

Abstract
The two fused five- and six-membered rings in the molecule of the title compound, C8H6BrN3O2, are approximately coplanar, the largest deviation from the mean plane being 0.011 (3) Å at the NH atom. The acetyl group is slightly twisted with respect to the imidazo[4,5-b]pyridine system, making a dihedral angle of 2.7 (2)°. In the crystal, adjacent molecules are linked by intermolecular N—H⋯N and C—H⋯O hydrogen bonds, forming infinite chains.
Related literature
For background information on the pharmacological activities of imidazo[4,5-b]pyridines, see: Kale et al. (2009 ▶); Silverman (2004 ▶); Cristalli et al. (1995 ▶); Cundy et al. (1997 ▶); Banie et al. (2007 ▶); Mader (2008 ▶); Janssens et al. (1985 ▶); Bavetsias et al. (2007 ▶); Coates et al. (1993 ▶).
Experimental
Crystal data
C8H6BrN3O2
M r = 256.07
Triclinic,
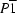
a = 4.8302 (15) Å
b = 9.645 (3) Å
c = 9.809 (3) Å
α = 81.542 (7)°
β = 85.735 (7)°
γ = 89.676 (8)°
V = 450.8 (2) Å3
Z = 2
Mo Kα radiation
μ = 4.53 mm−1
T = 298 K
0.41 × 0.16 × 0.11 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.423, T max = 0.607
2559 measured reflections
1685 independent reflections
1583 reflections with I > 2σ(I)
R int = 0.019
Refinement
R[F 2 > 2σ(F 2)] = 0.028
wR(F 2) = 0.074
S = 1.07
1685 reflections
128 parameters
H-atom parameters constrained
Δρmax = 0.43 e Å−3
Δρmin = −0.48 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEPIII (Burnett & Johnson, 1996 ▶) and ORTEP-3 for Windows (Farrugia, 1997 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811017004/dn2682sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811017004/dn2682Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811017004/dn2682Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N2—H2⋯N1i | 0.86 | 2.02 | 2.877 (3) | 175 |
| C3—H3⋯O2ii | 0.93 | 2.56 | 3.481 (3) | 172 |
Symmetry codes: (i) 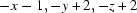 ; (ii)
; (ii) 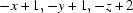 .
.
supplementary crystallographic information
Comment
Imidazo[4,5-b]pyridines represent the major backbone of numerous medical and biochemical agents possessing different chemical and pharmacological features (Kale et al., 2009; Silverman, 2004), which impart them diverse biological properties like antiviral (Cristalli et al.,1995; Cundy et al., 1997; Banie et al., 2007), and anti-inflammatory (Mader, 2008) activity. Substituted imidazo[4,5-b]pyridines have also been tested for their potential selective antihistamine (H1) agents (Janssens et al.,1985). Imidazo[4,5-b]pyridine derivatives were also reported as Aurora kinases (Bavetsias et al., 2007) and cyclic PDE inhibitors (Coates et al.,1993). Importantly,imidazo[4,5-b]pyridine is a structural analogue of purine whose derivatives easily interact with large biomolecules such as DNA, RNA or diverse proteins in vivo.
The molecular plot of the crystal structure of 3-Acetyl-6-bromo-1,3-dihydro- imidazo[4,5-b]pyridin-2-one is shown in Fig.1. The two fused five and six-membered rings building the molecule are nearly planar with the maximum deviation from the mean plane being -0.011 (3) A ° at N2. They form a dihedral angle of 2.7 (2)° with the acetyl group. In the crystal, adjacent molecules are linked by intermolecular N—H···N and C—H···O hydrogen bonding in the way to form infinite chains as shown in Fig. 2 and Table 1.
Experimental
To a stirred solution of 6-bromo-1,3-dihydro-imidazo[4,5 - b-]pyridin-2-one (0.2 g; 93.4 mmol), K2CO3 (0.38 g; 2.8 mmol), and tetra n-butyl ammonium bromide (0.03 g; 9.34 10–5 mol) in DMF, acetyl chloride (0.08 ml; 1.12 mmol) was added dropwise. The mixture was heated under reflux for 24 h. After completion of reaction (monitored by TLC), the salt was filtered and the solvent was removed under reduced pressure. The resulting residue was purified by column chromatography on silica gel using (ethylacetate/hexane) (1/1) as eluent. Crystals were isolated after the solvent was allowed to evaporate.
