels of HSPs after EC137 treatment of rat SCs (10, 50, 100, 250 or 500 nM) were analyzed in total cell lysates (20 μg/lane). Heat shock (HS) followed by 8 h chase at 37 °C is included as positive control. (C) For time-course studies, the levels of HSPs were analyzed after treatment with EC137 (50 nM) for 4, 8, 16, 24 or 48 h. (D) To assess the maintenance of chaperone expression, cells were treated with EC137 (50 nM) for 2, 3 and 4 h. After the 4 h treatment, EC137 was washed out and chaperone expression assayed at 4, 24 and 36 h chase time points. Molecular mass in kDa. Tubulin (A–C) or GAPDH (D) is shown as a protein loading control. (E) In cells treated with EC137 (50 nM, 16 h) the localization of HSP70 is detected with an anti-HSP70 (green) antibody. SCs treated with DMSO (control) exhibit low levels of HSP70-like immunoreactivity (upper left, inset). Hoechst dye was used to stain the nuclei. Scale bar, 10 μm.
To characterize the effect of EC137 on glial gene expression, the dose-dependent induction of a panel of chaperones was determined at five different concentrations, including 10, 50, 100, 250 and 500 nM (Fig. 3B). Compared to control levels, after a 16 h treatment with 10 nM EC137 the expression of HSP70, HSP27 and αB-crystallin are enhanced. However, the levels of these HSPs are elevated ~3-fold higher at the 50 nM dosage and are comparable to heat shock (HS; 45 °C for 20 min) preconditioning, followed by an 8 h chase (Fig. 3B). In agreement with known molecular targets of EC137, this concentration also enhanced the levels of HSP90 and HSP40 in the SCs. As treatment of the cells with higher dosages of EC137 did not appear to further stimulate chaperone expression, we chose the 50 nM concentration for subsequent studies. To optimize potential treatment paradigms for the myelinating samples, the induction of HSPs was analyzed after 4, 8, 16, 24 and 48 h incubation with 50 nM EC137 (Fig. 3C). As judged from the Western blot, the peak expression of HSPs is observed at 16 h. As seen previously (Fig. 2B), the levels of the ER chaperone calnexin are unaffected by EC137 (Fig. 3C). To further characterize the kinetics of HSP induction by EC137, SC cultures were treated for 4 h, followed by wash out and chase time points at 4, 24 and 36 h (Fig. 3D). As shown in the Western blot, a 4 h treatment with EC137 is associated with a sustained expression of HSP70 up to 36 h (Fig. 3D). In comparison, the influence on the levels of HSP27 is short-lived. The effects of EC137 on HSP70 in non-myelinating rat SCs were confirmed by immunostaining (Fig. 3E). As compared to DMSO controls, HSP70-like immunoreactivity is prominent and detected within the cell soma after EC137 treatment.
Inhibitors of HSP90, such as EC137 enhance the expression of chaperones by promoting the nuclear localization and phosphorylation of HSF1 (Westerheide and Morimoto, 2005). To investigate the activation of HSF1 in our SC model, cells were exposed to a brief HS (45 °C for 20 min) (Fortun et al., 2007), or 50 nM EC137 for 2, 4, 8 or 16 h, followed by analysis with an HSF1 antibody (Fig. 4A). HS preconditioning leads to rapid phosphorylation of HSF1, as detected by a shift in the mobility of the protein on SDS gels (Fig. 4A, arrow). The activation of HSF1 by HS is transient, as the non-phosphorylated form (Fig. 4A, arrowhead) becomes the prominent protein after a 0.5 h chase. In comparison, EC137 exposure promotes HSF1 phosphorylation starting from 2 h post-treatment and the active phosphorylated form remains for up to 8 h. The protein returns to baseline by 16 h (Fig. 4A). To corroborate the activation of HSF1 by EC137, we determined protein localization in cells post HS or after a 2 h compound exposure (Fig. 4B). In DMSO treated control cells, HSF1 is detected both in and around the nucleus (Fig. 4B, top panel, arrows). HS preconditioning of the cells promotes the translocation of HSF1 to the nucleus within 20 minutes (Fig. 4B, middle panel, arrows). Similarly, exposure of the cultures to EC137 for 2 h leads to prominent nuclear HSF1-like immunoreactivity (Fig. 4B, bottom panel, arrows). Together, these studies indicate that synthetic inhibitors of HSF1 are well tolerated by primary peripheral glial cells and EC137 is an effective inducer of the HS response, as judged from the enhanced and sustained expression of HSPs post-treatment. The results of the cellular toxicity and protein expression studies with EC137 from rat SCs (Figs. 2–4), were confirmed in mouse SC isolated from Wt pups, prior to the beginning of the studies with the explant cultures from transgenic mice.
Figure 4. Treatment with EC137 activates HSF1.
