

Abstract
Central core disease (CCD) is a human congenital myopathy characterized by fetal hypotonia and proximal muscle weakness that is linked to mutations in the gene encoding the type-1 ryanodine receptor (RyR1). CCD is thought to arise from Ca2+-induced damage stemming from mutant RyR1 proteins forming “leaky” sarcoplasmic reticulum (SR) Ca2+ release channels. A novel mutation in the C-terminal region of RyR1 (I4898T) accounts for an unusually severe and highly penetrant form of CCD in humans [Lynch, P. J., Tong, J., Lehane, M., Mallet, A., Giblin, L., Heffron, J. J., Vaughan, P., Zafra, G., MacLennan, D. H. & McCarthy, T. V. (1999) Proc. Natl. Acad. Sci. USA 96, 4164–4169]. We expressed in skeletal myotubes derived from RyR1-knockout (dyspedic) mice the analogous mutation engineered into a rabbit RyR1 cDNA (I4897T). Here we show that homozygous expression of I4897T in dyspedic myotubes results in a complete uncoupling of sarcolemmal excitation from voltage-gated SR Ca2+ release without significantly altering resting cytosolic Ca2+ levels, SR Ca2+ content, or RyR1-mediated enhancement of dihydropyridine receptor (DHPR) channel activity. Coexpression of both I4897T and wild-type RyR1 resulted in a 60% reduction in voltage-gated SR Ca2+ release, again without altering resting cytosolic Ca2+ levels, SR Ca2+ content, or DHPR channel activity. These findings indicate that muscle weakness suffered by individuals possessing the I4898T mutation involves a functional uncoupling of sarcolemmal excitation from SR Ca2+ release, rather than the expression of overactive or leaky SR Ca2+ release channels.
Several inherited human muscle diseases, including malignant hyperthermia (MH), Brody disease, and central core disease (CCD), arise from disruptions in Ca2+ homeostasis caused by mutations to proteins that serve essential roles in the regulation of intracellular Ca2+ in skeletal muscle (1, 2). CCD is a congenital myopathy with an autosomal dominant pattern of inheritance (3) that is characterized by hypotonia and proximal muscle weakness in infancy, as well as a variety of skeletal defects (4, 5). Diagnosis of CCD is typically confirmed through histochemical identification of amorphous central areas (cores) in type 1 muscle fibers. The core regions consist of unstructured myofibrils and a general lack (or absence) of mitochondria and oxidative enzymatic activity (6, 7). Electron microscopic analysis of the central cores reveals a disintegration of the contractile apparatus and alterations in the structure and amount of SR and transverse tubule membranes (7).
Patients with CCD are also susceptible to episodes of MH, a related pharmacogenetic disorder in which exposure to inhalation anesthetics and depolarizing muscle relaxants triggers uncontrolled SR Ca2+ release in skeletal muscle (8). Both MH and CCD are caused by mutations in the gene that encodes for the skeletal muscle isoform of the ryanodine receptor (RyR1), the protein that constitutes the tetrameric Ca2+ release channel of the SR (9). Currently, 25 missense mutations in RyR-1 have been shown to be associated with MH, of which nine are also associated with CCD (10–13). Recently, skeletal muscle of individuals possessing certain CCD mutations in RyR1 has also been shown to exhibit thread-like, rod-shaped structures characteristic of nemaline rod myopathy (11, 13). The number of mutations in RyR1 associated with MH and CCD are certain to increase as the search for new mutations continues. In addition, candidate genes other than RyR1 [including the CACNL1A3 gene, which encodes the skeletal muscle dihydropyridine receptor (DHPR); ref. 14] have also been proposed to account for some forms of MH. CCD, on the other hand, appears to be more tightly linked to mutations in the RyR1 gene.
