

Abstract
The title compound, C7H4ClFO2, is a twofold halogenated derivative of benzoic acid. The C—C—C angles within the aromatic moiety cover a range 116.11 (14)–123.96 (15)°, with the maximum and the minimum value next to each other. In the crystal, O—H⋯O hydrogen bonds form carboxylic acid dimers, which are further connected by C—H⋯F contacts into undulating sheets perpendicular to the a axis.
Related literature
For the crystal structure of benzoic acid (applying neutron radiation), see: Wilson et al. (1996 ▶). For the crystal structure of ortho-fluorobenzoic acid, see: Krausse & Dunken (1966 ▶) and of ortho-chlorobenzoic acid, see: Ferguson & Sim (1961 ▶); Polito et al. (2008 ▶). For graph-set analysis of hydrogen bonds, see: Etter et al. (1990 ▶); Bernstein et al. (1995 ▶).
Experimental
Crystal data
C7H4ClFO2
M r = 174.55
Monoclinic,
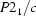
a = 3.7655 (2) Å
b = 13.9660 (7) Å
c = 13.2300 (7) Å
β = 98.034 (3)°
V = 688.92 (6) Å3
Z = 4
Mo Kα radiation
μ = 0.51 mm−1
T = 200 K
0.51 × 0.19 × 0.15 mm
Data collection
Bruker APEXII CCD diffractometer
11312 measured reflections
1671 independent reflections
1267 reflections with I > 2σ(I)
R int = 0.081
Refinement
R[F 2 > 2σ(F 2)] = 0.032
wR(F 2) = 0.081
S = 1.02
1671 reflections
101 parameters
H-atom parameters constrained
Δρmax = 0.23 e Å−3
Δρmin = −0.21 e Å−3
Data collection: APEX2 (Bruker, 2010 ▶); cell refinement: SAINT (Bruker, 2010 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶) and Mercury (Macrae et al., 2006 ▶); software used to prepare material for publication: SHELXL97 and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811016734/gw2099sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811016734/gw2099Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811016734/gw2099Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯O2i | 0.84 | 1.81 | 2.6436 (17) | 172 |
| C5—H5⋯F1ii | 0.95 | 2.46 | 3.175 (2) | 132 |
Symmetry codes: (i) 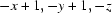 ; (ii)
; (ii) 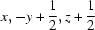 .
.
Acknowledgments
The authors thank Mrs Wilma Nelson for helpful discussions.
supplementary crystallographic information
Comment
Benzoic acid has found widespread use as a ligand in coordination chemistry for a variety of transition metals and elements from the s- and p-block of the periodic system of the elements. It can act as a neutral or – upon deprotonation – an anionic ligand and serve as mono- or bidentate ligand. By varying the substituents on the phenyl moiety, the acidity of the carboxylic acid group can be fine-tuned. Particular interest rests in benzoic acid derivatives showing an asymmetric pattern of substituents on the aromatic moiety due to different possible orientations of the ligand in coordination compounds and the possible formation of stereoisomeric products. At the beginning of a comprehensive study aimed at rationalizing the coordination behaviour of various benzoic acid derivatives towards a number of transition metals in dependence of the pH value of the reaction batches it seemed interesting to determine the crystal structure of the title compound to enable comparative studies.
C–C–C angles within the phenyl ring span a range of 116.11 (14) ° to 123.96 (15) ° with the smallest angle found on the C-atom bearing the carboxylic acid group. The biggest angle is found on the fluorine-bonded C-atom and thus directly adjacent to the smallest one (Fig. 1).
Possibly due to steric factors, the carboxylic acid group is not in plane with the phenyl ring. The least-squares plane defined by its C-atom and O-atoms encloses an angle of 47.83 (6) ° with the least-squares plane defined by the C-atoms of the carbocycle and the halogen-atoms.
In the crystal structure hydrogen bonds between the OH-group and the carbonylic O-atom of the carboxylic acid group give rise to the formation of dimeric units. These units are further connected by C–F···H contacts (whose ranges fall by more than 0.2 Å below the sum of van-der-Waals radii of the respective atoms) to wave-like sheets perpendicular to the crystallographic a axis. The hydrogen atom involved in the latter contacts is present in para-position to the carboxylic acid group on the aromatic carbocycle (Fig. 2). In terms of graph-set analysis, the descriptor for the hydrogen bonds on the unitary level is R22(8) while the C–F···H contacts necessitate a C11(5) descriptor on the same level. No π-stacking is observed in the crystal structure.
The packing of the title compound is shown in Figure 3.
Experimental
The compound was obtained commercially (fluorochem). Crystals suitable for X-ray diffraction were obtained upon slow cooling of a hot aqueous solution of the compound.
