

Abstract
Several new 2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides 12a–s and 17t–v were synthesized by stirring in pyridine the (E)-3-(2-R1-3-R2-4-R3-phenyl)acrylic acid chlorides 11c–k and 11t–v with the appropriate anthranilamide derivatives 10a–c or the 5-iodoanthranilic acid 13. Some of the synthesized compounds were evaluated for their in vitro antiproliferative activity against the full NCI tumor cell line panel derived from nine clinically isolated cancer types (leukemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate and breast). COMPARE analysis, effects on tubulin polymerization in cells and with purified tubulin, and effects on cell cycle distribution for 17t, the most active of the series, indicate that these new antiproliferative compounds act as antitubulin agents.
Keywords: 2-{[2E]-3-phenylprop-2-enoylamino}, benzamides, Antimitotic agents, Cytotoxic activity
1. Introduction
During a screening program to find antiproliferative compounds in our laboratory’s collection of small organic molecules, the 2-cinnamamido-5-iodobenzamide 1 was found at 10 μM to inhibit proliferation of the leukemic cell line K562 by 74%.
Compound 1 belongs to cinnamoyl anthranilates, which represent a class of biologically active substances of great importance in medicinal chemistry. Tranilast (Rizaben®) 2 is an antiallergic drug approved in 1982 for use in Japan and South Korea for bronchial asthma and was also investigated for use as an antiproliferative agent on drug-eluting stents. Its derivative (E)-2-(3-(3,4-dime-thoxyphenyl)acrylamido)benzamide 3 [1] is another member of this class of compounds and is more potent than the lead compound, Tranilast (Fig. 1).
Fig. 1.
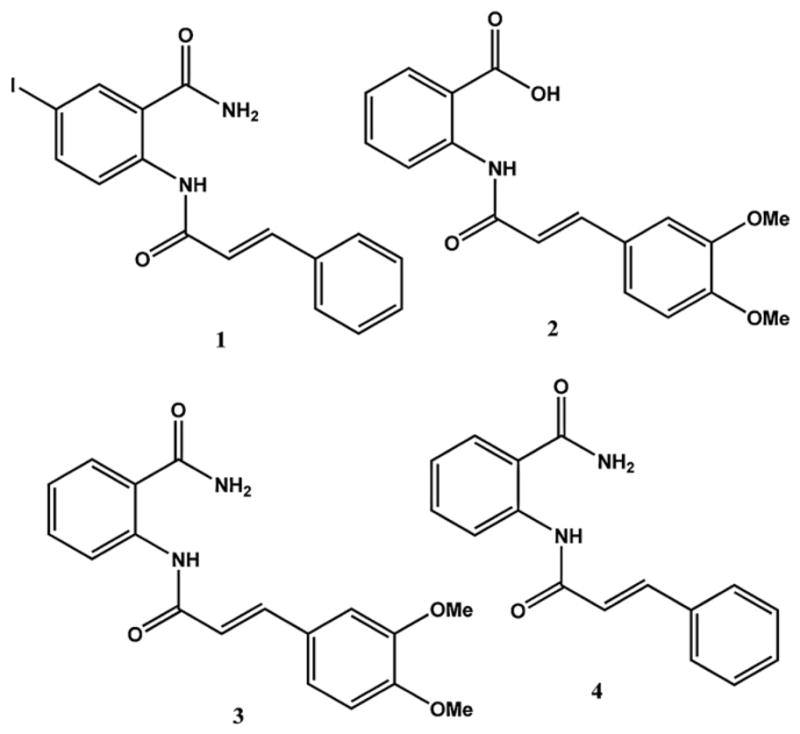
Structure of cinnamoyl anthranilates.
Other biological activities possessed by this class of compounds are antifibrotic and antinflammatory properties [2,3] and inhibition of cornea pterygium progression and blood vessel development [4,5]. Finally, cinnamoyl anthranilates are useful for prevention and treatment of glomerular diseases [6] and diseases caused by the excessive proliferation of vascular intimal cells [7].
However, despite their wide range of biological activities, a review of the literature revealed that no anticancer activity had been described forcinnamoyl anthranilates. Thus, theactivity ofcompound 1 as an inhibitor of K562 proliferation led us to explore the potential of this class of compounds as anticancer agents, and we therefore synthesized a series of novel cinnamoyl anthranilates and screened the compounds for antileukemic activity. Our work enabled us to perform an initial study of their structure-activity relationships and to determine their mechanism of action. Compounds 12a–s and 17t–v were initially tested in vitro for their antileukemic activity against the K562 (human chronic myelogenous leukemia) cell line (Table 1). Among these, 12a–c and 17t,u, which showed the best anti-proliferative activity, were selected by the National Cancer Institute (NCI) for evaluation in the 60 human tumor cell line screen of the NCI.
Table 1.
Percent growth inhibition obtained with the K562 cell line with compounds at 10 μM and IC50 values (μM) with the same cells.
| Com. | % inhibition | IC50 (μM) |
|---|---|---|
| 4 | 14.2 | >10 |
| 12a | 65.4 | 5.5 |
| 12b | 64.0 | 2.5 |
| 12c | 62.4 | 5.0 |
| 12d | 58.0 | 7.4 |
| 12e | 28.2 | >10 |
| 12f | 31.0 | >10 |
| 12g | 49.0 | 10 |
| 12h | 27.0 | >10 |
| 12i | 52.2 | 9.5 |
| 12j | 37.8 | >10 |
| 12k | 64.0 | 6.3 |
| 12l | 59.0 | 8.1 |
| 12m | 25.5 | >10 |
| 12n | 22.0 | >10 |
| 12o | 46.2 | >10 |
| 12p | 45.9 | >10 |
| 12q | 43.2 | >10 |
| 12r | 43.8 | >10 |
| 12s | 45.9 | >10 |
| 17t | 74.5 | 0.57 |
| 17u | 74.1 | 1.2 |
| 17v | 26.3 | >10 |
| 18 | 63.6 | 0.02 |
2. Results and discussion
2.1. Chemistry
A series of 2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides 12a–s and 17t–v was synthesized by stirring the (E)-3-(2-R1-3-R2-4-R3-phenyl)acrylic acid chlorides 11c–k, 11t–v and the appropriate anthranilamide derivatives 10a–c or the 5-iodoanthranilic acid 13 as described in Schemes 1 and 2.
Scheme 1.

Reagents and conditions: (a) SOCl2, reflux, 5 h; (b) acetonitrile, aqueous ammonia (25%), reflux, 8 h; (c) aqueous ammonia (25%), stirring, rt, 1 h; (d) SnCl2, HCl (35%), stirring, 0–5 °C, (24 h); (e) pyridine, stirring, rt, 24 h.
Scheme 2.

Reagents and conditions: (a) SOCl2, reflux, 5 h; (b) pyridine, stirring, 0–5 °C, 24 h; (c) aqueous NaPO3 (0.01 M), reflux, 20 h; (d) ethyl chloroformate, acetyl chloride reflux, (15 min + 30 min); (e) dioxane, acqueous ammonia (25%), reflux, 3 h.
Crude (E)-3-(2-R1-3-R2-4-R3-phenyl)acrylic acids 11c–k, 11t–v were obtained by refluxing the appropriately substituted acrylic acid 8c–k, 8t–v with thionyl chloride. The 5-methylanthranilamide 10a was obtained by reduction of 2-nitro-5-methyl-benzamideamide 7a as shown in Scheme 1. Compound 7 was obtained by reaction of the acid 5 with thionyl chloride to afford 6, followed by treatment of 6 with aqueous ammonia.
The anthranilamide derivatives 10b,c were obtained by stirring the appropriate 2H-3,1-benzoxazine-2,4(1H)-dione 9b,c in aqueous ammonia solution (Scheme 1) [8].
