

Abstract
Polymersomes are vesicles whose membranes are comprised of self-assembled block co-polymers. We recently showed that co-encapsulating conjugated multi-porphyrin dyes in a polymersome membrane with ferritin protein in the aqueous lumen confers photo-lability to the polymersome. In the present study, we illustrate that the photo-lability can be extended to vesicles containing dextran, an inert and inexpensive polysaccharide, as the luminal solute. Here we explore how structural features of the polymersome/porphyrin/dextran composite affect its photo-response. Increasing dextran molecular weight, decreasing block copolymer molecular weight, and altering fluorophore-membrane interactions results in increasing the photo-responsiveness of the polymersomes. Amphiphilic interactions of the luminal encapsulant with the membrane coupled with localized heat production in the hydrophobic bilayer likely cause differential thermal expansion in the membrane and the subsequent membrane rupture. This study suggests a general approach to impart photo-responsiveness to any biomimetic vesicle system without chemical modification, as well as a simple, bio-inert method for constructing photo-sensitive carriers for controlled release of encapsulants.
Keywords: polymersomes, membrane rupture, photo-activation, amphiphilic interaction
1. Introduction
The ability of a cell to manipulate membrane stability is pivotal for cell biological function. Processes like secretion and uptake, for example, are mediated by dynamically controlled membrane instability. Cell membranes are thus an inspiration for designing transport and release mechanisms in synthetic membranes. A key step in inducing membrane instability is creating membrane asymmetry. This idea, first introduced as the ‘Bilayer-couple hypothesis,’ states that two halves of a closed lipid bilayer can have differential responses to various perturbations, due to asymmetries between the bilayer leaflets.[1] Often, the asymmetry causes a relative increase in the surface area of one leaflet of a closed bilayer, which then increases the spontaneous curvature of that leaflet. Provided that leaflet is unopposed, it will conform to its spontaneous curvature to minimize free energy. Thus, deformation often initiates on the leaflet side with additional surface area (i.e. where the asymmetry was located). Membrane asymmetry can appear in bilayers for a variety of reasons that include differential (i) leaflet composition,[2] (ii) thermal expansivity coefficients,[3] and (iii) thermal gradients,[4] as well as particle binding to either a leaflet or a portion of the membrane;[5–7] this final case is often used to explain deformation and instability in biological membranes.
Binding-induced membrane asymmetry is not, however, restricted to biological components. Several studies have now shown that membrane instability and deformation should, in principle, be caused by any particle binding asymmetrically on a membrane provided that the adhering object is sufficiently adhesive and rigid.[2, 5, 8] Several other characteristics of bound particles have been found to be important for inducing membrane instability. These include amphiphilicity,[2, 9, 10] self association and clustering of the particle,[10–13] structure,[14] and charge and electrostatic interactions.[1, 15] Synthetic molecules with some of the previously-mentioned characteristics have successfully induced membrane instability by way of asymmetrically interacting with the membrane and changing its spontaneous curvature.[5, 16, 17]
Furthermore, membrane instability can be studied in synthetic membranes in which the relationship between membrane stability and structure can be systematically studied. By recreating instability in designed membranes, we gain insight into the requirements for constructing membranes to perform specific tasks, such as delivering drugs. Polymersomes are a class of synthetic vesicles, made from diblock co-polymers, that self-assemble into bilayer vesicles ranging in diameter from 100 nm to 30 μm.[18] These vesicles are more stable than liposomes, are fully synthetic and hence can be designed,[19–21] and have been shown to encapsulate a wide variety of both hydrophilic and hydrophobic solutes.[22] Recently, our group identified a polymersome composite with tunable sensitivity to light. Polymersomes with the luminal, protein-solute ferritin, and a meso-to-meso ethyne-bridged bis[(porphinato)zinc] (PZn2) chromophore membrane-solute, will degrade upon exposure to light.[16] Through systematic experimentation, we hypothesized that the encapsulated protein interacted with the luminal side of the membrane bilayer, thus creating an asymmetric membrane. We further hypothesized that differential stress between leaflets of the membrane, caused by the protein interaction and excitation of the PZn2 dye, which dissipates a significant fraction (> 80%) of the energy of absorbed photons as heat, led to asymmetric membrane deformation and rupture.
If this mechanism of failure is correct, it suggests that any luminal solute that associates with the inner leaflet of the membrane can, in conjunction with PZn2, induce polymersome instability. We have investigated dextran, a carbohydrate solute of tunable size, as a luminal encapsulant. Dextran offers further advantages because it is inexpensive, biocompatible, and has some solubility in hydrophobic solution, suggesting it might associate with the membrane. In addition, the currently accepted mechanism of membrane deformation suggests that the encapsulant and chromophore can be replaced and elicit the same instability. This ability to substitute components suggests particle binding-induced instability in tandem with photo-activated membrane rupture could be recreated in almost any biomimetic vesicle carrier.
Here, we show that dextran and PZn2 encapsulation results in vesicle deformation and destruction upon light absorption by the chromophore. The amphiphilic-driven dextran interactions with the membrane were studied by analyzing changes to mechanical properties of the membrane and changes in the frequency of photo-induced deformation that occurred by modifying the dextran molecular weight and concentration. Increasing the molecular weight or concentration of dextran leads to a higher incidence of polymersome deformation and decreased mechanical strength of the membrane, suggesting that vesicle-encapsulant interactions, and the resulting membrane stability, can be finely tuned by modifying properties of the encapsulant.