All of the identified MH and CCD point mutations in RyR1 occur within three relatively restricted regions of the RyR1 protein: MH/CCD region 1 (residues 35–614), MH/CCD region 2 (residues 2162–2458), and MH/CCD region 3 (C-terminal region, including residues 4637–4898) (10). MH/CCD regions 1 and 2 have been hypothesized to function as interacting domains that regulate ligand activation of the channel (15), whereas MH/CCD region 3 lies within the transmembrane and pore-forming region of the channel. The MH and CCD mutations in RyR1 are thought to lead to the formation of SR Ca2+ release channels that exhibit varying degrees of Ca2+ leak (8, 11, 16–18). According to this hypothesis, MH mutations result in release channels that exhibit lesser degrees of Ca2+ leak and an increased propensity for activation by RyR1 activators (e.g., caffeine and halothane). Conversely, the CCD mutations would result in release channels that exhibit larger SR Ca2+ leak that ultimately leads to Ca2+-induced damage to the core of the muscle fiber. Indeed, functional analysis of MH and CCD mutant RyR1 proteins expressed in HEK293 cells supports the notion that many of these mutations lead to leaky intracellular Ca2+ release channels (11, 16–18).
Recently, Lynch et al. (1) reported that a mutation within MH/CCD region 3 causes an unusually severe and highly penetrant form of CCD in humans (I4898T). Although all affected individuals in this family are heterozygous for the I4898T RyR1 mutation, the relative expression levels of wild-type and mutant RyR1 proteins in type 1 fibers of these individuals are unknown. Thus, individual differences in expression levels of wild-type and mutant RyR1 proteins may contribute to the clinical variability (from mild to severe) found within this kindred (and possibly others). Cotransfection of HEK-293 cells with cDNAs encoding both wild-type RyR1 and the analogous mutation introduced into the rabbit RyR1 protein (I4897T) increased cytosolic Ca2+ and reduced luminal Ca2+ levels. These results were interpreted to indicate that the symptoms of individuals heterozygous for the I4898T CCD mutation arise from the expression of severely leaky SR Ca2+ release channels, possibly caused by a reduction in Ca2+ required for channel activation (16).
However, the physiological effects of MH and CCD RyR1 mutations on muscle cell function remains to be fully elucidated. Because RyR1 behavior in HEK-293 cells may not accurately reflect activity within a muscle environment, we evaluated the functional consequences [on resting cytosolic Ca2+ levels, luminal SR Ca2+ content, and excitation–contraction coupling (EC coupling)] of homozygous and heterozygous expression of the I4897T RyR1 mutation in skeletal myotubes derived from RyR1-knockout (dyspedic) mice (19). Our results indicate that this form of CCD involves a functional uncoupling of excitation from SR Ca2+ release, rather than the expression of leaky SR Ca2+ release channels.
Methods
Preparation and cDNA Injections of Dyspedic Myotubes.
Primary cultures of myotubes were prepared from skeletal muscle of newborn normal and dyspedic mice (19, 20). The I4897T mutation was introduced into a rabbit RyR1 cDNA by using a standard two-step site-directed mutagenesis strategy. The entire PCR-modified cDNA portion was ultimately confirmed by sequence analysis. Nuclei of dyspedic myotubes were microinjected with cDNAs encoding CD8 (0.2 μg/μl) and either wild-type rabbit RyR1 (0.5 μg/μl), I4897T (0.5 μg/μl), or a 1:1 mixture of both (0.25 μg/μl each). Expressing myotubes were identified 2–4 days after cDNA microinjection following incubation with CD8 antibody beads or by the presence of electrically evoked (8.0 V, 10–30 ms) contractile activity following stimulation with an extracellular electrode (20).
Resting Ca2+ Measurements.