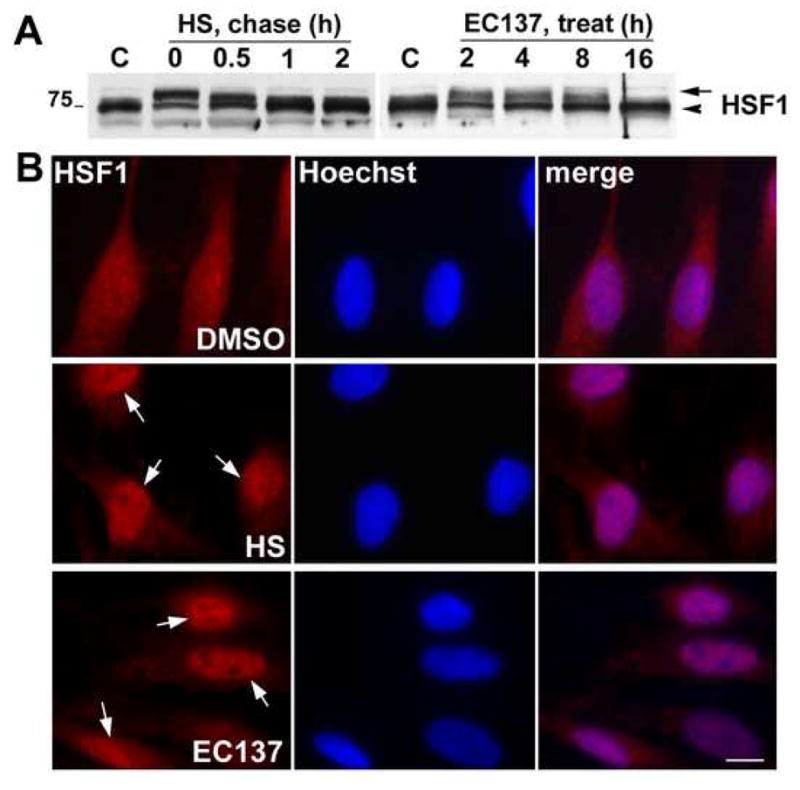
(A) The phosphorylation state of HSF1 after HS and EC137 (50 nM) treatment was assayed by Western blot (20 μg/lane). Phosphorylated HSF1 is indicated by the arrow and the arrowhead marks the migration position of the non-phosphorylated form. Molecular mass in kDa. (B) Cultured rat SCs were treated with HSP90 inhibitor, EC137 and the translocation of HSF1 from cytosol to the nucleus was monitored by staining with an anti-HSF1 (red) antibody. In untreated cells, HSF1 is predominantly cytosolic (upper panel). HS pre-conditioning (45 °C for 20 min) leads to rapid (0 h) nuclear localization of HSF1 (middle panel, arrows). The localization of HSF1 within the nucleus is detected at 2 h after treatment with EC137 (50 nM) (lower panel, arrows). Hoechst dye was used to stain the nuclei. Scale bar, 10μm.
Enhancement of HSPs promotes myelination in explant cultures from neuropathic mice
Next, we asked whether induction of HSPs could assist in the processing of glial proteins and improve myelination in samples from neuropathic mice. To test the influence of EC137 on myelination, DRGs from Wt and C22 embryos were incubated with DMSO as a control, or EC137 (50 nM), according to the paradigm shown (Fig. 5A). The treatment with EC137 was initiated after a 14-day period under myelination-promoting conditions (Fig. 5A). The explants were exposed to EC137 (50 nM) for a total of 96 h according to the schedule shown. This treatment paradigm is based on the data obtained from our dosage and time course experiments (Fig. 3B–D). In order to maximize the chaperone response, we chose 48 h exposure, during which time HSP levels are elevated in both SC and DRG explant cultures (Fig. 3C, 6D). The 48 h washout was selected based on the maintenance of chaperone expression for over 36 h (Fig. 3D). For the detection of compact myelin, samples were immunostained with antibodies to myelin basic protein (MBP) (Fig. 5B). As previously shown (Fortun et al., 2006; Amici et al., 2007), SCs in explant cultures from Wt embryos deposit numerous anti-MBP antibody reactive myelin segments, with or without compound treatment (Fig. 5B, top panels). At higher magnification, MBP-like immunoreactivity appears as the characteristic “rail-road tracks” of compact myelin (Fig. 5B, top panel, insets on bottom right). In comparison, in DRGs from C22 embryos, the DMSO control samples contain few, short MBP-positive myelin segments. Significantly, treatment of the neuropathic cultures with EC137 is associated with a pronounced improvement in the abundance of myelin internodes (Fig. 5B, bottom right). As indicated in the inset, myelin formed in response to EC137 exposure appears similar to those formed in cultures from Wt mice. To quantify the improvement in myelination in explant cultures from neuropathic mice, we measured MBP-reactive internode lengths (Fig. 5C). The average internode length of Wt DRGs treated with DMSO is 161.1 ± 4.818 μm (mean ± SEM). In Wt DRGs treated with EC137 there is a small, but statistically significant increase in internode lengths (178.8 ± 4.797 μm; p<0.05) (Fig. 5C). Strikingly, in DRGs from neuropathic mice, we found a 5-fold increase in internode lengths after treatment with EC137 (increase from 22.60 ± 1.384 μm to 90.26 ± 8.410 μm; p<0.001) (Fig. 5C). These results indicate that activation of HSPs has a positive influence on myelination by peripheral glial cells, particularly those from neuropathic mice.