2. Results and Discussion
2.1. Vesicle Deformation
Polymersomes were constructed with a meso-to-meso ethyne-bridged (porphinato)zinc(II) dimer (PZn2, 2123 Da)[23, 24] in the membrane and dextran in the lumen by thin film rehydration (see Methods). When both PZn2 and dextran were present, membranes deformed in response to laser illumination; these studies were carried out in an experimental configuration that allowed for confocal imaging and laser stimulation (λex = 458, 488, 515, 543, and 633 nm; laser power = 33 mW) to be carried out independently and virtually simultaneously. To investigate the effect of molecular weight and concentration of the solute, vesicles were either made with various size dextran solutes in the lumen, at a constant concentration (10 mg/mL) or made with varying concentrations of dextran of a specific molecular weight (500 kDa). Depending on the concentration and the molecular weight of dextran, a wide variety of membrane deformations were seen, including membrane budding, pearling instabilities, membrane stretching, and vesicle rupture. Rupture was the most frequent type of deformation, regardless of dextran size, and was often initiated at a single point in the membrane. Membrane rupture events also increased in frequency as the average molecular weight (MW) of dextran was increased (Figure 1B), indicating that the mechanism of rupture is influenced by dextran MW.
Figure 1. Membrane deformation of polymersomes observed following optical excitation.

(A) Distribution of types of polymersome deformation that occur for each MW of dextran encapsulated. (B) Membrane rupture of a dextran encapsulated polymersome following optical excitation (λex = 458, 488, 515, 543, and 63 nm; laser power = 33 mW) is the most common type of deformation that occurred. (C) Frequency of deformation events for different dextran MWs.
The method used to manufacture vesicles in this study, thin film rehydration, produces a variety of unilamellar and multilamellar structures. Of the unilamellar vesicles that deformed, membrane rupture was the predominant form of deformation. While multilamellar vesicles also experienced membrane rupture, they experienced a greater number of budding and membrane stretching events.
2.2 Modification of Dextran and Polymer
To explore the role of dextran on the failure of the membrane, we systematically varied the size of both dextran and the membrane polymer. Increasing the MW of dextran within a polymersome population made from a specific polymer results in an increased frequency of deformation (Figure 2A). Dextran has been shown to increase in amphiphilicity with increasing MW as it changes from a hydrophilic polysaccharide at low molecular weights (1000 Da) to becoming more hydrophobic at increasing molecular weights.[25] Increasing the molecular weight of dextran leads to decreased critical aggregation concentrations (CACs) which facilitates an increased rate of hydrophobic aggregate formation.[25] Thus, the increasing amphiphilicity and aggregation tendency of dextran with increasing molecular weight likely cause the increased incidence of polymersome deformation. When the hydyrophilic and inert PEO polymer (200,000 Da), known to have limited interactions with the membrane, was encapsulated in the polymersome in place of dextran, the photo response ceased (S2). Increasing the concentration of dextran also increases the frequency of deformation events. Furthermore, the exponential shape of the dextran concentration versus deformation frequency curve (Figure 2B) indicates that a critical aggregation concentration is required before the dextran affects the deformation rate. In a study of the effect of hydrophobic modification and MW of dextran on CAC, Filippov et al.[26] found that a 40,000 Da MW dextran had a CAC of 0.14 mg/mL. Since the dextran used in the current study had a higher MW (500,000 Da), we can assume the CAC of 500 kDa dextran (Figure 2B) is lower than 0.14 mg/mL. This estimated CAC corroborates our findings that deformation occurs at dextran encapsulation concentrations as low as 0.1 mg/mL and that a rapid increase in deformation events occurs with increasing dextran encapsulation concentration.
Figure 2. Effect of Dextran molecular weight on polymersome deformation.

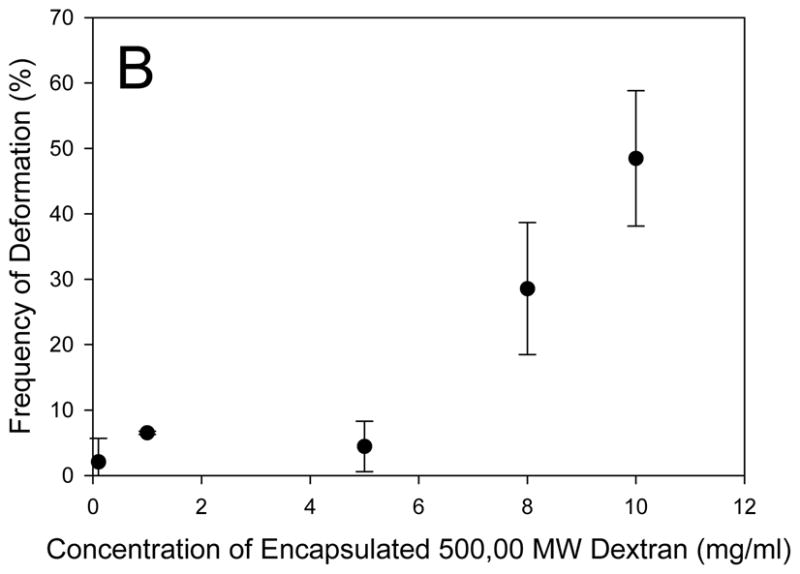
(A) Increasing the MW of encapsulated dextran results in an increased incidence of light-induced polymersome deformation (n = 3, error bars represent standard deviation (sd)). Deformation of OB18 polymersomes requires a higher MW dextran and occurs less frequently than OB29 polymersomes (p < 0.05 for dextran MWs of 5, 10, 100, 500, and 2,000 kDa). (B) A minimum concentration of dextran is required for deformation to occur in each vesicle population.