A different synthetic method was used to obtain the 2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides 17t–v (Scheme 2). The starting materials, 2-cinnamamido-5-iodobenzoic acids 15t–v, were obtained by stirring the 5-iodoanthranilic acid 13 and the cinnamoyl chlorides 11t–v. The reaction gave a mixture of (E)-6-iodo-2-styryl-4H-benzo[d] [1,3]oxazin-4-ones 14t–u and 2-cinnamamido-5-iodobenzoic acids 15t–u, and the mixtures were treated with aqueous Na3PO4 [9] to give the corresponding acids 15t [10], 15u,v as the only products. By treating the acids 15t–v with ethyl chloroformate, the 2H-3,1-benzoxazine-2,4-diones 16t–v were obtained [11], which, in turn, converted to the 2-{[2E]-3-phenyl-prop-2-enoylamino}benzamides 17t–v by refluxing them in an aqueous ammonia solution [8].
The structures of the new compounds were determined by analytical and spectroscopic measurements. In particular, 1H NMR spectra of compounds 12a–s and 17v–t were consistent with an E-olefinic structure. They showed signals attributable to the β-olefinic protons at 6.66–6.97 δ with coupling constants of 16.6–14.7 Hz, as required for E-structures [12], while the α-olefinic hydrogens were found along with aromatic multiplets. Moreover, their 1H NMR spectra showed both the NH and NH2 amidic signals; the cinnamamido NH proton appeared as a singlet at 11.77–11.92 δ, while, according to the literature [13], the presence of an intramolecular hydrogen bond renders the benzamido NH2 protons diastereotopic. Ha is easily exchangeable with D2O and appeared as a singlet at 8.24–8.44 δ. Hb was found at a lower field along with the aromatic multiplets.
2.2. Biology
Synthesized 2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides 12a–s and 17t–v were initially tested in vitro for their antileukemic activity against the K562 (human chronic myelogenous leukemia) cell line. Colchicine 18, whose antileukemic activity is well known, and the 2-cinnamamidobenzamide 4 [13] (Fig. 1), were used as reference compounds. The percent growth inhibition at a screening concentration of 10 μM and the IC50 values for compounds that exhibited at least 50% of growth inhibition at 10 μM are shown in Table 1. Compounds 12a–s and 17t–v had inhibitory activity against the K562 cells ranging from 22.0 to 74.5% at 10 μM, with 12a–d, 12k,l, and 17t,u (IC50 0.57–8.1 μM) being the most active compounds. Our data (Table 1) showed positive effects following substitution with halogens at the 5 position of the benzamido moiety, especially when the substituent was iodine (compounds 17t and 17u). As far as structure-activity relationships are concerned, it seems that the introduction of a substituent in both the benzamido and styryl moieties are favorable for inhibition of K562 cell growth relative to the unsubstituted 2-cinnamamido-benzamide 4 (Table 1). However, the best activity was obtained when the substitutions were present only on the benzamido moiety (compounds 12a–d, 17t). Compounds substituted in both the benzamido and styryl moieties (compounds 12e–s, 17v) were less active, even if antiproliferative activity was maintained in the ortho-styryl derivatives (12d,i,l,u).
Compounds 12a, 12b, 12c,17t and 17u were evaluated by the NCI for testing against a panel of approximately 60 human cell lines derived from seven clinically isolated cancer types (lung, colon, melanoma, renal, ovarian, brain, and leukemia) according to the NCI standard protocol [14] (Table 2). The data summarized in Table 2 showed that 12a, 12b, 12c, 17t and 17u caused 50% growth inhibition at micromolar (12a, 12b, 12c) and submicromolar concentrations (17t, 17u) against every type of tumor cell line investigated.
Table 2.
Results of multi-dose growth inhibition assay (GI50, μM).
| Cell line | 12a | 12b | 12c | 17t | 17u | |
|---|---|---|---|---|---|---|
| Leukemia | CCRF-CEM | 2.36 | 2.96 | 0.429 | 0.341 | 0.484 |
| HL-60 (TB) | 1.77 | 1.42 | 0.346 | 0.0542 | 0.165 | |
| K-562 | 1.11 | 0.845 | 0.415 | 0.920 | 0.370 | |
| MOLT-4 | 3.56 | 3.95 | 1.86 | 0.202 | 1.42 | |
| RPMI-8226 | nt | 2.79 | 1.03 | 0.347 | 0.772 | |
| Non-small cell lung cancer | A549/ATCC | 5.81 | 3.77 | 1.02 | 2.65 | 1.87 |
| EKVX | 5.18 | 2.83 | 2.41 | 0.734 | 1.00 | |
| HOP-62 | 7.74 | 3.15 | 1.51 | 0.800 | 2.30 | |
| HOP-92 | 11.4 | 2.87 | 1.96 | 21.2 | 13.2 | |
| NCI-H226 | 7.28 | 3.00 | 2.61 | 1.09 | 1.14 | |
| NCI-H23 | 3.38 | 3.37 | 1.09 | 0.840 | 1.39 | |
| NCI-H460 | 3.59 | 1.76 | 0.662 | 0.453 | 0.479 | |
| NCI-H522 | 2.83 | 2.08 | nt | 0.303 | 0.459 | |
| Colon cancer | COLO 205 | 1.84 | 1.68 | 0.917 | 0.343 | 0.797 |
| HCC-2998 | 6.45 | 0.487 | nt | 1.25 | 0.984 | |
| HCT-116 | 3.20 | 1.98 | 0.663 | 0.430 | 0.509 | |
| HCT-15 | 3.77 | 2.13 | 1.04 | 0.586 | 0.987 | |
| HT29 | 2.31 | 1.94 | 0.527 | 0.367 | 0.376 | |
| KM12 | 2.94 | 1.69 | 0.739 | 0.499 | 0.533 | |
| SW-620 | 3.30 | 1.99 | 0.705 | 0.541 | 0.605 | |
| CNS cancer | SF-268 | 10.9 | 2.90 | 1.83 | 2.31 | 0.778 |
| SF-295 | 3.14 | 1.40 | 0.625 | 0.350 | 0.415 | |
| SF539 | 2.05 | 2.05 | 0.819 | 0.445 | 0.444 | |
| SNB-19 | 5.69 | 3.18 | 2.00 | 0.586 | 1.41 | |
| SNB75 | 3.18 | 1.84 | 2.07 | 0.245 | 1.22 | |
| U251 | 4.13 | 2.14 | 0.887 | 0.501 | 0.663 | |
| Melanoma | LOX IMVI | 7.65 | 4.33 | 3.27 | 3.75 | 4.21 |
| MALME-3M | 7.59 | 2.68 | 1.21 | 1.60 | nt | |
| M14 | 2.35 | 1.71 | 0.482 | 0.429 | 0.698 | |
| MDA-MB-435 | 0.660 | 0.492 | 0.231 | nt | 0.234 | |
| SK-MEL-2 | 4.75 | 2.75 | nt | 1.40 | 0.678 | |
| SK-MEL-28 | 5.24 | 2.88 | 0.839 | 2.51 | nt | |
| SK-MEL-5 | 1.01 | 0.373 | 0.435 | 0.383 | 0.367 | |
| UACC-257 | 17.0 | 15.1 | nt | 14.3 | 38.7 | |
| UACC-62 | 3.77 | 2.52 | 0.782 | 0.632 | 0.388 | |
| Ovarian cancer | IGROV1 | 4.37 | 3.29 | 1023 | 0.917 | 0.352 |
| OVCAR-3 | 2.18 | 1.30 | 0.349 | 0.353 | 0.294 | |
| OVCAR-4 | 11.4 | 2.77 | nt | 1.84 | 2.37 | |
| OVCAR-5 | 8.20 | 2.74 | 2.92 | 3.52 | 4.02 | |
| OVCAR-8 | 7.49 | 3.89 | nt | 1.06 | 56.0 | |
| NCI/ADR-RES | 2.79 | 2.24 | 0.400 | nt | 0.719 | |
| SK-OV-3 | 2.79 | 2.30 | 1.41 | 0.744 | 0.539 | |
| Renal cancer | 786–0 | 4.00 | 2.32 | 1.76 | 1.43 | 0.937 |
| A498 | 5.56 | 3.61 | 1.70 | 2.70 | 0.880 | |
| ACHN | 13.5 | 4.75 | 3.36 | 3.70 | 9.86 | |
| CAKI-1 | 8.92 | 0.263 | 1.34 | 0.648 | 0.637 | |
| RXF 393 | nt | 1.71 | 0.444 | 3.02 | 0.425 | |
| SN12C | 9.63 | 3.96 | 2.96 | 0.635 | 1.55 | |
| TK-10 | 25.8 | 5.51 | 2.67 | 3.15 | 26.7 | |
| UO-31 | 9.62 | 3.57 | 1.85 | 3.53 | 1.63 | |
| Prostate cancer | PC-3 | 3.97 | 3.15 | 0.684 | 0.628 | 0.832 |
| DU-145 | 3.66 | 2.16 | 0.935 | 0.477 | 1.28 | |
| Breast cancer | MCF7 | 3.48 | 1.74 | 0.569 | 0.450 | 0.401 |
| MDA-MB-231 | 7.26 | 2.48 | 2.19 | 0.596 | 1.41 | |
| HS 578T | 1.94 | 2.04 | 0.624 | 0.375 | 0.362 | |
| BT-549 | 4.88 | 3.19 | 1.41 | 9.47 | 0.816 | |
| T-47D | 4.20 | 3.36 | 1.57 | 0.857 | 19.9 | |
| MDA-MB-468 | 3.12 | 1.17 | 0.279 | nt | 0.377 |
nt = not tested.