Refinement
H atoms were located in a difference map and treated as riding with C—H = 0.93 Å for all aromatic H atoms and 0.96 Å for the methyl with Uiso(H) = 1.2 Ueq and Uiso(H) = 1.5 Ueq for the aromatic and methyl respectively.
Figures
Fig. 1.

: Molecular view of the title compound with the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level. H atoms are represented as small circles of arbitrary radii.
Fig. 2.

: Partial packing view showing the N-H···N and C-H···O hydrogen bonds as dashed lines. H atoms not involved in hydrogen bonding have been omitted for clarity. [Symmetry codes: (i) -x-1, -y+2, -z+2; (ii) -x+1, -y+1, -z+2].
Crystal data
| C8H6BrN3O2 | Z = 2 |
| Mr = 256.07 | F(000) = 252 |
| Triclinic, P1 | Dx = 1.887 Mg m−3 |
| Hall symbol: -P 1 | Melting point: 507 K |
| a = 4.8302 (15) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 9.645 (3) Å | Cell parameters from 1685 reflections |
| c = 9.809 (3) Å | θ = 2.1–26.0° |
| α = 81.542 (7)° | µ = 4.53 mm−1 |
| β = 85.735 (7)° | T = 298 K |
| γ = 89.676 (8)° | Fiber, colourless |
| V = 450.8 (2) Å3 | 0.41 × 0.16 × 0.11 mm |
Data collection
| Bruker APEXII CCD diffractometer | 1685 independent reflections |
| Radiation source: fine-focus sealed tube | 1583 reflections with I > 2σ(I) |
| graphite | Rint = 0.019 |
| φ and ω scans | θmax = 26.0°, θmin = 2.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −5→5 |
| Tmin = 0.423, Tmax = 0.607 | k = −11→11 |
| 2559 measured reflections | l = 0→12 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.028 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.074 | H-atom parameters constrained |
| S = 1.07 | w = 1/[σ2(Fo2) + (0.0374P)2 + 0.2048P] where P = (Fo2 + 2Fc2)/3 |
| 1685 reflections | (Δ/σ)max = 0.001 |
| 128 parameters | Δρmax = 0.43 e Å−3 |
| 0 restraints | Δρmin = −0.48 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | −0.0666 (6) | 0.9228 (3) | 0.7492 (3) | 0.0412 (6) | |
| H1 | −0.0912 | 0.9789 | 0.6654 | 0.049* | |
| C2 | 0.1373 (5) | 0.8216 (3) | 0.7536 (2) | 0.0371 (5) | |
| C3 | 0.1881 (5) | 0.7340 (3) | 0.8759 (2) | 0.0377 (5) | |
| H3 | 0.3253 | 0.6658 | 0.8796 | 0.045* | |
| C4 | 0.0199 (5) | 0.7562 (3) | 0.9904 (2) | 0.0355 (5) | |
| C5 | −0.1838 (5) | 0.8602 (3) | 0.9766 (3) | 0.0372 (5) | |
| C6 | −0.2202 (6) | 0.7636 (3) | 1.2009 (3) | 0.0435 (6) | |
| C7 | 0.1715 (7) | 0.5870 (3) | 1.1871 (3) | 0.0466 (6) | |
| C8 | 0.1168 (8) | 0.5301 (3) | 1.3357 (3) | 0.0588 (8) | |
| H8A | 0.2348 | 0.4510 | 1.3590 | 0.088* | |
| H8B | −0.0740 | 0.5012 | 1.3534 | 0.088* | |
| H8C | 0.1541 | 0.6013 | 1.3909 | 0.088* | |
| N1 | −0.2322 (5) | 0.9436 (2) | 0.8618 (2) | 0.0426 (5) | |
| N2 | −0.3244 (5) | 0.8613 (3) | 1.1034 (2) | 0.0435 (5) | |
| H2 | −0.4601 | 0.9164 | 1.1191 | 0.052* | |