To confirm the role of dextran amphiphilicity and chain length in membrane rupture, we compared the response of two different diblock copolymer membranes, OB29 (3800 Da) and the higher MW, and more rigid OB18 (10400 Da).[27] Both polymersomes populations deform at increasing frequencies with increasing dextran MW, but an overall significantly lower frequency of OB18 vesicles deformed compared to OB29 vesicles. The lower deformation rate confirms the role of membrane cohesiveness in deformation, which has been discussed in previous theoretical and experimental studies as a parameter that reduces membrane deformation.[28]
Additionally, the photo-responsiveness is destroyed when the dextran MW falls below 1000 Da in OB29 vesicles and the vesicles remain stable even when the concentration of 1000 Da dextran is increased to 100 mg/mL. OB18 vesicles only become photo responsive with dextran of a MW higher than 5000 Da. The increased MW of dextran required to facilitate deformation in OB18 vesicles demonstrates that stronger amphiphilic membrane interactions between dextran and the membrane polymer are required to cause deformation for more cohesive polymer membranes. Specific interactions of dextran with the vesicle membrane are discussed in the following sections.
2.2.1. Fluorescence Recovery after Photobleaching (FRAP)
To assess whether dextran was directly interacting with the polymersome membrane, we measured dextran mobility within the polymersome by FRAP. Vesicles containing lumen-encapsulated Fluorescein isothiocyanate (FITC)-tagged dextran were first confirmed to deform upon exposure to laser light. For FRAP measurements, two locations within the vesicle, one near the membrane and the other at the center of the aqueous core, were photobleached (Figure 3A). The mobility of dextran in the vicinity of the membrane was observed to be restricted with no FITC fluorescence recovery in a nine second period after photobleaching (Figure 3B), indicating very slow or no diffusion of dextran at the membrane. In contrast, photobleaching of dextran in the core of the lumen led to complete fluorescent recovery within nine seconds, indicating that dextran located in the aqueous lumen is mobile. This hindered diffusion near the membrane suggests association of dextran with the membrane.
Figure 3. Fluorescence Recovery After Photobleaching (FRAP) of Dextran Encapsulated Polymersomes.


(A) FITC-labeled dextran (green) was encapsulated in the core of the polymersome and PZn2 (purple) was encapsulated in the membrane. A region of the membrane (red circle) and the aqueous core (blue circle) were photobleached to assess diffusion of dextran. (B) Fluorescence recovery of FITC occurs after photobleaching (blue) in the center of the polymersome while fluorescence does not recover near the membrane (red).
2.2.2. Micropipette Aspiration of Dextran Loaded Polymersomes
Because FRAP measurements suggested that dextran was associated with the membrane, we reasoned that dextran encapsulation should alter membrane material properties. Encapsulation of 500,000 Da dextran (10 mg/mL), which successfully caused deformation in approximately 50% of vesicles exposed to light, resulted in a decreased area elastic modulus (Ka) of membranes (Figure 4A). Encapsulation of 1000 Da dextran, which did not impart photo-responsiveness to vesicles, did not change the Ka. Additionally, vesicles without PZn2 were studied in order to confirm that the decreased Ka was due to dextran, not the membrane encapsulated chromophore. Figure 4B shows the area elastic modulus results of dextran encapsulation in different polymer vesicles. Encapsulation of dextran in both OB29 and OB18 vesicles led similarly to decreases in Ka, but the Ka of the OB18 vesicles remained higher than that of the OB29 vesicles when dextran was encapsulated in both. These measurements support the previous finding that OB18 vesicles require higher MW dextran than OB29 vesicles to facilitate light induced-deformation because of the increased strength of the OB18 membrane (Figure 2).
Figure 4. Effect of Dextran on Membrane Mechanical Strength.