Refinement
Carbon-bound H-atoms were placed in calculated positions (C—H 0.95 Å) and were included in the refinement in the riding model approximation, with U(H) set to 1.2Ueq(C). The H atom of the carboxylic acid group was allowed to rotate with a fixed angle around the C—O bond to best fit the experimental electron density (HFIX 147 in the SHELX program suite (Sheldrick, 2008)).
Figures
Fig. 1.

The molecular structure of the title compound, with atom labels and anisotropic displacement ellipsoids (drawn at 50% probability level).
Fig. 2.

Hydrogen bonds (blue dashed lines) and intermolecular C–F···H contacts (yellow dashed lines), viewed along [-1 0 0]. Symmetry operators: ix, -y + 1/2, z + 1/2; iix, -y + 1/2, z - 1/2; iii -x + 1, -y + 1, -z.
Fig. 3.

Molecular packing of the title compound, viewed along [-1 0 0] (anisotropic displacement ellipsoids drawn at 50% probability level).
Crystal data
| C7H4ClFO2 | F(000) = 352 |
| Mr = 174.55 | Dx = 1.683 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 4834 reflections |
| a = 3.7655 (2) Å | θ = 2.9–27.9° |
| b = 13.9660 (7) Å | µ = 0.51 mm−1 |
| c = 13.2300 (7) Å | T = 200 K |
| β = 98.034 (3)° | Needle, colourless |
| V = 688.92 (6) Å3 | 0.51 × 0.19 × 0.15 mm |
| Z = 4 |
Data collection
| Bruker APEXII CCD diffractometer | 1267 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.081 |
| graphite | θmax = 28.3°, θmin = 3.1° |
| φ and ω scans | h = −4→4 |
| 11312 measured reflections | k = −18→18 |
| 1671 independent reflections | l = −17→17 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.032 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.081 | H-atom parameters constrained |
| S = 1.02 | w = 1/[σ2(Fo2) + (0.0326P)2 + 0.237P] where P = (Fo2 + 2Fc2)/3 |
| 1671 reflections | (Δ/σ)max = 0.001 |
| 101 parameters | Δρmax = 0.23 e Å−3 |
| 0 restraints | Δρmin = −0.21 e Å−3 |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.56510 (12) | 0.54455 (3) | 0.33109 (3) | 0.03809 (14) | |
| F1 | 0.8294 (3) | 0.24457 (7) | 0.13846 (8) | 0.0484 (3) | |
| O1 | 0.4025 (4) | 0.39103 (9) | 0.06067 (9) | 0.0441 (3) | |
| H1 | 0.3547 | 0.4253 | 0.0084 | 0.066* | |
| O2 | 0.7380 (4) | 0.51658 (9) | 0.11476 (9) | 0.0418 (3) | |
| C1 | 0.6158 (4) | 0.43686 (11) | 0.12879 (12) | 0.0289 (3) | |
| C2 | 0.7161 (4) | 0.38510 (11) | 0.22715 (11) | 0.0254 (3) | |
| C3 | 0.7186 (4) | 0.42864 (11) | 0.32219 (12) | 0.0266 (3) | |
| C4 | 0.8310 (5) | 0.37956 (13) | 0.41170 (13) | 0.0359 (4) | |
| H4 | 0.8276 | 0.4100 | 0.4758 | 0.043* | |
| C5 | 0.9477 (5) | 0.28638 (14) | 0.40775 (13) | 0.0391 (4) | |
| H5 | 1.0304 | 0.2534 | 0.4694 | 0.047* | |
| C6 | 0.9461 (5) | 0.24039 (13) | 0.31564 (14) | 0.0362 (4) | |
| H6 | 1.0253 | 0.1760 | 0.3127 | 0.043* | |
| C7 | 0.8270 (4) | 0.29017 (12) | 0.22850 (12) | 0.0311 (4) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0461 (3) | 0.0291 (2) | 0.0393 (2) | 0.00636 (18) | 0.00668 (17) | −0.00432 (17) |
| F1 | 0.0872 (9) | 0.0278 (5) | 0.0328 (6) | 0.0057 (5) | 0.0178 (5) | −0.0044 (4) |
| O1 | 0.0638 (9) | 0.0349 (7) | 0.0284 (6) | −0.0170 (6) | −0.0120 (6) | 0.0065 (5) |
| O2 | 0.0631 (8) | 0.0291 (6) | 0.0295 (6) | −0.0149 (6) | −0.0066 (5) | 0.0066 (5) |