Figure 6. Induction of chaperones aids the processing and trafficking of PMP22.
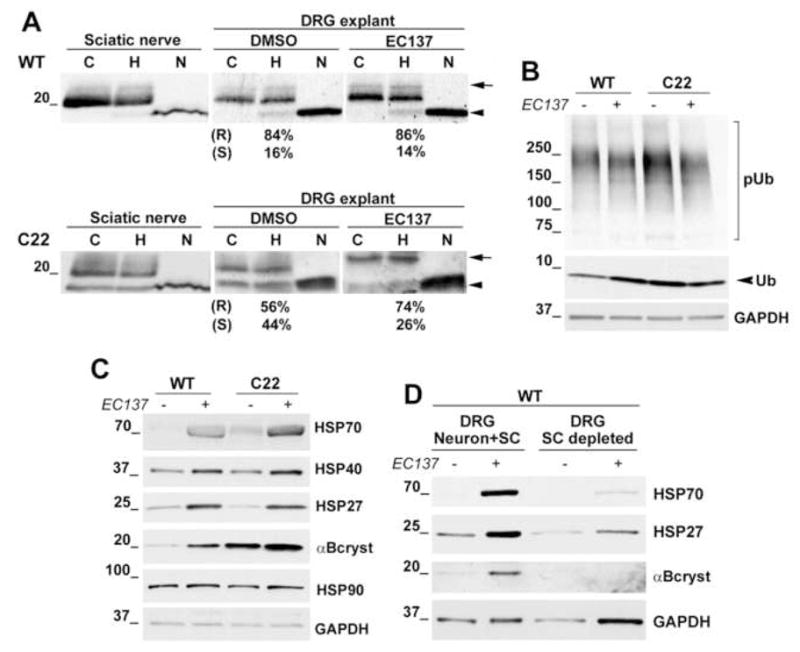
(A) Sciatic nerve lysates (10μg/lane) from 6 month old Wt and C22 mouse (n=3) were treated with endo H (H) or PNGase F (N) and blotted with anti-PMP22 antibody. Total lysates (20μg/lane) from Wt (top) and neuropathic (bottom) DRG explants treated with DMSO or EC137 (50nM) were incubated with endo H (H) or PNGase F (N) and PMP22 was detected. R denotes endo H-resistant and S denotes endo H-sensitive fractions (%) of PMP22. (B) A representative blot for the levels of poly-ubiquitinated proteins in lysates (20μg/lane) of SCs from Wt and C22 mice treated with DMSO and EC137 (50 nM). (C) The levels of HSP70, HSP40, HSP27, αB-crystallin and HSP90 in SC lysates (20μg/lane) from Wt and C22 mice (n=3) treated with DMSO and EC137 (50nM) were assayed. (D) The levels of HSP70, HSP27 and αB-crystallin in DRG explants, and SC-depleted DRG neurons from Wt mice, after treatment with DMSO or EC137 (50nM, 48 h) are shown. GAPDH serves as a constitutive marker for the blots. Molecular mass in kDa.
To corroborate the positive influence of EC137 on myelin protein expression, total protein lysates of DRGs from Wt and C22 embryos at the end of the treatment paradigms were analyzed by Western blots (Fig. 5D). In agreement with the improvement in myelin internode formation in EC137-treated samples (Fig. 5B, C), we found an increase in the steady-state level of myelin proteins, including MAG, P0 and MBP (Fig. 5D). In samples from Wt mice, there was a ~1.3–1.7-fold increase in the above mentioned myelin proteins, as determined by densitometric analysis, an effect that is statistically significant (p<0.05 for all the myelin proteins in Wt samples, n=3). In comparison, in samples from neuropathic mice, we detected a pronounced ~14-fold increase in the levels of MBP (p<0.01, n=3), ~4-fold increase in P0 (p<0.01, n=3) and ~3-fold increase in MAG (p<0.05, n=3) (Fig. 5D), subsequent to EC137 treatment. As expected, EC137 exposure is associated with a pronounced induction in HSP70 in both Wt and neuropathic samples. The blots shown are representative of at least three independent experiments for each condition and were reprobed with an antibody to GAPDH to monitor protein loading.