Measurements of resting Ca2+ were obtained in intact, Indo-1 AM-loaded myotubes (21). Cytosolic dye within a small rectangular region of myotubes was excited at 350 ± 7.5 nm by using a 75 W Xenon bulb and a high-speed DeltaRAM illuminator (Photon Technology International, Princeton). Fluorescence emission at 405 nm and 485 nm was collected by using a 40× (1.35 NA) oil-immersion fluorescence objective and a photomultiplier detection system (Photon Technology International). The ratio of fluorescence emission at 405 and 485 (F405/F485) was converted to cytosolic Ca2+ levels by using an in situ calibration approach (22). Briefly, after recording the resting fluorescence ratio in Indo-1 AM loaded myotubes, cells were subsequently perfused with patch pipette solutions containing (in mM): 145 CsCl, 5 MgCl2, 10 BAPTA, 0–10 CaCl2, 10 Hepes, 0.006 K5Indo-1 (pH 7.4). Values of free Ca2+ used in the patch pipette internal solutions were experimentally determined by extrapolating free Ca2+ levels from in vitro calibration curves (Kd = 253 ± 18 nM, n = 3) generated by using a Ca2+ calibration buffer kit (C-3009; Molecular Probes). Following equilibration of the pipette solution, a new ratio at the known free pipette Ca2+ level was recorded. If the concentration of Ca2+ in the pipette was lower than resting Ca2+, then a reduction in the ratio was observed and vice versa. Multiple experiments at four different free Ca2+ levels were used to generate a standard in situ resting Ca2+ calibration curve (Fig. 1A) and determine resting cytosolic Ca2+ levels in intact, Indo-1 AM-loaded myotubes (Fig. 1B). Although the Indo-1 fluorescence ratio obtained for dyspedic myotubes was significantly (P < 0.001) lower than that of normal myotubes, a more precise measurement of the true resting Ca2+ level of uninjected dyspedic myotubes will require the use of a higher affinity Ca2+ dye. Dyspedic myotubes injected with only CD8 cDNA exhibited resting Ca2+ levels (14 ± 7 nM, n = 18) that did not differ significantly (P > 0.4) from that of uninjected dyspedic myotubes, indicating that nuclear microinjection does not significantly disrupt long-term sarcolemmal integrity.
Figure 1.

The I4897T mutation in RyR1 reduces electrically evoked Ca2+ release without altering resting cytosolic Ca2+ levels. (A) In situ calibration (22) of Indo-1 obtained in intact myotubes. (Left) Three representative experiments in which Indo-1 ratios (F405/F485) were obtained in Indo-1 AM-loaded myotubes before and after (arrow) establishment of the whole cell configuration by using internal solutions containing different levels of free Ca2+. (Right) In situ Indo-1 calibration curve used to determine resting Ca2+ levels. This calibration curve was obtained by using four different BAPTA-buffered calibration solutions (closed triangles). The Indo-1 ratio of myotubes dialyzed with the internal solution (except fluo-3 was replaced with Indo-1) used in patch clamp experiments (Figs. 2 and 3) was superimposed on the fitted curve (open triangle). The free Ca2+ level of this internal solution (≈60 nM) is nearly identical to that of intact normal myotubes (Fig. 1B) and, thus, should be sufficient for maintaining the loading state of the SR. (B) Resting Ca2+ levels determined by using the in situ calibration obtained from normal myotubes (NML), uninjected dyspedic myotubes (DYP), and dyspedic myotubes expressing RyR1 alone (RyR-1), RyR1 and I4897T (RyR-1/I4897T), or I4897T alone (I4897T). (C) Electrically evoked (arrows) Ca2+ transients recorded from Indo-1 AM-loaded myotubes in the presence and absence of extracellular Ca2+.
Measurements of Electrically Evoked Ca2+ Transients.
Indo-1 AM-loaded myotubes were stimulated (8.0 V, 10–30 ms) by using an extracellular pipette. Myotubes were bathed in either normal rodent Ringer containing (in mM) 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, and 10 Hepes (pH 7.4 with NaOH), or in Ca2+-free Ringer (made by equimolar substitution of Mg2+ for Ca2+ in the normal rodent Ringer). Electrically evoked Ca2+ transients were not converted to free Ca2+ because Ca2+ levels above ≈150 nM resulted in contractions that could introduce movement artifacts into the calibration.
Simultaneous Measurements in Macroscopic Ca2+ Currents and Ca2+ Transients.