GI50 = Growth inhibition of 50%; [(Ti−Tz)/(C−Tz)] × 100 = 50 where Tz = absorbance at t = 0, Ti = absorbance at t = 48 h, C = absorbance of control at t = 48 h.
Moreover, a mean graph midpoint (MG_MID) is calculated for the GI50, TGI and LC50 parameters, giving an average activity parameter for all the cell lines. For the calculation of the MG_MID, insensitive cell lines are included and assigned as their values the highest concentration tested. Considering the MG_MID values (Table 3), the most active compound of the series was derivative 17t, at both the GI50 and TGI levels, followed by 17u and 12c.
Table 3.
Overview of the results of the in vitro antitumor screening for compounds 12a–c and 17t,u.a
| Comp | no. Studiede | pGI50b |
pTGIc |
pLC50d |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| no. giving positive resultse | range | MG_MIDf | no. giving positive resultse | range | MG_MIDf | no. giving positive resultse | range | MG_MIDf | ||
| 12a | 58 | 58 | 6.18–4.59 | 5.36 | 30 | 5.53–4.00 | 4.31 | 2 | 4.44–4.00 | 4.01 |
| 12b | 58 | 58 | 6.58–4.82 | 5.65 | 29 | 6.06–4.00 | 4.53 | 7 | 5.19–4.00 | 4.06 |
| 12c | 52 | 52 | 6.64–5.47 | 5.99 | 23 | 6.18–4.00 | 4.58 | 10 | 5.02–4.00 | 4.07 |
| 17t | 59 | 59 | 7.27–4.67 | 6.05 | 44 | 6.41–4.00 | 4.71 | 17 | 4.94–4.00 | 4.10 |
| 17u | 57 | 57 | 6.78–4.41 | 5.97 | 32 | 6.17–4.00 | 4.62 | 8 | 4.90–4.00 | 4.05 |
Data obtained from the NCI’s in vitro disease-oriented human tumor cells screen.
pGI50 is the –log of the molar concentration that inhibits 50% net cell growth.
pTGI is the –Log of the molar concentration giving total growth inhibition.
pLC50 is the –Log of the molar concentration leading to 50% net cell death.
Refers to the number of cell lines.
MG_MID = mean graph midpoint = arithmetical mean value for all tested cancer cell lines. If the indicated effect was not attainable within the used concentration interval, the highest tested concentration was used for the calculation.
Compound 17t was selected by the NCI for evaluation in its vivo toxicity assay, with the finding that the compound was nontoxic at a dose of 400 mg/kg in nontumored mice. Compound 17t was also selected by the NCI for testing in the hollow fiber assay, a preliminary in vivo screening tool, but the compound was not sufficiently active for evaluation in xenograft models.
To predict the probable mechanism of action, the NCI’s bio-informatic tool COMPARE analysis [15] was performed for the most active compound 17t. When tested as seed against the NCI “standard agents” Database, the compound showed Pearson Correlation Coefficients (PCC) of 0.492 and 0.476 at the GI50 level and higher values, 0.677, 0.600, 0.597, at the TGI level. In all cases the highest PCC’s were with compounds NSC 332598 (rhizoxin), NSC 125973 (paclitaxel), NSC 49842 (vinblastine sulfate) and NSC 153858 (maytansine), which are all antimitotic agents directed against tubulin.
To verify the prediction of the COMPARE algorithm, the effects of 17t on cell cycle distribution as determined by flow cytometry and on tubulin polymerization were evaluated.
As shown in Fig. 2, 17t caused a dose dependant increase of K562 cells in the G2-M phase of the cell cycle and a decrease of cells in G0–G1 after a 24 h treatment. This is the typical flow cytometric cell cycle distribution observed with drugs targeting tubulin (see panel b in Fig. 2), which is the main component of the mitotic spindle.
Fig. 2.

Effects of compound 17t on DNA content/cell following treatment of K562 cells for 24 h. The cells were cultured without compound (control, a), with an antimitotic drug used as internal standard (60 nM taxol (b)) and with 1 μM (c), 2 μM (d), or 4 μM (e) 17t. Cell cycle distribution was analyzed by the standard propidium iodide procedure as described in Materials and Methods. Sub-G0–G1 (A), G0–G1, S, and G2-M cells are indicated in (a).
After 48 h 17t caused extensive apoptosis in K562 cells, with an AC50 (concentration inducing 50% apoptosis) of 3.7 μM (Fig. 3). Apoptosis in this experiment was measured as described in the Experimental Section.
Fig. 3.
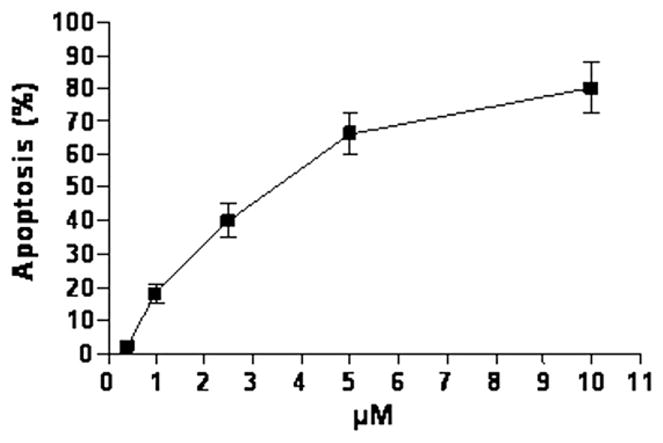
Percentage of apoptosis induced by compound 17t in K562 cells. Cells were cultured with different concentrations of 17t. Apoptosis was evaluated after 48 h of treatment as described in the experimental section. Bars: ±SE.
To confirm the ability of 17t to act on microtubules, the percentage of cells blocked in mitosis (M) was morphologically determined after staining cells with ethidium bromide and acridine orange. The percentage of K562 cells blocked in the M phase after 24 h of exposure to 1 μM 17t was 35 ± 6 (Fig. 4A, untreated K562 cell as control <1%). Of interest, concentrations of 17t higher than 2 μM induced morphologic alterations in almost all treated cells, suggesting an interaction between 17t and the cytoskeleton of K562 cells (Fig. 4C and D).