In nerves of C22 neuropathic mice, by 35S pulse-chase analysis we detected an accumulation of endo H-sensitive PMP22 and an increase in PMP22 levels, when we used a combination of antibodies against the mouse and the human protein (Fortun et al., 2006). Therefore, rather than comparing the overall levels of PMP22 between our Wt and neuropathic samples, we asked whether the subcellular trafficking of PMP22 is influenced by EC137 treatment (Fig. 6A). Accumulation of endo H-resistant PMP22 within SCs correlates with myelination and represents the long-lived membrane form of the protein (Pareek et al., 1997). The fraction of endo H-resistant PMP22 in DMSO-treated DRGs from Wt and C22 embryos is comparable to that seen in sciatic nerve lysates from 6-month old Wt and C22 mice, respectively (Fig. 6A) (also see Fortun et al., 2006), thus mimicking the in vivo situation closely. With EC137 treatment, the endo H-resistant fraction in Wt DRGs is slightly increased from 84 to 86% (p=0.12; n=3). In comparison, in DRGs from neuropathic mice the endo H-resistant pool of PMP22 is significantly improved from 56.03 ± 1.23 % to 73.97 ± 0.92 % (mean ± SEM, p<0.001; n=3). Compare the levels of endo H-resistant ~22 kDa (arrow) and endo H-sensitive 18 kDa forms (arrowhead)] (Fig. 6A). The slowed mobility of the endo H-resistant PMP22 in EC137-treated samples, as compared to DMSO controls, is consistent among independent samples and may reflect altered glycosylation and/or folding of the protein.
The intracellular retention of PMP22 in samples from C22 mice is associated with an accumulation of poly-ubiquitinated proteins and an impairment of proteasome activity (Fortun et al., 2006). To test if EC137 (50 nM) treatment impacts the accumulation of such slow-migrating poly-ubiquitinated proteins in SCs from C22 mice, cell lysates with or without compound treatment were analyzed with an ubiquitin antibody (Fig. 6B). In agreement with our initial studies (Fig. 3A), a 16 h treatment with EC137 is associated with a reduction in high molecular weight poly-ubiquitinated substrates in SCs from C22 mice, as well as Wt mice. This reduction in poly-ubiquitinated substrates is statistically significant in SCs from C22 mice (p<0.01; n=3) but not in SCs from Wt mice (p=0.29; n=3) (Fig. 6B). GAPDH is shown as a protein loading control.
Improvement in myelination by neuropathic samples could be mediated by an influence of EC137 on glial, as well as neuronal genes. Therefore, we tested purified Wt and C22 mouse SCs (Fig. 6C), and DRG neurons with or without glia (Fig. 6D), for their response to EC137 treatment. As shown on the Western blots for treatment of DRGs from Wt and C22 mice (Fig. 5D), EC137 enhances the steady-state levels of HSP70 and αB-crystallin in SCs from Wt and C22 mice, alike (Fig. 6C). In agreement with our previous studies (Ryan et al., 2002; Fortun et al., 2003) and also seen previously (Fig. 3A), the basal levels of HSP70 and αB-crystallin are elevated in neuropathic mouse SCs as compared to Wt (compare DMSO controls in Fig. 6C), likely as a response to accumulated poly-ubiquitinated substrates. The basal levels of HSP27 and HSP40 in DMSO-treated SCs from C22 mice are comparable to that of SCs from Wt mice and are enhanced in response to EC137 treatment. The steady-state expression of HSP90 (Fig. 6C), calnexin and Bip/Grp78 (data not shown) are largely unaffected by EC137. DRG explants from Wt mice containing SCs respond to EC137 by induction of HSP70, HSP27 and αB-crystallin after a 48 h treatment (50 nM), while DRG neurons without SCs show an attenuated reaction (Fig. 6D). The same experiment was also done for 24 h treatment with EC137 (50 nM) and essentially showed a similar pattern of induction (data not shown). This result indicates that EC137 primarily influences chaperone synthesis in peripheral glial cells, as compared to sensory neurons.
Discussion
Enhancement of the HS response by natural or synthetic compounds is of therapeutic interest for protein misfolding disorders (Westerheide and Morimoto, 2005). Hereditary neuropathies linked to the misexpression of PMP22 share characteristics with such disorders including the formation of cytosolic protein aggregates (Fortun et al., 2003; Fortun et al., 2006). Here we tested if enhancement of the chaperone pathway through inhibition of HSP90 would be beneficial for myelin formation by SCs from neuropathic mice with PMP22 misexpression. The chosen synthetic, small molecule HSP90 inhibitors offer a favorable approach as they exhibit low cellular toxicity, and induce sustained expression of HSPs (Dickey et al., 2005; Dickey et al., 2007). Our results indicate that non-myelinating and myelinating glial cells respond to EC137 by increased expression of chaperones, including HSP70, HSP27 and αB-crystallin. Significantly, the enhancement of chaperones is associated with a pronounced improvement in myelination in neuron-glia explant cultures from neuropathic mice, as compared to untreated controls. These results suggest that peripheral glial cells are amenable to pharmacologic modulation of the HS response and recommend further studies with these compounds.
While the precise molecular mechanism by which inhibition of HSP90 aids myelin formation by SCs from neuropathic mice is unclear, it likely involves the assistance of chaperones in the folding and processing of myelin proteins, including PMP22 (Fig. 7). Studies in cultured cells and neuropathic nerves indicate that PMP22 is prone to aggregation and accumulates in the cytoplasm of SCs when the proteasome is inhibited or the protein is misexpressed (Fig. 7A) (Notterpek et al., 1999; Fortun et al., 2003; Fortun et al., 2007). These intracellular PMP22 aggregates retain cytosolic chaperones and MBP, which alters protein homeostasis within SCs. The sustained enhancement of the available pool of chaperones by EC137 likely aids the correct folding of newly-synthesized PMP22 and other glial proteins, and promotes their trafficking to the plasma membrane (Fig. 7B). The observed increase in the endo H-resistant fraction of PMP22 in EC137 treated cultures (Fig. 6A) indeed supports a primary influence of this compound on protein folding. The treatment with EC137 also decreased the levels of poly-ubiquitinated proteins within SCs from C22 neuropathic mice (Fig. 6B), which may suggest an effect on protein degradation. Based on our current results, a potential role for chaperones in aiding the removal of misfolded PMP22 cannot be ruled out.