The whole-cell patch clamp technique was used to simultaneously measure L-type Ca2+ currents (L-currents) and Ca2+ transients in expressing myotubes as described (19, 21). Peak L-currents were normalized to cell capacitance (pA/pF), plotted as a function of the membrane potential (I–V curves in Fig. 3B), and fitted according to:
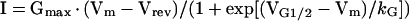 |
1 |
where Vrev is the extrapolated reversal potential of the Ca2+ current, Vm is the membrane potential during the test pulse, Gmax is the maximum L-channel conductance, VG1/2 is the voltage for half activation of Gmax, and kG is a slope factor. Relative changes in cytosolic Ca2+ in patch clamp experiments (Figs. 2 C and D and 3) were recorded in Fluo-3 dialyzed myotubes by using a 40× (0.6 NA) dry fluorescence objective. Fluorescence traces are expressed as ΔF/F and amplitudes at the end of each test pulse were plotted as a function of the membrane potential, and fitted according to:
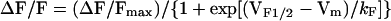 |
2 |
where ΔF/Fmax is the calculated maximal fluorescence change during the test pulse, VF1/2 is the midpoint potential, and kF is a slope factor. The internal solution contained (in mM): 145 Cs-Aspartate, 10 CsCl, 0.1 Cs2-EGTA, 1.2 MgCl2, 5 Mg-ATP, 0.2 K5-Fluo-3, and 10 Hepes (pH 7.4). The external solution contained (in mM): 145 TEA-Cl, 10 CaCl2, 0.003 TTX, and 10 Hepes (pH 7.4).
Figure 3.

The I4897T mutation in RyR1 disrupts voltage-gated SR Ca2+ release but not RyR1-mediated enhancement of DHPR channel activity. (A) Representative L-currents (lower traces) and Ca2+ transients (upper traces) obtained following 30-ms depolarizations to the indicated potentials obtained from uninjected dyspedic myotubes (first column) and dyspedic myotubes expressing RyR1 alone (second column), RyR1/I4897T (third column), or I4897T alone (fourth column). The outward deflection at the beginning of the current traces represents charge movement, which did not differ between uninjected and RyR1-expressing dyspedic myotubes (19, 20). For clarity, the ionic current during the voltage step to +40 mV for the uninjected dyspedic myotube (Inset) and all of the Ca2+ transients for the I4897T-expressing myotube were amplified ten times. Under these recording conditions (0.1 mM EGTA internal solution and 10-s interpulse duration), the baseline fluorescence was similar for all pulses within a sequence, even following pulses that elicit large Ca2+ release. (B–D) Average voltage dependence of peak L-current density (B) and Ca2+ transients (C) for dyspedic myotubes expressing RyR1 alone (black circles), RyR1/I4897T (white squares), or I4897T alone (white triangles). (D) Voltage dependence of the Ca2+ transients normalized to their respective peak values. The U-shaped curve through the data for the I4897T-expressing myotubes was obtained by inverting, and normalizing to one, the I–V curve shown in B.
Figure 2.

Homozygous and heterozygous expression of I4897T in dyspedic myotubes does not alter luminal SR Ca2+ levels monitored with Fluo-3. (A) CPA-induced Ca2+ transients in intact, fluo-3 AM-loaded dyspedic myotubes expressing RyR1 alone (Left), RyR1/I4897T (Center), or I4897T alone (Right). (B) Average maximal values of slow CPA-induced Ca2+ transients obtained from intact myotubes (mean ± SEM). Control experiments revealed that 30 μM CPA induced a similar fluorescence increase in intact normal (0.98 ± 0.30, n = 7) and dyspedic (1.07 ± 0.20, n = 7) myotubes. These values were not significantly different from those of RyR1-, I4897T-, or RyR1/I4897T-expressing myotubes (P > 0.3). In addition, RyR1- and I4897T-expressing myotubes exhibited CPA-induced Ca2+ transients of similar magnitude even when elicited in a nominally Ca2+-free external solution. (C) CPA-induced Ca2+ transients measured in whole-cell patch clamped dyspedic myotubes expressing RyR1 alone (Left), RyR1/I4897T (Center), or I4897T alone (Right). (D) Average maximal values of CPA-induced Ca2+ transients obtained from C (mean ± SEM).
Results
RyR1- and I4897T-Expressing Dyspedic Myotubes Exhibit Similar Resting Cytosolic Ca2+ Levels.