Fig. 4.
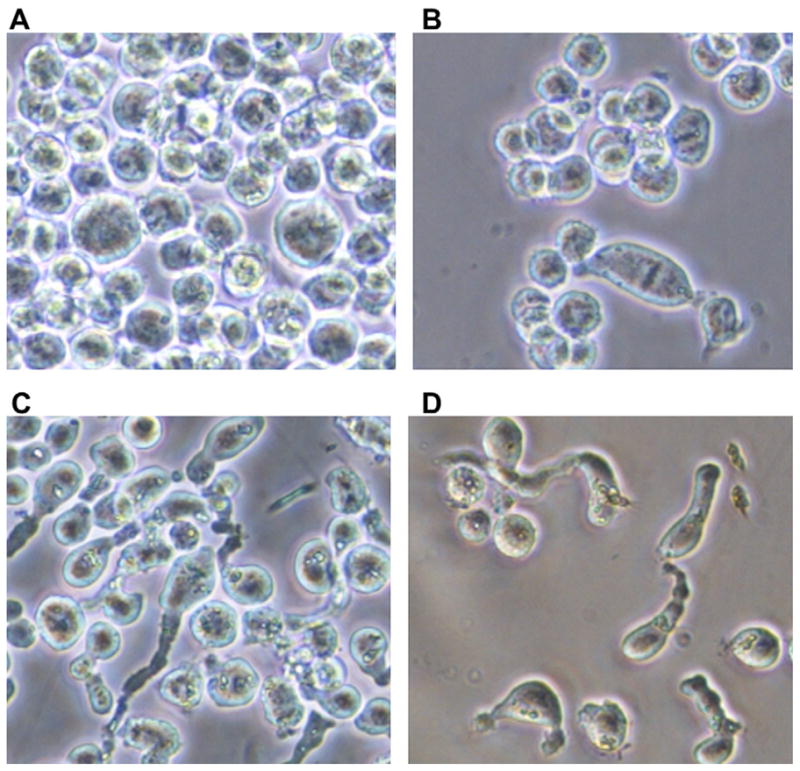
Morphology of K562 cells after 24 h exposure to 2 μM (B), 5 μM (C) and 10 μM (D) compound 17t. (A) Untreated K562 cells (control). Living cells in culture medium were observed by using a phase contrast invertoscope (40× magnification).
To examine the effects of 17t on microtubules (tubulin) and microfilaments (actin), PtK2 cells were examined by direct immunofluorescence. These cells were selected for this examination since their flattened morphology yields high quality images of cytoskeletal elements, particularly the microtubule and microfilament networks. As shown in Fig. 5, the microtubules disappeared while the microfilaments persisted with micromolar 17t. In addition, we found that 17t partially inhibited the polymerization of purified tubulin, but this effect was relatively weak, as shown in Fig. 6, with a comparison to the much stronger inhibition observed with colchicine 18. With 17t, we observed a concentration dependent inhibition of the rate of microtubule assembly, beginning at about 10 μM. However, within the concentration range we were able to examine (up to 40 μM), there was no effect on the extent of assembly. It should be pointed out that 17t appeared to partially precipitate at 40 μM. With the classic antitubulin agent colchicine 18, the rate of assembly was 50% inhibited at about 2 μM, and the extent of assembly at about 5 μM. At 7 and 10 μM colchicine 18, tubulin polymerization was essentially completely inhibited, an effect that we were unable to achieve with 17t.
Fig. 5.

Panels A, C, E, and G, PtK2 cells stained for tubulin with an antibody to β-tubulin conjugated to Cy3. Panels B, D, F, and H, PtK2 cells stained for actin with an antibody to β-actin conjugated to FTIC. A and B, untreated cells. C and D, cells treated with 10 μM 17t. E and F, cells treated with 1 μM combretastatin A-4 (a potent antitubulin drug). G and H, cells treated with 1 μM latrunculin A (a potent inhibitor of actin assembly).
Fig. 6.
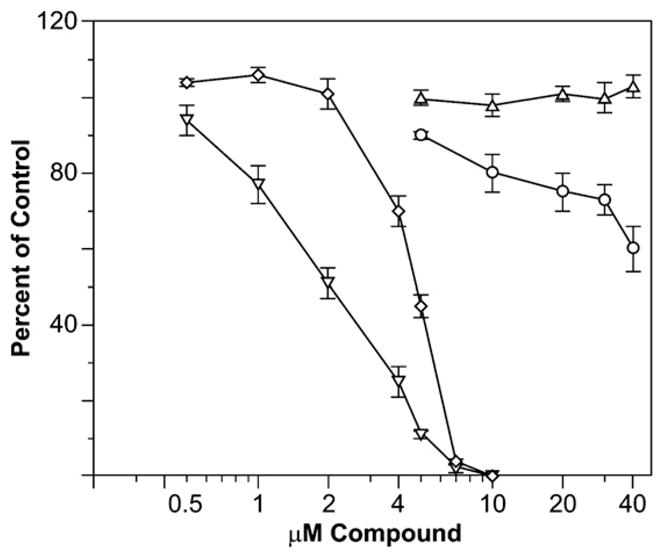
Comparison of the effects of 17t with those of colchicine as an inhibitor of the polymerization of purified tubulin. Assembly was followed in Gilford model 250 recording spectrophotometers equipped with electronic temperature controllers. Reaction mixtures were preincubated at 30 °C without GTP and chilled on ice. GTP (0.4 mM) was added to the samples, which were transferred to cuvettes held at 0 °C. Baselines were established, and the temperature was jumped to 30 °C over about 60 s and held there for 20 min. Maximal reaction rates and extent of assembly were determined for each sample and compared to a control reaction mixture in the same experiment. Each data point was obtained in triplicate, and standard deviations are shown. Symbols as follows: o, maximum rates obtained with 17t; ▲, extents of reaction obtained with 17t; ▽, maximum rates obtained with colchicine; ◇, extents of reaction obtained with colchicine.
Taken together, our data suggest that 17t interacts with tubulin, preventing formation of the mitotic spindle and thereby caused the block in M phase. Inhibition of tubulin assembly would also disrupt the integrity of the microtubule cytoskeleton in interphase cells, causing extensive alterations in cell morphology. Although its effect on in vitro tubulin assembly is relatively weak, the cellular effects obtained with 17t are most consistent with those observed with more potent antitubulin agents. Furthermore, disruption of cellular microtubules invariably results in activation of cellular apoptosis.
3. Conclusions
The data reported here show that the 2-{[2E]-3-phenylprop-2-enoylamino}benzamides 12a–s and 17t–v caused growth inhibition against many tumor cell lines. The best agents inhibited proliferation at low micromolar (12a, 12b, 12c) and submicromolar concentrations (17t, 17u) against every tumor cell line investigated. The best activity was obtained when the 5 position of the benzamido moiety was substituted with an iodine atom. COMPARE analysis, effects on tubulin polymerization in cells and with purified tubulin, and effects on cell cycle distribution, including induction of apoptosis, indicate that these new antiproliferative compounds act as antitubulin agents.
4. Experimental
4.1. Chemistry
4.1.1. General
Reaction progress was monitored by TLC on silica gel plates (Merck 60, F254, 0.2 mm). Organic solutions were dried over Na2SO4. Evaporation refers to the removal of solvent on a rotary evaporator under reduced pressure. All melting points were determined on a Büchi 530 capillary melting point apparatus and are uncorrected. IR spectra were recorded with a Perkin Elmer Spectrum RXI FT-IR System spectrophotometer, with compound as a solid in a KBr disc. 1H NMR spectra were obtained using a Bruker AC-E 300 MHz spectrometer (tetramethylsilane as internal standard): chemical shifts are expressed in δ values (ppm). Merck silica gel (Kiesegel 60/230–400 mesh) was used for flash chromatography columns. Microanalyses data (C, H, N) were obtained by an Elemental Vario EL III apparatus and were within ±0.4% of the theoretical values. Yields refer to purified products and are not optimized. The names of the products were obtained using the ACD/I-Lab Web service (ACD/IUPAC Name Free 8.05).