Figure 7. Working model: HSP90 inhibitor aids PMP22 processing and improves myelination in neuropathic samples.
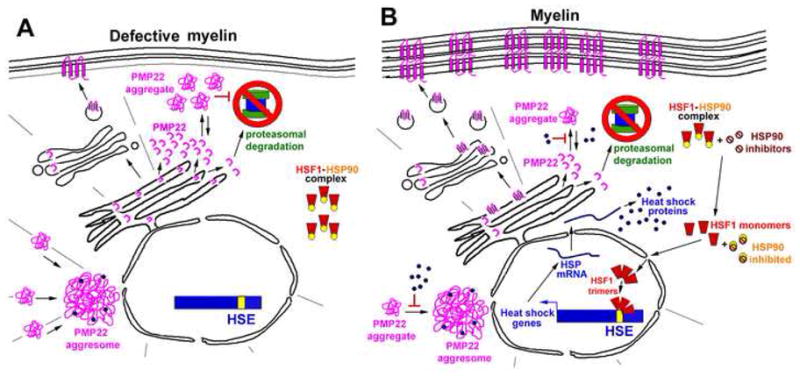
(A) In SCs from neuropathic mice, there is an accumulation of PMP22 in cytosolic aggregates, which is associated with an impairment of protein degradation by the proteasome. Only a small fraction of PMP22 is transported to the plasma membrane, which leads to defects in myelination. (B) Exposure of SCs to HSP90 inhibitors promotes the activation and nuclear translocation of HSF1. The induction of heat shock genes and the expression of HSPs prevent the aggregation and promote the correct folding and processing of PMP22, as well as other myelin proteins. Restoration of subcellular protein homeostasis improves myelin formation.
A protective role for chaperones in preventing the misfolding and subsequent aggregation of PMP22 is supported by our previous in vitro studies (Notterpek et al., 1999; Fortun et al., 2007). In normal non-myelinating rat SCs, under conditions of proteasome inhibition, over ninety percent of the cells form PMP22 aggregates. When these studies were performed in conjunction with HS preconditioning or GA treatment, the misfolding of PMP22 was significantly reduced (Fortun et al., 2007). The decrease in protein aggregate formation in this pharmacologic model was likely due to the enhancement of cytosolic molecular chaperones, which aid the processing of newly-synthesized PMP22 and/or refolding of small aggregates before the assembly of large inclusions. In the same assay, GA was more effective in preventing protein aggregate formation, as compared to HS (Fortun et al., 2007). GA binds to the ATP site on HSP90 and blocks its interaction with HSF1, and thus promotes HSF1 activation and the synthesis of HSPs (Prodromou et al., 1997; Zou et al., 1998). However, extended or high dose treatment with GA is associated with cellular toxicity which limits the potential therapeutic use of this compound (Miyata, 2005). EC137 a synthetic small molecule inhibitor of HSP90 used in this study has suitable pharmacokinetic profile (Figs. 2–4) for potential therapeutic use and enhances the levels of HSP70 for over 36 h in SCs, as compared to vehicle control (Fig. 3D). Intraperitoneal injection of EC102, a small synthetic HSP90 inhibitor, and EC72, a derivative of GA, in mouse models of tauopathy and experimental autoimmune encephalomyelitis, respectively, induced high levels of HSP70 with low toxicity and was associated with an amelioration of disease (Dello Russo et al., 2006; Dickey et al., 2007). It is yet to be determined if EC137 identified in this study could be administered intraperitoneally to neuropathic mice.
The modulation of the HSR in preventing the aggregation of cytosolic and nuclear disease-linked proteins has been studied extensively (Westerheide and Morimoto, 2005). For example, live cell imaging experiments show that HSP70 associates transiently with huntingtin aggregates, with association-dissociation kinetics identical to chaperone interactions with unfolded polypeptides (Kim et al., 2002). On the contrary, the protection observed in Drosophila models of Parkinson’s and polyglutamine expansion in response to overexpression of HSP70 is not accompanied by a reduction in the number of inclusions (Warrick et al., 1999; Auluck et al., 2002; Kazemi-Esfarjani and Benzer, 2002). A multidomain glycoprotein whose misfolding is associated with disease is cystic fibrosis transmembrane regulator (CFTR) (Amaral and Kunzelmann, 2007). CFTR and PMP22 share similarities in their high propensity for aggregation and they are both substrates for proteasomal degradation (Johnston et al., 1998; Notterpek et al., 1999). In cells stably transfected with Wt CFTR, the overexpression of HSP70 and its co-chaperone HSP40 was associated with an increased stability of the immature CFTR, but had no influence on the maturation of the protein (Farinha et al., 2002). Currently it is unknown if the beneficial effects of elevated HSPs on PMP22 processing are mediated by direct or indirect interactions between these proteins. The only chaperone so far identified to interact with PMP22 is calnexin (Dickson et al., 2002).