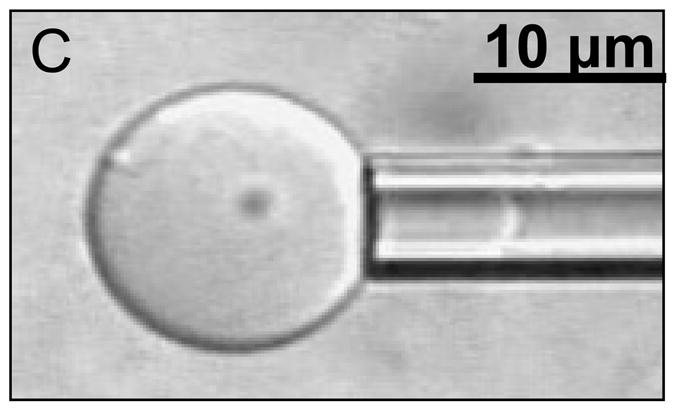
(A) Dextran encapsulation causes a significant decrease in the Ka of polymersomes for both membranes without PZn2 (p < 5*10−5) and in polymersomes membranes containing PZn2 (p < 0.05). Encapsulation of 1000 MW dextran, which does not result in deformation, does not significantly change the Ka of the membrane (p > 0.05). Error bars represent standard error of the mean (sem). (B) Dextran encapsulation significantly decreases the Ka of the higher molecular weight (and more rigid) OB18 polymersomes (p < 0.005). The resulting dextran encapsulated OB18 polymersome has a higher Ka (p = 0.05) than dextran encapsulated OB29 polymersomes. (C) Snapshot of a polymersome area expansion test in a micropipette
The fact that the membrane is made more compliant by the encapsulation of dextran suggests that the polysaccharide interacts substantially with it. Unlike ferritin, whose encapsulation causes an increased Ka,[16] this change in mechanical properties rules out the possibility that the interaction of dextran with the membrane is merely physisorption on the exterior PEO corona. Rather, the surfactant-like behavior of dextran suggests that the amphiphilic saccharide penetrates the corona, at least to the point of the PEO-PBD interface. Intercalation of dextran at this interface would reduce the interfacial tension, thereby reducing the Ka.[9, 29, 30] Dextran has previously demonstrated a capacity to penetrate a vesicle bilayer and destabilize the membrane,[8, 17, 31] yet this instability has been insufficient to cause complete membrane failure.
2.3. Role of Porphyrin Dimer (PZn2)
2.3.1. PZn2 loading
To investigate the role of PZn2 in the photo-activated polymersome deformation, we varied the amount of chromophore in the membrane, and measured the resulting frequency of deformation. Relative to conventional NIR fluorophores, PZn2 and related structures are unusual in that they feature strongly attenuated inter-system crossing yields; hence PZn2 excited states decay via fluorescence (φF = 16%) or via fast internal conversion to produce heat and the chromophore ground state, and do not give rise to significant yields of electronically excited triplets.[23] Localized heat production leads to thermal expansion of the membrane, which because of membrane asymmetry resulting from physisorbed molecules on the inner leaflet, leads to membrane instability. Thus, the role of the chromophore was thought to be limited to energy conversion in order to induce membrane thermal expansion. Decreasing the loading of PZn2 below a 1:7 (chromophore:polymer) molar ratio resulted in a decrease in the frequency of polymersome deformation (Figure 5A). This finding is consistent with the hypothesis that decreased PZn2 loading leads to decreased heat production and the resulting incidence of vesicle deformation. It was found, however, that increasing the loading of PZn2 from a 1:7 molar ratio in the membrane to a 2:7 and 3:7 molar ratio also decreased the frequency of polymersome deformation (Figure 5A). To determine if increased membrane rigidity, caused by increased PZn2 loading, caused the decreased number of deformation events, we conducted mechanical analysis of the polymersome membranes. Micropipette results revealed no significant change in the Ka of the membranes, indicating no mechanical stabilization of the membranes results from increased PZn2 loading (Figure 5B).
Figure 5. Increasing loading of PZn2 in membrane.



(A) Increasing the concentration of PZn2 in the membrane results in a decreased incidence of polymersome deformation for polymersomes encapsulating 500,000 MW dextran (error bars represent sd). (B) Polymersome membranes do not experience a significant change in rigidity with increasing amounts of PZn2. (C) Fluorescence emission of PZn2 decreases as loading in polymersomes increases, consistent with concentration-dependent self-quenching.
Next, we measured the effect of differential loading of PZn2 on chromophore emission. As PZn2 loading increases, the chromophore emission decreases (Fig 5C) and the total emission from PZn2 at the highest loadings (2:7 and 3:7) plateaus. The decrease in emission per molecule indicates that PZn2 self quenches due to close contact between chromophores. Previous studies show that PZn2 loading as low as 0.4:7 (chromophore:polymer) molar ratio results in measurable self quenching.[22] As the number of available pathways for excited-state non-radiative decay generally increases with fluorophore aggregation, self quenching often gives rise to increased heat dissipation.[32] Yet, a decreased incidence of polymersome deformation occurs in polymersomes that display increased PZn2 self quenching. The fraction of excited-states that decay via non-radiative pathways, the non-radiative decay rate (which is normally fast in PZn2 and PZn3 [23]), the heating rate, and the nature of the chromophore vibrational modes are all affected by chromophore packing. These factors are likely to be crucial in facilitating light-induced membrane rupture. We hypothesize that these factors may be more important in producing the photo-response than the total amount of heat released over a long time period. It is difficult, however, to resolve the changes in photo-physical dynamics of the chromophores due to the complexity of the polymersome membrane environment.
Another possible explanation, that we can explore, is the ability of the chromophore to stabilize the membrane though cohesive interactions with the polymer chains. Chromophore-membrane interactions are expected to change upon changes in PZn2 loading and could serve to stabilize the membrane as PZn2 is increasingly loaded in the membrane. By replacing the current chromophore with one that has similar excited-state properties but different amphiphilicity we can explore the role of non-covalent interactions between the chromophore and the polymer chains in modulating membrane stability and membrane photo response.