| C1 | 0.0364 (8) | 0.0252 (8) | 0.0241 (8) | −0.0017 (6) | 0.0006 (6) | 0.0004 (6) |
| C2 | 0.0282 (8) | 0.0244 (7) | 0.0231 (7) | −0.0018 (6) | 0.0021 (6) | 0.0028 (6) |
| C3 | 0.0255 (7) | 0.0256 (8) | 0.0285 (8) | 0.0010 (6) | 0.0036 (6) | −0.0006 (6) |
| C4 | 0.0411 (9) | 0.0422 (10) | 0.0239 (8) | 0.0015 (8) | 0.0033 (7) | 0.0017 (7) |
| C5 | 0.0442 (10) | 0.0412 (10) | 0.0315 (9) | 0.0074 (8) | 0.0039 (7) | 0.0133 (7) |
| C6 | 0.0417 (9) | 0.0274 (9) | 0.0407 (10) | 0.0078 (7) | 0.0096 (7) | 0.0093 (7) |
| C7 | 0.0399 (9) | 0.0272 (8) | 0.0274 (8) | −0.0010 (7) | 0.0093 (7) | 0.0007 (6) |
Geometric parameters (Å, °)
| Cl1—C3 | 1.7285 (16) | C3—C4 | 1.383 (2) |
| F1—C7 | 1.3519 (18) | C4—C5 | 1.377 (3) |
| O1—C1 | 1.2902 (19) | C4—H4 | 0.9500 |
| O1—H1 | 0.8400 | C5—C6 | 1.377 (3) |
| O2—C1 | 1.2287 (19) | C5—H5 | 0.9500 |
| C1—C2 | 1.490 (2) | C6—C7 | 1.367 (2) |
| C2—C7 | 1.389 (2) | C6—H6 | 0.9500 |
| C2—C3 | 1.395 (2) | ||
| C1—O1—H1 | 109.5 | C5—C4—H4 | 120.1 |
| O2—C1—O1 | 123.63 (14) | C3—C4—H4 | 120.1 |
| O2—C1—C2 | 121.11 (14) | C4—C5—C6 | 120.84 (16) |
| O1—C1—C2 | 115.24 (13) | C4—C5—H5 | 119.6 |
| C7—C2—C3 | 116.11 (14) | C6—C5—H5 | 119.6 |
| C7—C2—C1 | 120.86 (13) | C7—C6—C5 | 118.00 (16) |
| C3—C2—C1 | 122.98 (14) | C7—C6—H6 | 121.0 |
| C4—C3—C2 | 121.23 (15) | C5—C6—H6 | 121.0 |
| C4—C3—Cl1 | 118.11 (12) | F1—C7—C6 | 117.50 (15) |
| C2—C3—Cl1 | 120.62 (12) | F1—C7—C2 | 118.51 (14) |
| C5—C4—C3 | 119.81 (16) | C6—C7—C2 | 123.96 (15) |
| O2—C1—C2—C7 | −131.11 (17) | Cl1—C3—C4—C5 | 178.59 (14) |
| O1—C1—C2—C7 | 47.3 (2) | C3—C4—C5—C6 | −1.6 (3) |
| O2—C1—C2—C3 | 46.4 (2) | C4—C5—C6—C7 | 0.3 (3) |
| O1—C1—C2—C3 | −135.22 (17) | C5—C6—C7—F1 | 179.46 (15) |
| C7—C2—C3—C4 | 0.9 (2) | C5—C6—C7—C2 | 1.7 (3) |
| C1—C2—C3—C4 | −176.64 (15) | C3—C2—C7—F1 | 179.95 (14) |
| C7—C2—C3—Cl1 | −176.68 (12) | C1—C2—C7—F1 | −2.4 (2) |
| C1—C2—C3—Cl1 | 5.7 (2) | C3—C2—C7—C6 | −2.3 (2) |
| C2—C3—C4—C5 | 0.9 (3) | C1—C2—C7—C6 | 175.35 (16) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2i | 0.84 | 1.81 | 2.6436 (17) | 172 |
| C5—H5···F1ii | 0.95 | 2.46 | 3.175 (2) | 132 |
Symmetry codes: (i) −x+1, −y+1, −z; (ii) x, −y+1/2, z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: GW2099).
References
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl. 34, 1555–1573.
- Bruker (2010). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565.
- Ferguson, G. & Sim, G. A. (1961). Acta Cryst. 14, 1262–1270.
- Krausse, J. & Dunken, H. (1966). Acta Cryst. 20, 67–73.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- Polito, M., D’Oria, E., Maini, L., Karamertzanis, P. G., Grepioni, F., Braga, D. & Price, S. L. (2008). CrystEngComm, 10, 1848–1854.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Wilson, C. C., Shankland, N. & Florence, A. J. (1996). J. Chem. Soc. Faraday Trans. pp. 5051–5057.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811016734/gw2099sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811016734/gw2099Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811016734/gw2099Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report