4.1.2. General procedure for preparation of 5-methyl-2-nitrobenzoyl chloride 6a and 3-phenylacryloyl chlorides 11c–k, 11t–v
Substituted benzoyl and acryloyl chlorides 6a, 11c–k and 11t–v were obtained by refluxing for 5 h the appropriate acid derivatives 5a, 8c–k and 8t–v (0.01 mol) with thionyl chloride (7.25 ml) [16]. After evaporation under reduced pressure, the crude liquid residue was used for subsequent reactions without purification.
4.1.3. Preparation of 5-methyl-2-nitrobenzamide 7a
To 0.01 mol of 5-methyl-2-nitrobenzoyl chloride 6a 10 ml of aqueous ammonia solution (25%) and 33 ml of acetonitrile were added. The solution was first refluxed for 8 h, then evaporated under reduced pressure to give pure 7a.
4.1.4. Preparation of 5-methyl-2-aminobenzamide 10a
To a magnetically stirred suspension of stannous chloride (0.038 mol) in concentrated HCl (37%) (15 ml), 0.013 mole of 7a was added at a rate so that the temperature of the slurry was maintained below 5 °C (about 1 h). After addition was complete, the mixture was stirred for 24 h. The white slurry thus obtained was diluted with cold water (150 ml), and aqueous sodium hydroxide (40%) was added until the tin salt dissolved. The solution was extracted with ethyl acetate (3 × 150 ml), and the extracts dried and evaporated in vacuo to obtain pure 10a.
4.1.5. General procedure for preparation of aminobenzamides 10b,c
A mixture of 0.01 mol of 2H-3,1-benzoxazine-2,4(1H)-diones 9b,c and 25 ml of aqueous ammonia solution (25%) was stirred for 1 h. The solid precipitate was removed by filtration, washed with an aqueous ammonia solution (5%) and crystallized from ethanol.
4.1.6. General procedure for preparation of 2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides 12a–s
To a cold (0–5 °C) stirred suspension of aminobenzamides 10a–c (0.016 mol) in pyridine (13 ml), 0.016 mol of the appropriate 3-phenylacryloyl chloride 11c–k was added over 30 min. After addition was complete, the solution was stirred for 24 h and then poured onto crushed ice. The precipitate was removed by filtration, washed with water, and crystallized from ethanol.
5-Methyl-2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamide (12a)
yield 85%; mp 228–230 °C (dioxane); I.R. (KBr) cm−1 3400-3258 (NH, NH2), 1681, 1651 (2XCO); 1H NMR (DMSO) δ 2.31 (s, 3H, CH3); 6.89 (d, 1H, J = 15.6 Hz, olefinic CH); 7.32–8.49 (a set of signals, 9H, aromatic protons and NH-H); 7.58 (d, 1H, J = 15.6Hz, olefinic CH); 8.24 (s, 1H, NH–H, exchangeable); 11.80 (s, 1H, NH, exchangeable). Anal. (C17H16N2O2) C,H,N
5-Chloro-2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides (12b)
yield 67%; mp 218–220 °C (dioxane); I.R. (KBr) cm−1 3384, 3218 (NH, NH2), 1682, 1659 (2XCO); 1H NMR (DMSO) δ 6.83 (d, 1H, J = 14.9 Hz, olefinic CH); 7.43–8.63 (a set of signals, 10H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.82 (s, 1H, NH, exchangeable). Anal. (C16H13ClN2O2) C,H,N
5-Bromo-2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides (12c)
yield 98%; mp 229–231 °C (dioxane); I.R. (KBr) cm−1 3388, 3217 (NH, NH2), 1682, 1659 (2XCO); 1H NMR (DMSO) δ 6.82 (d, 1H, J = 14.9 Hz, olefinic CH); 7.43–8.58 (a set of signals, 10H, aromatic protons, olefinic CH and NH–H); 8.44 (s, 1H, NH–H, exchangeable); 11.84 (s, 1H, NH, exchangeable). Anal. (C16H13BrN2O2) C,H,N
5-Chloro-2-{[(2E)-3-(2-chlorophenyl)prop-2-enoyl]amino}benzamides (12d)
yield 36%; mp 268–270 °C (dioxane); I.R. (KBr) cm−1 3365, 3155 (NH, NH2), 1686, 1661 (2XCO); 1H NMR (DMSO) δ 6.90 (d, 1H, J = 15.2 Hz olefinic CH); 7.38–8.63 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.43 (s, 1H, NH–H, exchangeable); 11.92 (s, 1H, NH, exchangeable). Anal. (C16H12Cl2N2O2) C,H,N
5-Chloro-2-{[(2E)-3-(3-chlorophenyl)prop-2-enoyl]amino}benzamides (12e)
yield 88%; mp 260–261 °C (dioxane); I.R. (KBr) cm−1 3351, 3157 (NH, NH2), 1681, 1662 (2XCO); 1H NMR (DMSO) δ 6.97 (d, 1H, J = 16.6 Hz, olefinic CH); 7.45–8.63 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.79 (s, 1H, NH, exchangeable). Anal. (C16H12Cl2N2O2) C,H,N
5-Chloro-2-{[(2E)-3-(4-chlorophenyl)prop-2-enoyl]amino}benzamides (12f)
yield 58%; mp 243–244 °C (dioxane); I.R. (KBr) cm−1 3356, 3283, 3177 (NH, NH2), 1675, 1661 (2XCO); 1H NMR (DMSO) δ 6.87 (d, 1H, J = 14.9 Hz, olefinic CH); 7.47–8.62 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H). 8.43 (s, 1H, NH–H, exchangeable); 11.82 (s, 1H, NH, exchangeable). Anal. (C16H12Cl2N2O2) C,H,N
2-{[(2E)-3-(2-bromophenyl)prop-2-enoyl]amino}benzamides (12g)
yield 66%; mp 261–262 °C (dioxane); I.R. (KBr) cm−1 3366, 3157 (NH, NH2), 1686, 1662 (2XCO); 1H NMR (DMSO) δ 6.87 (d, 1H, J = 14.8 Hz, olefinic CH); 7.35–8.63 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.43 (s, 1H, NH–H, exchangeable); 11.90 (s, 1H, NH, exchangeable). Anal. (C16H12BrClN2O2) C,H,N
2-{[(2E)-3-(3-bromophenyl)prop-2-enoyl]amino}benzamides (12h)
yield 91%; mp 251–252 °C (dioxane); I.R. (KBr) cm−1 3349, 3167 (NH, NH2), 1678, 1662 (2XCO); 1H NMR (DMSO) δ 6.96 (d, 1H, J = 15.1 Hz, olefinic CH); 7.34–8.64 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.43 (s, 1H, NH–H, exchangeable); 11.80 (s, 1H, NH, exchangeable). Anal. (C16H12BrClN2O2) C,H,N
5-Chloro-2-{[(2E)-3-(2-methylphenyl)prop-2-enoyl]amino}benzamides (12i)
yield 42%; mp 231–232 °C (dioxane); I.R. (KBr) cm−1 3383, 3165 (NH, NH2), 1687, 1660 (2XCO); 1H NMR (DMSO) δ 2.34 (s, 3H, CH3); 6.66 (d, 1H, J = 15.6 Hz, olefinic CH); 7.28–8.42 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.89 (s, 1H, NH, exchangeable). Anal. (C17H15ClN2O2) C,H,N
5-Chloro-2-{[(2E)-3-(3-methylphenyl)prop-2-enoyl]amino}benzamides (12j)