2.3.2. PZn2 membrane stabilization
As chromophore loading is increased in the membrane, we found that PZn2 molecules appear to stabilize the membrane against photo-disruption through changes in interactions between PZn2 and polymer chains. In order to investigate the stabilizing effect of PZn2, it is important to understand the specific localization of this hydrophobic chromophore within the membrane. The porphyrin dimer contains two chromophoric cores, each possessing 3′5′-disubstituted pendant aryl groups. The ancillary substituents on the aryl groups can be varied, altering the hydrophobicity of the porphyrin molecule and its interactions with the membrane. The PZn2 molecule used in the current study contains a 9-methoxy-1,4,7-trioxanonyl substituent (essentially a pegylated group) on one aryl group and a more hydrophobic 3,3-dimethyl-1-butyloxy substituent on the other.[23] The presence of the pegylated substituent reduces porphyrin hydrophobicity and causes PZn2 localization near the interface of the hydrophilic PEO and the hydrophobic PBD blocks.[33, 34] Increased loading of PZn2 increases interactions between the chromophore and the membrane chains.
To explore the possibility that PZn2 may stabilize the membrane through interactions with the polymer chains, the porphyrin dimer was replaced with the more hydrophobic PZn3 trimer. Containing an additional chromophoric core with the hydrophobic 3,3-dimethyl-1-butyloxy ancillary substituents, this dye was expected to fully localize within the hydrophobic membrane core and have enhanced interactions with the polymer chains. There was little difference in deformation frequency between PZn2 and PZn3 in the OB29 series (PEO-PBD, 3800 Da) vesicles, but a significant difference resulted in the OB18 series (PEO-PBD, 10400 Da) vesicles, which have thicker hydrophobic membrane cores (Fig. 6). Fewer than 5% of OB18 vesicles deformed when PZn3 was incorporated. While the effect of porphyrin-polymer interactions will be further investigated in a future study, initial results indicate that the localization of the chromophore within the membrane (the PEO-PBD interface versus the core of the hydrophobic phase) affects chromophore-polymer interactions and thus, the extent of membrane instability upon PZn2 light absorption. The peak frequency of deformation that results from differential porphyrin loading (Fig. 5A) demonstrates that a balance exists between PZn2-induced heat production and PZn2-induced membrane stabilization.
Figure 6. Replacing the membrane fluorophore.

When the membrane fluorophore is changed from PZn2 (black) to the more hydrophobic PZn3 (white) there is no noticeable change in frequency of deformation in the OB29 polymer vesicles, however, deformation is significantly reduced in the OB18 polymer (p < 0.005, error bars represent sd).
2.4. Mechanism of Dextran-induced membrane destabilization
We consider various possibilities for the mechanism of membrane destabilization. Recently, Mabrouk et al. [35] showed that polymersomes with one UV-responsive inner leaflet similarly burst in response to UV light exposure. The authors proposed that the mechanism of membrane bursting was due to asymmetric stresses on the membrane. In response to UV light, the responsive leaflet changed conformation introducing excess surface area in one leaflet and increasing the spontaneous curvature there. Upon thermal activation, the additional surface area of the inner leaflet led to the formation of a pore, whose growth enabled relaxation of surface elastic energy (caused by the increased spontaneous curvature) and ultimately membrane rupture. In the present study, we believe this proposed mechanism applies to the dextran-induced polymersome bursting since our studies show dextran enters the PEO corona and decreases interfacial tension, thereby increasing the surface area of the inner leaflet. This mechanism is consistent with the decrease in membrane Ka upon dextran encapsulation. The decrease in interfacial tension should lead to a relaxation of the polymer chains in the inner leaflet and a subsequent increased inner leaflet surface area due to chain relaxation[29] (Fig. 7A). As dextran chain length and membrane interactions increase with MW, dextran both physically entangles itself in and chemically interacts with the inner membrane leaflet. Along with the increase in the inner leaflet surface area, the inner leaflet would be restricted in movement upon heating while the outer leaflet would be free to move. This differential expansion upon localized PZn2 heating would finally cause pore growth and ultimately membrane rupture.
Figure 7. Schematic of proposed photo-activated polymersome deformation.

(A) Proposed Mechanism (a) Polymersome membranes without encapsulant have chains that balance interfacial tension and chain entropy to exist in a semi-coiled state (b) Dextran penetration into corona reduces interfacial tension and causes further coiling of chains (c) Excess area is created in inner leaflet changing the spontaneous curvature. Dextran chemical and physical interaction with inner leaflet restricts chain movement and thermal activation initiates the formation of a pore. (d) Continued localized heating by porphyrin initiates pore growth which reduces spontaneous curvature. Pore growth without self healing of the polymers results in membrane bursting and curling. (B) Alternate mechanism (a) same as above (b) Dextran absorption on the outer surface of the PEO brush causes a change in the spontaneous curvature (curvature denoted by white dotted line). (c) Localized heating provides the energy for the membrane to diffuse and achieve its intrinsic curvature. This membrane movement leads to budding and membrane stretching and is not believed to describe the current system.