Fig. 1 illustrates the effects of homozygous and heterozygous expression of the I4897T mutation in RyR1 in dyspedic myotubes on both resting cytosolic Ca2+ levels and electrically evoked intracellular Ca2+ transients. Resting cytosolic Ca2+ levels were monitored by using an in situ calibration approach (22) and the Ca2+-sensitive dye, Indo-1 (Fig. 1). These experiments revealed that resting cytosolic Ca2+ levels did not differ significantly (P > 0.5) for normal myotubes and dyspedic myotubes expressing either RyR1 alone, I4897T alone, or coexpressing both RyR1 and I4897T (RyR1/I4897T) (Fig. 1B). Interestingly, the presence of RyR1 exerts a profound effect on resting Ca2+ because uninjected dyspedic myotubes exhibited significantly (P < 0.001) reduced resting cytosolic Ca2+ levels. Because the long-term regulation of cytosolic Ca2+ depends on the balance between net influx and efflux of Ca2+ across the plasma membrane, the presence of RyR1 presumably influences the functional activity and/or expression of one or more essential sarcolemmal Ca2+ transport processes (20).
I4897T Expression Reduces Electrically Evoked Ca2+ Release.
To evaluate the effect of the I4897T mutation on EC coupling under conditions that would minimize the amount of exogenous Ca2+ buffering, the magnitude of electrically evoked intracellular Ca2+ transients was monitored in intact myotubes loaded with Indo-1 AM (Fig. 1C). Both normal myotubes and RyR1-expressing dyspedic myotubes exhibited visible contractions and large intracellular Ca2+ transients following electrical stimulation. In both cases, these Ca2+ transients reflected a skeletal-type EC coupling mechanism (9, 23) because the depolarization-evoked Ca2+ transients were observed even in the absence of extracellular Ca2+. Uninjected dyspedic myotubes and I4897T-expressing dyspedic myotubes never contracted in response to electrical stimulation and completely lacked electrically evoked intracellular Ca2+ transients (Fig. 1C). Because individuals with CCD arising from the I4897T mutation in RyR1 are heterozygous for this mutation (16), we also determined the effect of coexpression of the I4897T mutant with wild-type RyR1. Electrically evoked Ca2+ transients in dyspedic myotubes coinjected with both RyR1 and I4897T were significantly (P < 0.005) diminished compared with Ca2+ transients recorded from either normal myotubes or RyR1-expressing dyspedic myotubes [Rpeak − Rbaseline was 0.38 ± 0.03, 0.46 ± 0.06, and 0.23 ± 0.03 for normal (n = 16), RyR1-expressing dyspedic myotubes (n = 22), and RyR1/I4897T-expressing dyspedic myotubes (n = 35), respectively]. Nevertheless, the small electrically evoked transients recorded from RyR1/I4897T-expressing dyspedic myotubes reflected a skeletal-type EC coupling mechanism because they persisted in the absence of extracellular Ca2+ (Fig. 1C). These data suggest that the I4897T mutation in RyR1 exerts a dominant negative or “loss-of-function” action on the activity of the resulting SR Ca2+ release channels, consistent with the autosomal dominant pattern of inheritance of this human myopathy (3).
Releasable SR Ca2+ Stores Are Similar for RyR1- and I4897T-Expressing Dyspedic Myotubes.