yield 82%; mp 238–239 °C (dioxane); I.R. (KBr) cm−1 3337, 3163 (NH, NH2), 1681, 1660 (2XCO); 1H NMR (DMSO) δ 2.34 (s, 3H, CH3); 6.80 (d, 1H, J = 15.4 Hz, olefinic CH); 7.21–8.65 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.43 (s, 1H, NH–H, exchangeable); 11.83 (s, 1H, NH, exchangeable). Anal. (C17H15ClN2O2) C,H,N
5-Chloro-2-{[(2E)-3-(4-methylphenyl)prop-2-enoyl]amino}benzamides 12k)
yield 82%; mp 238–239 °C (dioxane); I.R. (KBr) cm−1 3379, 3212 (NH, NH2),1682,1660 (2XCO); 1H NMR (DMSO) δ 2.34 (s, 3H, CH3); 6.78 (d, 1H, J = 15.2 Hz, olefinic CH); 7.23–8.62 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.43 (s, 1H, NH–H, exchangeable); 11.77 (s, 1H, NH, exchangeable). Anal. (C17H15ClN2O2) C,H,N
5-Bromo-2-{[(2E)-3-(2-chlorophenyl)prop-2-enoyl]amino}benzamides (12l)
yield 73%; mp 250–251 °C (dioxane); I.R. (KBr) cm−1 3366, 3153 (NH, NH2), 1685, 1661 (2XCO); 1H NMR (DMSO) δ 6.91 (d, 1H, J = 15.1 Hz, olefinic CH); 7.42–8.02 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.90 (s, 1H, NH, exchangeable). Anal. (C16H12BrClN2O2) C,H,N
5-Bromo-2-{[(2E)-3-(3-chlorophenyl)prop-2-enoyl]amino}benzamides (12m)
yield 56%; mp 260–261 °C (dioxane); I.R. (KBr) cm−1 3352, 3154 (NH, NH2), 1680, 1662 (2XCO); 1H NMR (DMSO) δ 6.97 (d, 1H, J = 16.0 Hz, olefinic CH); 7.45–8.58 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.44 (s, 1H, NH–H, exchangeable); 11.81 (s, 1H, NH, exchangeable). Anal. (C16H12BrClN2O2) C,H,N
5-Bromo-2-{[(2E)-3-(4-chlorophenyl)prop-2-enoyl]amino}benzamides (12n)
yield 57%; mp 255–256 °C (dioxane); I.R. (KBr) cm−1 3357, 3285, 3178 (NH, NH2), 1673, 1660 (2XCO); 1H NMR (DMSO) δ 6.87 (d, 1H, J = 14.7 Hz, olefinic CH); 7.47–8.55 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.79 (s, 1H, NH, exchangeable). Anal. (C16H12BrClN2O2) C,H,N
5-Bromo-2-{[(2E)-3-(2-bromophenyl)prop-2-enoyl]amino}benzamides (12o)
yield 81%; mp 258–259 °C (dioxane); I.R. (KBr) cm−1 3366, 3153 (NH, NH2), 1685, 1661 (2XCO); 1H NMR (DMSO) δ 6.87 (d, 1H, J = 16.5 Hz, olefinic CH); 7.71–8.52 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.88 (s, 1H, NH, exchangeable). Anal. (C16H12Br2N2O2) C,H,N
5-Bromo-2-{[(2E)-3-(3-bromophenyl)prop-2-enoyl]amino}benzamides (12p)
yield 81%; mp 258–259 °C (dioxane); I.R. (KBr) cm−1 3348, 3156 (NH, NH2), 1678, 1661 (2XCO); 1H NMR (DMSO) δ 6.97 (d, 1H, J = 15.6 Hz, olefinic CH); 7.35–8.56 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.43 (s, 1H, NH–H, exchangeable); 11.78 (s, 1H, NH, exchangeable). Anal. (C16H12Br2N2O2) C,H,N
5-Bromo-2-{[(2E)-3-(2-methylphenyl)prop-2-enoyl]amino}benzamides (12q)
yield 98%; mp 238–240 °C (dioxane); I.R. (KBr) cm−1 3384, 3160 (NH, NH2), 1686, 1659 (2XCO); 1H NMR (DMSO) δ 2.32 (s, 3H, CH3); 6.74 (d, 1H, J = 15.4 Hz, olefinic CH); 7.25–8.56 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.89 (s, 1H, NH, exchangeable). Anal. (C17H15BrN2O2) C,H,N
5-Bromo-2-{[(2E)-3-(3-methylphenyl)prop-2-enoyl]amino}benzamides (12r)
yield 71%; mp 268–270 °C (dioxane); I.R. (KBr) cm−1 3338, 3163 (NH, NH2), 1681, 1661 (2XCO); 1H NMR (DMSO) δ 2.34 (s, 3H, CH3); 6.80 (d, 1H, J = 16.5 Hz, olefinic CH); 7.24–8.63 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.80 (s, 1H, NH, exchangeable). Anal. (C17H15BrN2O2) C,H,N
5-Bromo-2-{[(2E)-3-(4-methylphenyl)prop-2-enoyl]amino}benzamides (12s)
yield 88%; mp 236–237 °C (dioxane); I.R. (KBr) cm−1 3353, 3163 (NH, NH2), 1678,1660 (2XCO); 1H NMR (DMSO) δ 2.33 (s, 3H, CH3); 6.75 (d, 1H, J = 14.8 Hz, olefinic CH); 7.22–8.56 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.78 (s, 1H, NH, exchangeable). Anal. (C17H15BrN2O2) C,H,N
4.1.7. General procedure for preparation of 2-cinnamamido-5-iodobenzoic acids 15t–v
To an ice cooled (0–5 °C) stirred solution of 2-amino-5-iodo-benzoic acid 13 (3 mmol) in anhydrous pyridine (20 ml) 2 mmol of cinnamoyl chlorides 11t–v was added. The solution was left stirring overnight, then poured into cold water. The precipitate was collected as a mixture of (E)-6-iodo-2-styryl-4H-benzo[d] [1,3] oxazin-4-ones 14t–u and 2-cinnamamido-5-iodobenzoic acids 15t–v. The mixture was refluxed in 0.01 M aqueous Na3PO4 for 20 h. The solution was allowed to cool to room temperature and, after filtration, was acidified with HCl (0.1 M) to pH 2 to give the corresponding acids 15t [10] and 15u as the only products. Finally, the precipitate was removed by filtration and washed with cold chloroform to obtain pure 15t,u. In the case of cinnamoyl chloride 11v, treatment with 2-amino-5-iodobenzoic acid 13 (3 mmol) in anhydrous pyridine (20 ml) directly gave pure (E)-5-iodo-2-(3-o-tolylacrylamido)benzoic acid 15v.
(E)-2-(3-(2-chlorophenyl)acrylamido)-5-iodobenzoic acid (15u)
yield 80%; mp 250–252 °C (ethanol); I.R. (KBr) cm−1 3448, 3117 (NH, OH), 1703, 1688 (2XCO); 1H NMR (DMSO) δ 6.94 (d, 1H, J = 15.3 Hz, olefinic CH); 7.44–8.43 (a set of signals, 8H, aromatic protons, olefinic CH); 11.37 (s, 1H, NH–H, exchangeable); 13.98 (s, 1H, OH, exchangeable). Anal. (C16H11ClINO3) C,H,N
(E)-5-iodo-2-(3-o-tolylacrylamido)benzoic acid (15v)
yield 83%; mp 239–240 °C (ethanol); I.R. (KBr) cm−1 3353, 3163 (NH, NH2), 1678, 1660 (2XCO); 1H NMR (DMSO) δ 2.37 (s, 3H, CH3); 6.82 (d, 1H, J = 15.3 Hz, olefinic CH); 7.27–8.48 (a set of signals, 8H, aromatic protons, olefinic CH); 11.28 (s, 1H, NH–H, exchangeable); 13.90 (s, 1H, OH, exchangeable). Anal. (C17H14INO3) C,H,N
4.1.8. General procedure for preparation of 6-iodo-1H-benzo[d] [1,3]oxazine-2,4-diones 16t–v
A mixture of 2-cinnamamido-5-iodobenzoic acids 15t–v (4.6 mmol) and ethylchloroformate (25 ml) was refluxed for 15 min. After this time, acetyl chloride (1.5 ml) was added, and reflux continued for an additional 30 min. On cooling, the product precipitated, was removed by filtration, and was washed with chloroform to give pure 16t–v. Compound 16v easily decomposed and was used as is without purification.