A second possible mechanism of membrane destabilization, that we do not believe describes our system, would be dextran-induced curvature strain at the membrane (Fig. 7B). Several studies have shown that membrane interacting polymers can induce a spontaneous curvature in both leaflets of the membrane by simply exerting pressure against them.[5–7, 10] Opposed to insertion into the polymer membrane and thereby causing an increased leaflet area, polymers or proteins act as ‘curvactants,’[6] binding or interacting with the membrane and leading to intrinsic curvature strain at the membrane. Any imposed deformation at this location can lead to reduction in the interfacial energy through membrane displacement or reorganization to deal with the imposed curvature at this point. This mechanism would require dextran to interact more passively at the PEO brush and would more likely lead to local membrane changes like budding and pearling instabilities. As a result of this type of curvactant-induced stress on the membrane, the membrane Ka often increases due to decreased membrane mobility induced by the curvactant.[36, 37] For this reason, this passive binding and curvature strain-induced deformation is not likely the mechanism in our current system, since dextran causes a decrease in membrane Ka (Fig 4). In our previous ferritin study, however, in which ferritin protein was the luminal encapsulant, the Ka did increase, suggesting we might have been observing curvactant-induced deformation (i.e. budding and membrane stretching). As ferritin is much larger and more rigid than dextran, it is likely that ferritin did not enter into the PEO corona, but rather physisorbed onto the outer edge of the membrane. The finding that the Ka increased upon ferritin encapsulation is consistent with this type of interaction.
The results of the present study suggest the former mechanism of polymersome rupture dictates our photo-labile polymersome/porphyrin/dextran composite system. Due to dextran intercalation into the membrane, interfacial tension in the inner leaflet is reduced and the inner leaflet increases in surface area. Since the outer leaflet does not come into as much contact with dextran, due to the lumen encapsulation, the inner leaflet experiences a relative increase in surface area, and the membrane bilayer becomes asymmetric with a differential stress exerted across it. The instability that this asymmetry imparts to the membrane is exacerbated and thus, controllable by light exposure. Upon light activation, PZn2 provides, via nonradiative decay, the thermal energy required to increase membrane motion and relieve the differential stress created by the asymmetric membrane. This membrane motion either leads to membrane shape changes or pore formation and growth, ultimately leading to membrane rupture.
3. Conclusions
We show that polymersomes with a conjugated multi-porphyrin chromophore solute in the hydrophobic core and dextran in the aqueous lumen are photo-responsive. The extent of photo-response increases with the molecular weight and concentration of dextran. The dynamic response of vesicles, as well as the decrease in area expansion modulus with dextran, suggests dextran associates with the inner leaflet of the vesicles and thus causes a differential response of the two leaflets upon illumination of the low quantum-yield dye, leading to membrane deformation and rupture. Since dextran is an innocuous biological solute, these membrane composites allow the construction of photo-sensitive structures with biological applications.
4. Experimental
Materials
Two molecular weight polyethylene oxide-polybutadiene diblock copolymers (PEO30-PBD46, denoted OB29 and PEO80-BD125, denoted OB18) were purchased from Polymer Source, Inc (Montreal, Quebec, Canada). A molecular weight series of Dextran (Leuconostoc mesenteroides and Leuconostoc spp.), fluorescein isothiocyanate (FITC)-labeled dextan (5000 MW), and phosphate buffered saline (PBS) tablets were purchased from Sigma Aldrich (St. Louis, MO). Polyethylene oxide (200,000 MW) was purchased from Polyscience. Methylene chloride (HPLC grade) and sucrose were purchased from Fisher Chemicals (Pittsburgh, PA). The meso-to-meso ethyne bridged (porphinato)zinc(II) dimer (PZn2) and trimer (PZn3) were synthesized in the Therien laboratory following methodology and photo-physical characterization previously described[23].
Vesicle Preparation
The near infrared chromophore, porphyrin (PZn2), was dissolved in methylene chloride and added to the diblock copolymer PEO30-b-PBD46 (MW= 3800 g mol−1) at a 7.64 M:1 M ratio (polymer:fluorophore). The resulting solution was uniformly coated on the rough side of a Teflon strip and the solvent was evaporated for > 24 h. A 10 mg/mL dextran-sucrose solution was created by dissolving dextran of the desired MW in 2 mL of sucrose solution (290 mOsm). After hydration of the polymer-porphyrin film with the dextran-sucrose solution, the system was heated at 60°C for >24 h and vortexed, resulting in spontaneous budding of giant NIR emissive polymersomes off the Teflon into the surrounding aqueous solution. The resulting giant polymersomes contained porphyrin loaded in the membrane (7.64:1 molar ratio of polymer to porphyrin) and dextran loaded in the aqueous core of the polymersomes (10 mg/mL). Vesicles were separated from free dextran by dilution of polymersomes in PBS. The diluted polymersome sample was placed in a centrifuge tube, which contained a cushion of sucrose buffer + density gradient medium (Optiprep, Sigma-Aldrich) and was spun (15,000 rpm, 1 hr, 4°C) to separate the vesicles. The separated vesicles were then dialyzed into PBS (290 mOsm, 12 hr, 4°C).