In general, a critical role for chaperones in myelination is supported by multiple studies, in distinct model systems. The association of HSP70 with MBP in an ATP-dependent manner in the normal human brain (Lund et al., 2006) implies that HSP70 is involved in the proper folding and trafficking (Hartl, 1996) of this cytosolic myelin protein. Indeed, constitutive expression of HSC70 appears to be essential for the correct expression of MBP during the differentiation of oligodendrocytes (Aquino et al., 1998). When HSC70 expression was shut-down the steady-state levels of MBP dramatically decreased (Aquino et al., 1998). In agreement, we observed elevated expression of MBP and significant increase in MBP-positive myelin internodes in response to EC137 treatment, as compared to control samples (Fig. 5). This functional improvement correlates with induction of HSP70 and HSP27 within SCs (Fig. 5D, 6C). In an independent study using the transgenic approach, the deletion of hsf1 resulted in a demyelinating phenotype, possibly due to defects in oligodendrocyte differentiation, or myelin synthesis and assembly (Homma et al., 2007). These results support our working model (Fig. 7), in which induction of HSPs via HSF1 activation in myelinating neuropathic SCs enhances the cytosolic chaperone pool, reduces the accumulation of poly-ubiquitinated substrates, and aids the trafficking of PMP22.
Studies in CMT1A pedigrees and genetically engineered neuropathic models underscore the importance of adequate levels of correctly-folded PMP22 for myelin formation and stability (Kuhlenbaumer et al., 2002; Robertson et al., 2002). In C22 mice, the secretory pathway appears to be overwhelmed as we detected an accumulation of newly-synthesized PMP22 within the cytosol and a decrease in the endo H-resistant PMP22 at the plasma membrane (Fortun et al., 2006). In agreement, studies of sural nerve biopsies from CMT1A patients with PMP22 gene duplication or point mutations show PMP22-like immunoreactivity in the myelin sheath, as well as within the SC cytoplasm (Nishimura et al., 1996; Hanemann et al., 2000). However, the PMP22-like myelin staining is thin (Nishimura et al., 1996) and there is a reduction in the number of PMP22-positive myelinated fibres (Hanemann et al., 1994), implying that only a small fraction of PMP22 is incorporated into myelin. Previously published therapeutic approaches to correct the myelin defects in PMP22-associated neuropathies include the use of progesterone antagonists and ascorbic acid (Sereda et al., 2003; Passage et al., 2004). In both of these studies, the neuropathic phenotype was substantially ameliorated by the interventions, and ascorbic acid is now in clinical trial for CMT 1A (Pareyson et al., 2006). Our current findings, while posing to be promising in culture, await further testing in neuropathic mice where optimal bioavailability of these compounds in peripheral nerves can be established. Based on data presented here, the tested small molecule inhibitors of HSP90, particularly EC137, could potentially offer a new approach for the treatment of demyelinating neuropathies.
Acknowledgments
The authors wish to thank Doug Smith for assistance with confocal microscopy. These studies in part were supported by the National Muscular Dystrophy Association, a Pilot Project Award by the NMSS and the NIH-NINDS (LN).
Abbreviations
- PMP22
peripheral myelin protein 22
- Schwann cell
SC
- Wt
wild type
- HSP
heat shock protein
- DMSO
dimethylsulfoxide
- GA
geldanamycin
- HS
heat shock
- CMT1A
Charcot–Marie–Tooth disease type 1A
- P
postnatal
- DRG
dorsal root ganglion
- HSF1
heat shock factor 1
- MBP
myelin basic protein
- MAG
myelin-associated glycoprotein
- P0
protein zero
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amaral MD, Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol Sci. 2007;28:334–341. doi: 10.1016/j.tips.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Amici SA, Dunn WA, Jr, Notterpek L. Developmental abnormalities in the nerves of peripheral myelin protein 22-deficient mice. J Neurosci Res. 2007;85:238–249. doi: 10.1002/jnr.21118. [DOI] [PubMed] [Google Scholar]
- Aquino DA, Peng D, Lopez C, Farooq M. The constitutive heat shock protein-70 is required for optimal expression of myelin basic protein during differentiation of oligodendrocytes. Neurochem Res. 1998;23:413–420. doi: 10.1023/a:1022473904335. [DOI] [PubMed] [Google Scholar]
- Archelos JJ, Roggenbuck K, Schneider-Schaulies J, Toyka KV, Hartung HP. Detection and quantification of antibodies to the extracellular domain of P0 during experimental allergic neuritis. J Neurol Sci. 1993;117:197–205. doi: 10.1016/0022-510x(93)90174-w. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Cosgaya JM, Chan JR, Shooter EM. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science. 2002;298:1245–1248. doi: 10.1126/science.1076595. [DOI] [PubMed] [Google Scholar]