1-Cinnamoyl-6-iodo-1H-benzo[d] [1,3] oxazine-2,4-dione (16t)
yield 83%; mp 201–204 ° C (CHCl3); I.R. (KBr) cm−1 1760, 1670 (3XCO); 1H NMR (DMSO) δ 6.96 (d, 1H, J = 16 Hz, olefinic CH); 7.39–8.42 (a set of signals, 9H, aromatic protons and olefinic C). Anal. (C17H10INO4) C,H,N
(E)-1-(3-(2-chlorophenyl)acryloyl)-6-iodo-1H-benzo[d] [1,3]oxazine-2,4-dione (16u)
yield 83%; mp 201–204 °C (CHCl3); I.R. (KBr) cm−1 1760, 1660 (3XCO); 1H NMR (DMSO) δ 7.01 (d, 1H, J = 16 Hz, olefinic CH); 7.38–8.33 (a set of signals, 8H, aromatic protons and olefinic C). Anal. (C17H9IClNO4) C,H,N
4.1.9. General procedure for preparation of 2-cinnamamido-5-iodo-benzamides 17t-v
A solution of 6-iodo-1H-benzo[d] [1,3]oxazine-2,4-diones 16t–v (2.3 mmol), 25% aqueous ammonia solution (28 ml) and dioxane (5 ml) was left under reflux for 3 h. After this time, the solution was kept in a freezer to allow the product to precipitate. It was removed by filtration and crystallized from dioxane.
5-iodo-2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamide (17t)
yield 88%; mp 260–261 °C (dioxane); I.R. (KBr) cm−1 3393, 3215 (NH, NH2), 1680, 1656 (2XCO); 1H NMR (DMSO) δ 6.82 (d, 1H, J = 15.6 Hz, olefinic CH); 7.43–8.42 (a set of signals, 10H, aromatic protons, olefinic CH and NH–H); 8.15 (s, 1H, NH–H, exchangeable); 11.80 (s, 1H, NH, exchangeable). Anal. (C16H13IN2O2) C,H,N
5-Iodo-2-{[(2E)-3-(2-chlorophenyl)prop-2-enoyl]amino}benzamides (17u)
yield 88%; mp 260–261 °C (dioxane); I.R. (KBr) cm−13381, 3155 (NH, NH2), 1681, 1660 (2XCO); 1H NMR (DMSO) δ 6.97 (d, 1H, J = 16.6 Hz, olefinic CH); 7.45–8.63 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.42 (s, 1H, NH–H, exchangeable); 11.79 (s, 1H, NH, exchangeable). Anal. (C16H12ClIN2O2) C,H,N
5-iodo-2-{[(2E)-3-(2-methylphenyl)prop-2-enoyl]amino}benzamide (17v)
yield 88%; mp 260–261 °C (dioxane); I.R. (KBr) cm−1 3368, 3294, 3192 (NH, NH2), 1673, 1658 (2XCO); 1H NMR (DMSO) δ 2.34 (s, 3H, CH3); 6.97 (d,1H, J = 16.6 Hz, olefinic CH); 7.45–8.63 (a set of signals, 9H, aromatic protons, olefinic CH and NH–H); 8.14 (s, 1H, NH–H, exchangeable); 11.77 (s, 1H, NH, exchangeable). Anal. (C17H15IN2O2) C,H,N
4.2. Biology
4.2.1. Antiproliferative activity
Compounds 12a–s and 17t–v were initially tested in vitro for antiproliferative activity against the K562 (human chronic myelogenous leukemia) cell line. These cell lines were grown at 37°C in a humidified atmosphere containing 5% CO2, in RPMI-1640 medium supplemented with 10% fetal bovine serum and antibiotics.
K562 cells were suspended at a density of 1 × 105 cells/ml in growth medium, transferred to a 24-well plate (1 ml/well), cultured with or without (in the case of control wells) a screening concentration of 10 μM compounds and incubated at 37 °C for 48 h. Numbers of viable cells were determined by counting in a hematocytometer after dye exclusion with trypan blue [17]. The anti-proliferative effects of the compounds were estimated in terms of % growth inhibition, comparing cell viability of treated and untreated cells. We determined IC50 values (test agent concentration at which the cell proliferation was inhibited by 50% as compared with the untreated control) for compounds that exhibited the best activity at the screening concentration.
4.2.2. Tumor cell line screening
The in vitro antiproliferative activity values were obtained by the Developmental Therapeutics Program, National Cancer Institute (USA) [18]
The human tumor cell lines of the cancer screening panel are grown in RPMI 1640 medium containing 5% fetal bovine serum and 2 mM L-glutamine. For a typical screening experiment, cells are inoculated into 96 well microtiter plates in 100 μl at plating densities ranging from 5000 to 40,000 cells/well, depending on the doubling time of the individual cell lines. After cell inoculation, the microtiter plates are incubated at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of experimental drugs. After 24 h, two plates of each cell line are fixed in situ with TCA, to represent a measurement of the cell population for each cell line at the time of drug addition (Tz). Experimental drugs are solubilized in dimethyl sulfoxide at 400-fold the desired final maximum test concentration and stored frozen prior to use. At the time of drug addition, an aliquot of frozen concentrate is thawed and diluted to twice the desired final maximum test concentration with complete medium containing 50 μg/ml gentamicin. Additional four, 10-fold or ½ log serial dilutions are made to provide a total of five drug concentrations plus control. Aliquots of 100 μl of these different drug dilutions are added to the appropriate microtiter wells already containing 100 μl of medium, resulting in the required final drug concentrations. Following drug addition, the plates are incubated for an additional 48 h at 37 °C, 5% CO2, 95% air, and 100% relative humidity. For adherent cells, the assay is terminated by the addition of cold TCA. Cells are fixed in situ by the gentle addition of 50 μl of cold 50% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 min at 4 °C. The supernatant is discarded, and the plates are washed five times with tap water and air dried. Sulforhodamine B (SRB) solution (100 μl) at 0.4% (w/v) in 1% acetic acid is added to each well, and plates are incubated for 10 min at room temperature. After staining, unbound dye is removed by washing five times with 1% acetic acid, and the plates are air dried. Bound stain is subsequently solubilized with 10 mM trizma base, and the absorbance is read on an automated plate reader at a wavelength of 515 nm. For suspension cells, the methodology is the same except that the assay is terminated by fixing settled cells at the bottom of the wells by gently adding 50 μl of 80% TCA (final concentration, 16% TCA). Using the seven absorbance measurements [time zero (Tz), control growth (C), and test growth in the presence of drug at the five concentration levels (Ti)], the percentage growth is calculated at each of the drug concentration levels. Percentage growth inhibition is calculated as:
Three dose response parameters are calculated for each experimental agent. Growth inhibition of 50% (GI50) is calculated from [(Ti−Tz)/(C−Tz)] × 100 = 50, which is the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells during the drug incubation. The drug concentration resulting in total growth inhibition (TGI) is calculated from Ti = Tz. The LC50 (concentration of drug resulting in a 50% reduction in the measured protein at the end of the drug treatment as compared to that at the beginning) indicating a net loss of cells following treatment is calculated from [(Ti−Tz)/Tz] × 100 = −50. Values are calculated for each of these three parameters if the level of activity is reached; however, if the effect is not reached or is exceeded, the value for that parameter is expressed as greater or less than the maximum or minimum concentration tested.