Rupture and Release of Dextran Encapsulated Polymersomes
Confocal laser scanning microscopy (CLSM) was used to expose dextran polymersomes to light at wavelengths of 458, 488, 543, and 633 nm for 3 min. An Olympus Fluoview FV1000 confocal microscope (Center Valley, PA) with a UPLFLN 40x oil objective lens was used to image the vesicles with a scan speed of 2.0 μs/pixel (2.213 s/frame) for a total time of 3 min 26 ms. The resulting membrane rupture and deformation of the polymersome sample that occurred during this time was observed and the percent of polymersomes that deformed for a given MW of dextran (n=3) was recorded.
Fluorescence Recovery After Photobleaching (FRAP)
The Olympus Fluoview FV1000 confocal microscope, with 40x oil objective lens (described above), was used for the FRAP study. FITC conjugated dextran was encapsulated in the aqueous core of polymersomes that contained membrane encapsulated PZn2. Two regions of the polymersome, at the membrane-core interface and in the center of the polymersomes, were chosen for imaging. Following a period of imaging, the selected region of the polymersome was photobleached with 351 and 364 nm lasers at 80% power for 8 s. The polymersome area beyond the bleaching area was not imaged in order to reduce photobleaching effects across the entire vesicle. The fluorescence intensities of the regions were tracked before, during, and after the bleaching using a 488 nm laser for FITC.
Micropipete Aspiration
Mircopipet aspiration of polymersomes followed similar procedures to those described by Evans et al [38]. Briefly, micropipets made of borosilicate glass tubing (Griedrich and Dimmock, Milville, NJ) were prepared using a needle/pipet puller (model 730, David Kopf Instruments, Tujunga, CA) and microforged using a glass bead to give the tip a smooth, flat edge. Inner diameters of pipets used ranged from 5 to 7 μm and were measured using computer imaging software. Pipets were filled with PBS solution and connected to an aspiration station mounted on the side of a Zeiss inverted microscope, equipped with a manometer, Validyne pressure transducer (models DP 15–32 and DP 103–14, Validyne Engineering Corp., Northridge, CA), digital pressure read-outs, micromanipulators (model WR-6, Narishige, Tokyo, Japan), and MellesGriot millimanipulators(course x, y, z contro). Suction pressure was applied via a syringe connected to the manometer. Both dextran encapsulated and unloaded vesicles were picked up by the micropipets and pressure was increased stepwise in 2–5 cm H2O increments. The membrane was allowed 10 s after each pressure change to equilibrate. The resulting membrane extensions and membrane diameter were measured with ImageJ software [39] and used to calculated the area expansion modulus (Ka) of the different polymersomes.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant R01CA115229; NSF MRSEC program under award number DMR05-20020, and a NSF Graduate Fellowship for N.P.K. We would also like to thank Eric Johnston for technical assistance and Joshua Katz for helpful discussions.
Footnotes
Supporting Information is available online from Wiley InterScience or from the author.
Contributor Information
Neha P. Kamat, Departments of Bioengineering and Department of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA 19103 (USA)
Gregory P. Robbins, Departments of Bioengineering and Department of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA 19103 (USA)
Jeffrey S. Rawson, Department of Chemistry, Duke University, Durham, NC 27708 (USA)
Michael J. Therien, Department of Chemistry, Duke University, Durham, NC 27708 (USA)
Ivan J. Dmochowski, Department of Chemistry, University of Pennsylvania, Philadelphia, PA 19104 (USA)
Daniel A. Hammer, Email: hammer@seas.upenn.edu, Departments of Bioengineering and Department of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA 19103 (USA)
References
- 1.Sheetz MP, Singer SJ. Proc Natl Acad Sci U S A. 1974;71:4457–61. doi: 10.1073/pnas.71.11.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMahon HT, Gallop JL. Nature. 2005;438:590–6. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 3.Berndl K, Lipowsky KJR, Sackmann E, Seifert U. Europhys Lett. 1990;13:659–664. [Google Scholar]
- 4.Ivanitskii GR, Krest’eva IB, Khizhnyak EP, Deev AA, Rudneva OA. Dokl Biophys. 2000;373–375:41–4. doi: 10.1023/a:1026640410526. [DOI] [PubMed] [Google Scholar]
- 5.Koltover I, Radler JO, Safinya CR. Phys Rev Lett. 1998;82:1991–1994. [Google Scholar]