- Dello Russo C, Polak PE, Mercado PR, Spagnolo A, Sharp A, Murphy P, Kamal A, Burrows FJ, Fritz LC, Feinstein DL. The heat-shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin suppresses glial inflammatory responses and ameliorates experimental autoimmune encephalomyelitis. J Neurochem. 2006;99:1351–1362. doi: 10.1111/j.1471-4159.2006.04221.x. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Eriksen J, Kamal A, Burrows F, Kasibhatla S, Eckman CB, Hutton M, Petrucelli L. Development of a high throughput drug screening assay for the detection of changes in tau levels -- proof of concept with HSP90 inhibitors. Curr Alzheimer Res. 2005;2:231–238. doi: 10.2174/1567205053585927. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Dunmore J, Lu B, Wang JW, Lee WC, Kamal A, Burrows F, Eckman C, Hutton M, Petrucelli L. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. Faseb J. 2006;20:753–755. doi: 10.1096/fj.05-5343fje. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson KM, Bergeron JJ, Shames I, Colby J, Nguyen DT, Chevet E, Thomas DY, Snipes GJ. Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for “gain-of-function” ER diseases. Proc Natl Acad Sci U S A. 2002;99:9852–9857. doi: 10.1073/pnas.152621799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S, Milner TA, Giancotti F, Salzer JL. Axonal regulation of Schwann cell integrin expression suggests a role for alpha 6 beta 4 in myelination. J Cell Biol. 1993;123:1223–1236. doi: 10.1083/jcb.123.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, Nogueira P, Mendes F, Penque D, Amaral MD. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem J. 2002;366:797–806. doi: 10.1042/BJ20011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J, Go JC, Li J, Amici SA, Dunn WA, Jr, Notterpek L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol Dis. 2006;22:153–164. doi: 10.1016/j.nbd.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Fortun J, Verrier JD, Go JC, Madorsky I, Dunn WA, Notterpek L. The formation of peripheral myelin protein 22 aggregates is hindered by the enhancement of autophagy and expression of cytoplasmic chaperones. Neurobiol Dis. 2007;25:252–265. doi: 10.1016/j.nbd.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanemann CO, D’Urso D, Gabreels-Festen AA, Muller HW. Mutation-dependent alteration in cellular distribution of peripheral myelin protein 22 in nerve biopsies from Charcot-Marie-Tooth type 1A. Brain. 2000;123 (Pt 5):1001–1006. doi: 10.1093/brain/123.5.1001. [DOI] [PubMed] [Google Scholar]
- Hanemann CO, Stoll G, D’Urso D, Fricke W, Martin JJ, Van Broeckhoven C, Mancardi GL, Bartke I, Muller HW. Peripheral myelin protein-22 expression in Charcot-Marie-Tooth disease type 1a sural nerve biopsies. J Neurosci Res. 1994;37:654–659. doi: 10.1002/jnr.490370513. [DOI] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Madorsky I, Paiva M, Siler-Marsiglio KI. Vitamin E amelioration of ethanol neurotoxicity involves modulation of apoptotis-related protein levels in neonatal rat cerebellar granule cells. Brain Res Dev Brain Res. 2004;150:117–124. doi: 10.1016/j.devbrainres.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Herbst M, Wanker EE. Small molecule inducers of heat-shock response reduce polyQ-mediated huntingtin aggregation. A possible therapeutic strategy. Neurodegener Dis. 2007;4:254–260. doi: 10.1159/000101849. [DOI] [PubMed] [Google Scholar]
- Homma S, Jin X, Wang G, Tu N, Min J, Yanasak N, Mivechi NF. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J Neurosci. 2007;27:7974–7986. doi: 10.1523/JNEUROSCI.0006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellissier JF, Fontes M. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet. 1996;5:563–569. doi: 10.1093/hmg/5.5.563. [DOI] [PubMed] [Google Scholar]
- Huxley C, Passage E, Robertson AM, Youl B, Huston S, Manson A, Saberan-Djoniedi D, Figarella-Branger D, Pellissier JF, Thomas PK, Fontes M. Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum Mol Genet. 1998;7:449–458. doi: 10.1093/hmg/7.3.449. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi-Esfarjani P, Benzer S. Suppression of polyglutamine toxicity by a Drosophila homolog of myeloid leukemia factor 1. Hum Mol Genet. 2002;11:2657–2672. doi: 10.1093/hmg/11.21.2657. [DOI] [PubMed] [Google Scholar]
- Kim HR, Kang HS, Kim HD. Geldanamycin induces heat shock protein expression through activation of HSF1 in K562 erythroleukemic cells. IUBMB Life. 1999;48:429–433. doi: 10.1080/713803536. [DOI] [PubMed] [Google Scholar]
- Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002;4:826–831. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- Kincaid MM, Cooper AA. ERADicate ER stress or die trying. Antioxid Redox Signal. 2007;9:2373–2387. doi: 10.1089/ars.2007.1817. [DOI] [PubMed] [Google Scholar]