| N3 | −0.0003 (5) | 0.6957 (2) | 1.1302 (2) | 0.0395 (5) | |
| O1 | −0.2966 (5) | 0.7398 (2) | 1.3223 (2) | 0.0595 (6) | |
| O2 | 0.3528 (6) | 0.5462 (3) | 1.1138 (2) | 0.0757 (8) | |
| Br1 | 0.35286 (6) | 0.80290 (3) | 0.58804 (2) | 0.04632 (13) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0412 (14) | 0.0477 (14) | 0.0338 (13) | 0.0047 (11) | −0.0012 (10) | −0.0033 (10) |
| C2 | 0.0392 (14) | 0.0439 (13) | 0.0276 (11) | −0.0007 (10) | 0.0057 (10) | −0.0072 (9) |
| C3 | 0.0387 (14) | 0.0428 (12) | 0.0310 (12) | 0.0059 (10) | 0.0035 (10) | −0.0064 (10) |
| C4 | 0.0369 (13) | 0.0405 (12) | 0.0283 (11) | 0.0025 (10) | 0.0032 (9) | −0.0050 (9) |
| C5 | 0.0330 (13) | 0.0449 (13) | 0.0341 (12) | 0.0037 (10) | 0.0031 (10) | −0.0100 (10) |
| C6 | 0.0405 (15) | 0.0523 (15) | 0.0366 (14) | 0.0057 (11) | 0.0085 (11) | −0.0088 (11) |
| C7 | 0.0598 (18) | 0.0451 (14) | 0.0325 (13) | 0.0098 (12) | 0.0069 (12) | −0.0034 (10) |
| C8 | 0.078 (2) | 0.0572 (17) | 0.0349 (15) | 0.0131 (15) | 0.0123 (14) | 0.0039 (12) |
| N1 | 0.0397 (13) | 0.0504 (12) | 0.0371 (12) | 0.0090 (10) | −0.0002 (9) | −0.0059 (9) |
| N2 | 0.0382 (12) | 0.0550 (13) | 0.0359 (12) | 0.0105 (10) | 0.0076 (9) | −0.0074 (9) |
| N3 | 0.0417 (12) | 0.0461 (11) | 0.0287 (10) | 0.0075 (9) | 0.0083 (8) | −0.0045 (8) |
| O1 | 0.0640 (14) | 0.0745 (14) | 0.0354 (11) | 0.0152 (11) | 0.0188 (9) | −0.0049 (9) |
| O2 | 0.0976 (19) | 0.0822 (16) | 0.0386 (11) | 0.0532 (15) | 0.0185 (11) | 0.0060 (10) |
| Br1 | 0.0522 (2) | 0.0565 (2) | 0.02805 (16) | 0.00477 (12) | 0.00836 (11) | −0.00472 (11) |
Geometric parameters (Å, °)
| C1—N1 | 1.354 (3) | C6—O1 | 1.209 (3) |
| C1—C2 | 1.381 (4) | C6—N2 | 1.363 (4) |
| C1—H1 | 0.9300 | C6—N3 | 1.433 (3) |
| C2—C3 | 1.398 (4) | C7—O2 | 1.193 (4) |
| C2—Br1 | 1.894 (2) | C7—N3 | 1.409 (4) |
| C3—C4 | 1.379 (3) | C7—C8 | 1.486 (4) |
| C3—H3 | 0.9300 | C8—H8A | 0.9600 |
| C4—C5 | 1.401 (4) | C8—H8B | 0.9600 |
| C4—N3 | 1.406 (3) | C8—H8C | 0.9600 |
| C5—N1 | 1.319 (3) | N2—H2 | 0.8600 |
| C5—N2 | 1.374 (3) | ||
| N1—C1—C2 | 122.6 (2) | N2—C6—N3 | 105.9 (2) |
| N1—C1—H1 | 118.7 | O2—C7—N3 | 118.5 (2) |
| C2—C1—H1 | 118.7 | O2—C7—C8 | 123.7 (3) |
| C1—C2—C3 | 122.0 (2) | N3—C7—C8 | 117.8 (2) |
| C1—C2—Br1 | 118.30 (19) | C7—C8—H8A | 109.5 |
| C3—C2—Br1 | 119.66 (19) | C7—C8—H8B | 109.5 |
| C4—C3—C2 | 115.1 (2) | H8A—C8—H8B | 109.5 |
| C4—C3—H3 | 122.5 | C7—C8—H8C | 109.5 |
| C2—C3—H3 | 122.5 | H8A—C8—H8C | 109.5 |
| C3—C4—C5 | 119.2 (2) | H8B—C8—H8C | 109.5 |
| C3—C4—N3 | 134.3 (2) | C5—N1—C1 | 115.0 (2) |
| C5—C4—N3 | 106.5 (2) | C6—N2—C5 | 110.9 (2) |
| N1—C5—N2 | 125.6 (2) | C6—N2—H2 | 124.6 |
| N1—C5—C4 | 126.0 (2) | C5—N2—H2 | 124.6 |
| N2—C5—C4 | 108.4 (2) | C4—N3—C7 | 124.2 (2) |
| O1—C6—N2 | 127.0 (3) | C4—N3—C6 | 108.4 (2) |