- 6.Sens P, Johannes L, Bassereau P. Curr Opin Cell Biol. 2008;20:476–82. doi: 10.1016/j.ceb.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Yim H, Kent MS, Sasaki DY, Polizzotti BD, Kiick KL, Majewski J, Satija S. Phys Rev Lett. 2006;96:198101. doi: 10.1103/PhysRevLett.96.198101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsafrir I, Sagi D, Arzi T, Guedeau-Boudeville MA, Frette V, Kandel D, Stavans J. Phys Rev Lett. 2001;86:1138–41. doi: 10.1103/PhysRevLett.86.1138. [DOI] [PubMed] [Google Scholar]
- 9.Longo ML, Waring AJ, Hammer DA. Biophys J. 1997;73:1430–9. doi: 10.1016/S0006-3495(97)78175-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farsad K, De Camilli P. Curr Opin Cell Biol. 2003;15:372–81. doi: 10.1016/s0955-0674(03)00073-5. [DOI] [PubMed] [Google Scholar]
- 11.Leibler S. J Phys. 1986;47:507–516. [Google Scholar]
- 12.Solon J, Gareil O, Bassereau P, Gaudin Y. J Gen Virol. 2005;86:3357–63. doi: 10.1099/vir.0.81129-0. [DOI] [PubMed] [Google Scholar]
- 13.Hristova K, Dempsey CE, White SH. Biophys J. 2001;80:801–11. doi: 10.1016/S0006-3495(01)76059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cornelissen J, Fischer M, Sommerdijk N, Nolte RJM. Science (New York, NY) 1998;280:1427–30. doi: 10.1126/science.280.5368.1427. [DOI] [PubMed] [Google Scholar]
- 15.Kadurugamuwa JL, Beveridge TJ. J Antimicrob Chemother. 1997;40:615–21. doi: 10.1093/jac/40.5.615. [DOI] [PubMed] [Google Scholar]
- 16.Robbins GP, Jimbo M, Swift J, Therien MJ, Hammer DA, Dmochowski IJ. J Am Chem Soc. 2008;131:3872–4. doi: 10.1021/ja808586q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsafrir I, Caspi Y, Guedeau-Boudeville MA, Arzi T, Stavans J. Phys Rev Lett. 2003;91:138102. doi: 10.1103/PhysRevLett.91.138102. [DOI] [PubMed] [Google Scholar]
- 18.Discher BM, Won YY, Ege DS, Lee JC, Bates FS, Discher DE, Hammer DA. Science (New York, NY) 1999;284:1143–6. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- 19.Blanazs A, Massignani M, Battaglia G, Armes SP, Ryan AJ. Adv Funct Mater. 2009:1–9. [Google Scholar]
- 20.Lomas H, Canton I, MacNeil S, Du J, Armes SP, Ryan AJ, Lewis AL, Battaglia G. Adv Mater. 2007;19:4238–4243. [Google Scholar]
- 21.Discher DE, Eisenberg A. Science (New York, NY) 2002;297:967–73. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- 22.Ghoroghchian PP, Lin JJ, Brannan AK, Frail PR, Bates FS, Therien MJ, Hammer DA. Soft Matter. 2006;2:973–980. doi: 10.1039/b604212k. [DOI] [PubMed] [Google Scholar]
- 23.Duncan TV, Susumu K, Sinks LE, Therien MJ. J Am Chem Soc. 2006;128:9000–1. doi: 10.1021/ja061897o. [DOI] [PubMed] [Google Scholar]
- 24.Ghoroghchian PP, Frail PR, Susumu K, Blessington D, Brannan AK, Bates FS, Chance B, Hammer DA, Therien MJ. Proc Natl Acad Sci U S A. 2005;102:2922–7. doi: 10.1073/pnas.0409394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vieira NAB, Moscardini MS, Tiera VAdO, Tiera MJ. Carb Polym J. 2002;53:137–143. [Google Scholar]
- 26.Filippov SK, Lezov AV, Sergeeva OY, Olifirenko AS, Lesnichin SB, Domnina NS, Komarova EA, Almgren M, Karlsson G, Stepanek P. Eur Polym J. 2008;44:3361–3369. [Google Scholar]
- 27.Bermudez H, Brannan AK, Hammer DA, Bates FS, Discher DE. Macromolecules. 2002;35:8203–8208. [Google Scholar]
- 28.Evans E, Needham D. J Phys Chem. 1987;91:4219–4228. [Google Scholar]
- 29.Santore MM, Discher DE, Won Y-Y, Bates FS, Hammer DA. Langmuir. 202(14):2385–2395. [Google Scholar]
- 30.Fa N, Lins L, Courtoy PJ, Dufrene Y, Van Der Smissen P, Brasseur R, Tyteca D, Mingeot-Leclercq MP. Biochim Biophys Acta. 2007;1768:1830–8. doi: 10.1016/j.bbamem.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 31.Ringsdorf H, Schlarb B, Venzmer J. Angew Chem Int Ed Engl. 1988;27:113–158. [Google Scholar]
- 32.Turro NJ. Modern Molecular Photochemistry. Benjamin/Cummings; Menlo Park: 1978. [Google Scholar]
- 33.Ghoroghchian PP, Frail PR, Li G, Zupancich JA, Bates FS, Hammer DA, Therien MJ. Chem Mater. 2007;19:1309–1318. doi: 10.1021/cm062427w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duncan TV, Ghoroghchian PP, Rubtsov IV, Hammer DA, Therien MJ. J Am Chem Soc. 2008;130:9773–84. doi: 10.1021/ja711497w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mabrouk E, Cuvelier D, Brochard-Wyart F, Nassoy P, Li MH. Proc Natl Acad Sci U S A. 2009;106:7294–8. doi: 10.1073/pnas.0813157106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohing K, Iglic A, May S. Europhys Lett. 2005;71:145–151. [Google Scholar]
- 37.Rawicz W, Olbrich KC, McIntosh T, Needham D, Evans E. Biophy J. 2000;79:328–39. doi: 10.1016/S0006-3495(00)76295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans EA, Skalak R. CRC Crit Rev Bioeng. 1979;3:181–330. [PubMed] [Google Scholar]
- 39.Rasband WS. 1997–2009 http://rsb.info.nih.gov/ij/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.