- Kuhlenbaumer G, Young P, Hunermund G, Ringelstein B, Stogbauer F. Clinical features and molecular genetics of hereditary peripheral neuropathies. J Neurol. 2002;249:1629–1650. doi: 10.1007/s00415-002-0946-3. [DOI] [PubMed] [Google Scholar]
- Lund BT, Chakryan Y, Ashikian N, Mnatsakanyan L, Bevan CJ, Aguilera R, Gallaher T, Jakowec MW. Association of MBP peptides with Hsp70 in normal appearing human white matter. J Neurol Sci. 2006;249:122–134. doi: 10.1016/j.jns.2006.05.066. [DOI] [PubMed] [Google Scholar]
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, Chakravarti A, Patel PI. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- Magyar JP, Martini R, Ruelicke T, Aguzzi A, Adlkofer K, Dembic Z, Zielasek J, Toyka KV, Suter U. Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J Neurosci. 1996;16:5351–5360. doi: 10.1523/JNEUROSCI.16-17-05351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean PJ, Klucken J, Shin Y, Hyman BT. Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem Biophys Res Commun. 2004;321:665–669. doi: 10.1016/j.bbrc.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Miyata Y. Hsp90 inhibitor geldanamycin and its derivatives as novel cancer chemotherapeutic agents. Curr Pharm Des. 2005;11:1131–1138. doi: 10.2174/1381612053507585. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yoshikawa H, Fujimura H, Sakoda S, Yanagihara T. Accumulation of peripheral myelin protein 22 in onion bulbs and Schwann cells of biopsied nerves from patients with Charcot-Marie-Tooth disease type 1A. Acta Neuropathol. 1996;92:454–460. doi: 10.1007/s004010050546. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Paskevich PA, Sihag RK, Thayer CY. Phosphorylation on carboxyl terminus domains of neurofilament proteins in retinal ganglion cell neurons in vivo: influences on regional neurofilament accumulation, interneurofilament spacing, and axon caliber. J Cell Biol. 1994;126:1031–1046. doi: 10.1083/jcb.126.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterpek L, Snipes GJ, Shooter EM. Temporal expression pattern of peripheral myelin protein 22 during in vivo and in vitro myelination. Glia. 1999;25:358–369. [PubMed] [Google Scholar]
- Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberte J, Iacampo S, Suter U, Shooter EM, Murphy RA. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci. 1997;17:7754–7762. doi: 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D, Schenone A, Fabrizi GM, Santoro L, Padua L, Quattrone A, Vita G, Gemignani F, Visioli F, Solari A. A multicenter, randomized, double-blind, placebo-controlled trial of long-term ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL): the study protocol [EudraCT no.: 2006–000032–27] Pharmacol Res. 2006;54:436–441. doi: 10.1016/j.phrs.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, Robaglia-Schlupp A, Pellissier JF, Fontes M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10:396–401. doi: 10.1038/nm1023. [DOI] [PubMed] [Google Scholar]
- Perea J, Robertson A, Tolmachova T, Muddle J, King RH, Ponsford S, Thomas PK, Huxley C. Induced myelination and demyelination in a conditional mouse model of Charcot-Marie-Tooth disease type 1A. Hum Mol Genet. 2001;10:1007–1018. doi: 10.1093/hmg/10.10.1007. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Robertson AM, Huxley C, King RH, Thomas PK. Development of early postnatal peripheral nerve abnormalities in Trembler-J and PMP22 transgenic mice. J Anat. 1999;195 (Pt 3):331–339. doi: 10.1046/j.1469-7580.1999.19530331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AM, Perea J, McGuigan A, King RH, Muddle JR, Gabreels-Festen AA, Thomas PK, Huxley C. Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A. J Anat. 2002;200:377–390. doi: 10.1046/j.1469-7580.2002.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MC, Shooter EM, Notterpek L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis. 2002;10:109–118. doi: 10.1006/nbdi.2002.0500. [DOI] [PubMed] [Google Scholar]
- Sanders CR, Ismail-Beigi F, McEnery MW. Mutations of peripheral myelin protein 22 result in defective trafficking through mechanisms which may be common to diseases involving tetraspan membrane proteins. Biochemistry. 2001;40:9453–9459. doi: 10.1021/bi010894f. [DOI] [PubMed] [Google Scholar]
- Sereda M, Griffiths I, Puhlhofer A, Stewart H, Rossner MJ, Zimmerman F, Magyar JP, Schneider A, Hund E, Meinck HM, Suter U, Nave KA. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- Sereda MW, Meyer zu Horste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A) Nat Med. 2003;9:1533–1537. doi: 10.1038/nm957. [DOI] [PubMed] [Google Scholar]
- Shy ME, Balsamo J, Lilien J, Kamholz J. A molecular basis for hereditary motor and sensory neuropathy disorders. Curr Neurol Neurosci Rep. 2001;1:77–88. doi: 10.1007/s11910-001-0079-6. [DOI] [PubMed] [Google Scholar]
- Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem. 2005;280:33097–33100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]