| O1—C6—N3 | 127.1 (3) | C7—N3—C6 | 127.4 (2) |
| N1—C1—C2—C3 | −0.5 (5) | N1—C5—N2—C6 | −178.9 (3) |
| N1—C1—C2—Br1 | 179.6 (2) | C4—C5—N2—C6 | 0.7 (3) |
| C1—C2—C3—C4 | 0.1 (4) | C3—C4—N3—C7 | 0.3 (5) |
| Br1—C2—C3—C4 | −179.98 (19) | C5—C4—N3—C7 | −179.4 (3) |
| C2—C3—C4—C5 | 0.5 (4) | C3—C4—N3—C6 | −179.8 (3) |
| C2—C3—C4—N3 | −179.2 (3) | C5—C4—N3—C6 | 0.5 (3) |
| C3—C4—C5—N1 | −0.8 (4) | O2—C7—N3—C4 | 2.8 (5) |
| N3—C4—C5—N1 | 178.9 (3) | C8—C7—N3—C4 | −177.5 (3) |
| C3—C4—C5—N2 | 179.5 (2) | O2—C7—N3—C6 | −177.1 (3) |
| N3—C4—C5—N2 | −0.7 (3) | C8—C7—N3—C6 | 2.6 (5) |
| N2—C5—N1—C1 | −179.9 (3) | O1—C6—N3—C4 | −179.6 (3) |
| C4—C5—N1—C1 | 0.5 (4) | N2—C6—N3—C4 | 0.0 (3) |
| C2—C1—N1—C5 | 0.2 (4) | O1—C6—N3—C7 | 0.3 (5) |
| O1—C6—N2—C5 | 179.1 (3) | N2—C6—N3—C7 | 179.8 (3) |
| N3—C6—N2—C5 | −0.4 (3) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N2—H2···N1i | 0.86 | 2.02 | 2.877 (3) | 175. |
| C3—H3···O2ii | 0.93 | 2.56 | 3.481 (3) | 172. |
Symmetry codes: (i) −x−1, −y+2, −z+2; (ii) −x+1, −y+1, −z+2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: DN2682).
References
- Banie, H., Sinha, A., Thomas, R. J., Sircar, J. C. & Richards, M. L. (2007). J. Med. Chem. 50, 5984–5993. [DOI] [PubMed]
- Bavetsias, V., Sun, C., Bouloc, N., Reynisson, J., Workman, P., Linardopoulos, S. & McDonald, E. (2007). Bioorg. Med. Chem. Lett. 17, 6567–6571. [DOI] [PubMed]
- Bruker (2009). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Burnett, M. N. & Johnson, C. K. (1996). ORTEPIII Report ORNL-6895. Oak Ridge National Laboratory, Tennessee, USA.
- Coates, W. J., Connolly, B., Dhanak, D., Flynn, S. T. & Worby, A. (1993). J. Med. Chem. 36, 1387–1392. [DOI] [PubMed]
- Cristalli, G., Vittori, S., Eleuteri, A., Volpini, R., Camaioni, E., Lupidi, G., Mohmoud, N., Bevilacqua, F. & Palu, G. (1995). J. Med. Chem. 38, 4019–4025. [DOI] [PubMed]
- Cundy, D. J., Holan, G., Otaegui, M. & Simpson, G. W. (1997). Bioorg. Med. Chem. Lett. 7, 669–674.
- Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565.
- Janssens, F., Torremans, J., Janssen, M., Stokbroekx, R. A., Luyckx, M. & Janssen, P. A. J. (1985). J. Med. Chem. 28, 1943–1947. [DOI] [PubMed]
- Kale, R. P., Shaikh, M. U., Jadhav, G. R. & Gill, C. H. (2009). Tetrahedron Lett. 50, 1780–1782.
- Mader, M. (2008). Bioorg. Med. Chem. Lett. 18, 179–183.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Silverman, R. B. (2004). The Organic Chemistry of Drug Design and Drug Action, 2nd ed. Amsterdam: Elsevier Academic Press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811017004/dn2682sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811017004/dn2682Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811017004/dn2682Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report