4.2.3. Cytotoxicity assays
To evaluate the number of live and dead neoplastic cells, the cells were stained with trypan blue and counted on a hemocytometer. To determine the growth inhibitory activity of the drugs tested, 2 × 105 cells were plated into 25 mm wells (Costar, Cambridge, UK) in 1 ml of complete medium and treated with different concentrations of each drug. After 48 h of incubation, the number of viable cells was determined and expressed as percent of control proliferation.
4.2.4. Morphological evaluation of apoptosis
Drug induced apoptosis was determined morphologically after labeling with acridine orange and ethidium bromide. Cells (2 × 105) were centrifuged (300 × g), and the pellet was resuspended in 25 μl of the dye mixture. Ten μl of the mixture was examined under oil immersion with a 100× objective using a fluorescence microscope. Live cells were determined by the uptake of acridine orange (green fluorescence) and exclusion of ethidium bromide (red fluorescence) stain. Live and dead apoptotic cells were identified by perinuclear condensation of chromatin stained by acridine orange (100 μg/ml) or ethidium bromide (100 μg/ml), respectively, and by the formation of apoptotic bodies. The percentage of apoptotic cells was determined after counting at least 300 cells.
4.2.5. Determination of apoptosis by annexin-V
Cells (1 × 106) were washed with phosphate-buffered saline (PBS) and centrifuged at 200 × g for 5 min. Cell pellets were suspended in 100 μl of staining solution containing FITC-conjugated annexin-V and propidium iodide (Annexine-V-Fluos Staining Kit, Roche Molecular Biochemicals, Mannheim, Germany) and incubated for 15 min at 20 °C. Annexin-V positive cells were evaluated by flow cytometry (Becton–Dickinson.
4.2.6. Flow cytometric analysis of cell cycle and apoptosis
Cells were washed once in ice-cold PBS and resuspended at 1 × 106 ml in a hypotonic fluorochrome solution containing propidium iodide (Sigma) 50 μg/ml in 0.1% sodium citrate plus 0.03% (v/v) nonidet P-40 (Sigma). After 30 min of incubation, the fluorescence of each sample was analyzed as a single-parameter frequency histogram using a FACScan flow cytometer (Becton–Dickinson, San Jose, CA). Distribution of cells in the cell cycle was determined using the ModFit LT program (Verity Software House, Inc.). Apoptosis was determined by evaluating the percentage of hypodiploid nuclei accumulated in the sub-G0–G1 peak after labeling with propidium iodide.
4.2.7. Immunofluorescence
PtK2 cells (Potorus tridactylis kidney epithelial cells) were obtained from the American Type Tissue Collection and grown as recommended by the supplier. The technique was described in detail previously [19,20]. Cells were treated with compounds for 24 h at 37 °C prior to fixation and staining with antibodies (Cy3 conjugate of anti-β-tubulin clone TUB2.1 and FTIC conjugate of anti-β-actin clone Ac-15 monoclonal antibodies from Sigma). Cells were examined with a Nikon Eclipse E800 microscope equipped with epifluorescence and appropriate filters, and images were collected with a Spot digital camera.
4.2.8. Tubulin assembly
The procedure with purified bovine brain tubulin was described in detail previously [21].
Acknowledgments
Financial support from MIUR (40% funding) is gratefully acknowledged. The authors wish to thank the Developmental Therapeutics Program of the National Cancer Institute of the United States of America for performing the antiproliferative screening of compounds.
References
- 1.Ogita H, Isobe Y, Takaku H, Sekine R, Goto Y, Misawa S, Hayashi H. Bioorg Med Chem Lett. 2001;11:549–551. doi: 10.1016/s0960-894x(00)00717-4. [DOI] [PubMed] [Google Scholar]
- 2.Williams SJ, Stapleton D, Zammit S, Kelly DJ, Gilbert RE, Krum H. Patent WO2008003141. Chem Abst. 2008;148:144415.
- 3.Bo Y, Chakrabarti PP, Chen N, Doherty EM, Fotsch CH, Han N, Kelly MG, Liu Q, Norman MH, Wang X, Zhu J, Ognynanov V. Patent WO03049702. Chem Abst. 2003;139:53025.
- 4.Harada H, Isaji M, Miyata H, Kusama H, Nonaka Y, Kamata K, Yazaki T, Hotei Y. Patent JP10330254. Chem Abst. 1998;130:105331.
- 5.Harada H, Isaji M, Kusama H, Taketana Y, Nonaka Y, Kamata T, Futai Y. Patent JP10259129. Chem Abst. 1998;129:321163.
- 6.Harada H, Kusama H, Nonaka Y, Kamata K, Fotei Y. Patent JP10360124. Chem Abst. 1998;130:47477.
- 7.Harada H, Kusama H, Nonaka Y, Kamata K, Hotei Y, Iyobe A, Fujikura H, Satoh F. Patent WO9709301. Chem Abst. 1997;126:250990.
- 8.Dolzhenco-Podchezertseva AV, Korkodinova LM, Vasilyuc MV, Kotegov VP. Pharm Chem J. 2002;36:647–648. [Google Scholar]
- 9.Bratt K, Sunnerheim K, Bryngelsson S, Fagerlund A, Engman L, Andersson RE, Dimberg LH. J Agr Food Chem. 2003;51:594–600. doi: 10.1021/jf020544f. [DOI] [PubMed] [Google Scholar]
- 10.Rani P, Srivastava VK, Kumar A. Ind J Chem B Org. 2003;42B:1729–1733. [Google Scholar]
- 11.Coppola GM. Synthesis. 1980;7:505–536. [Google Scholar]
- 12.Chimichi S, De Sio F, Donati D, Fina G, Pepino R, Sarti-Fantoni P. Heterocycles. 1983;20:263–267. [Google Scholar]
- 13.Hanusek J, Sedlák M, Šimůnek P, Štĕrba V. Eur J Org Chem. 2002;11:1855–1863. [Google Scholar]
- 14.Boyd MR, Paull KD. Drug Dev Res. 1995;34:91–109. [Google Scholar]
- 15.Paull KD, Lin CM, Malspeis L, Hamel E. Cancer Res. 1992;52:3892–3900. [PubMed] [Google Scholar]
- 16.Palmer MH, McVie GJ. J Chem Soc B. 1968:745–751. [Google Scholar]
- 17.Manfredini S, Bazzanini R, Baraldi PG, Guarneri M, Simoni D, Marongiu ME, Pani A, La Colla P, Tramontano E. J Med Chem. 1992;35:917–924. doi: 10.1021/jm00083a017. [DOI] [PubMed] [Google Scholar]
- 18.The methodology can be found at http://dtp.nci.nih.gov/.
- 19.Bai R, Verdier-Pinard P, Gangwar S, Stessman CC, McClure KJ, Sausville EA, Pettit GR, Bates RB, Hamel E. Mol Pharmacol. 2001;59:462–469. doi: 10.1124/mol.59.3.462. [DOI] [PubMed] [Google Scholar]
- 20.Cruz-Monserrate Z, Vervoort HC, Bai R, Newman DJ, Howell SB, Los G, Mullaney JT, Williams MD, Pettit GR, Fenical W, Hamel E. Mol Pharmacol. 2003;63:1273–1280. doi: 10.1124/mol.63.6.1273. [DOI] [PubMed] [Google Scholar]
- 21.Hamel E. Cell Biochem Biophys. 2003;38:1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]