Although the I4897T mutation in RyR1 leads to a depletion of intracellular Ca2+ stores when expressed in HEK-293 cells (16), the effect of this mutation on SR Ca2+ content within a muscle environment is unknown. To clarify this point, we determined the effect on SR Ca2+ stores of homozygous and heterozygous I4897T expression in Fluo-3-loaded dyspedic myotubes (Fig. 2). For these experiments, luminal SR Ca2+ stores were assessed following treatment with cyclopiazonic acid (CPA), an agent that increases cytosolic Ca2+ by reversibly inhibiting SR Ca2+-ATPase (SERCA) pumps, and thereby prevents the reuptake of Ca2+ lost through passive leak pathways.§ Application of CPA (30 μM) induced reversible increases in cytosolic Ca2+ in intact dyspedic myotubes expressing RyR1 alone, I4897T alone, or RyR1/I4897T (Fig. 2 A and B). For wild-type RyR1-expressing dyspedic myotubes, a rapid, presumably Ca2+-induced–Ca2+-release (CICR) transient was often (9 of 10 experiments) observed during the initial CPA-induced increase in fluorescence. Rapid CICR transients were also observed for most RyR1/I4897T-expressing myotubes (9 of 10 experiments), but never for myotubes expressing I4897T alone (0 of 10 experiments). Nevertheless, a similar time course and magnitude of slow SR Ca2+ leak following application of CPA was observed under each condition. Similar results were observed for myotubes monitored under whole-cell patch clamp conditions (Fig. 2 C and D). For these experiments, rapid CICR transients were occasionally observed for dyspedic myotubes expressing RyR1 (7 of 14 experiments), but not RyR1/I4897T (0 of 4 experiments) or I4897T alone (0 of 8 experiments). The reduced occurrence of CICR transients in patch clamped myotubes is presumably due to the presence of an additional Ca2+ buffer (EGTA) in the internal solution. The lack of an effect of the I4897T mutation on resting Ca2+ levels (Fig. 1B) and SR Ca2+ content (Fig. 2) indicates that the reductions in electrically evoked Ca2+ release observed following homozygous and heterozygous expression of I4897T (Fig. 1C) does not arise from the introduction of severely leaky SR Ca2+ release channels.
Expression of I4897T Fully Restores L-Current Density but not Voltage-Gated SR Ca2+ Release.
Skeletal muscle EC coupling involves a unique, bidirectional signaling interaction between sarcolemmal DHPRs and RyR1 proteins of the SR (19). Specifically, during EC coupling in skeletal muscle, sarcolemmal depolarization induces voltage-driven conformational changes in the DHPR that result in the opening of nearby SR Ca2+ release channels (termed voltage-gated SR Ca2+ release or “orthograde coupling”) (9, 23). In addition, the presence of RyR1 proteins enhances the Ca2+ conducting activity of the skeletal muscle DHPR (termed “retrograde coupling”), demonstrating the reciprocal nature of the DHPR-RyR1 gating interaction (19). Effects of CCD mutations in RyR1 on orthograde and retrograde coupling have not been investigated. We used the whole-cell patch clamp technique in conjunction with the Ca2+-sensitive dye, fluo-3, to simultaneously monitor macroscopic L-currents and voltage-gated SR Ca2+ release in uninjected dyspedic myotubes and dyspedic myotubes expressing RyR1, I4897T, or RyR1/I4897T (Fig. 3). Uninjected dyspedic myotubes (Fig. 3A, first column) completely lack voltage-gated SR Ca2+ release and exhibit a low density of L-current (Gmax = 38 ± 4 nS/nF, n = 14) (19, 20). RyR1-expressing dyspedic myotubes (Fig. 3A, second column) exhibit a higher L-current density (Gmax = 133 ± 10 nS/nF, n = 22) and significant voltage-gated SR Ca2+ release (ΔF/Fmax = 3.3 ± 0.5, n = 22). Thus, reintroduction of RyR1 into dyspedic myotubes restores both the orthograde and retrograde signals of skeletal muscle EC coupling (19, 20).
Homozygous expression of I4897T in dyspedic myotubes (Fig. 3A, fourth column) fully restored L-currents (Gmax = 156 ± 13 nS/nF, n = 14), but not SR Ca2+ release (maximal ΔF/F < 0.1). I4897T-expressing dyspedic myotubes exhibited Ca2+ transients that were only barely detectable. These Ca2+ transients apparently resulted from Ca2+ influx through L-channels during the pulse because they exhibited a U-shaped voltage-dependence (Fig. 3D), closely followed the time course of the ionic current, and were blocked by the addition of extracellular Cd2+ (0.5 mM) and La3+ (0.2 mM). The I4897T mutant was likely to be functionally expressed within junctions between the SR and the sarcolemma because L-channel activity was restored to a similar degree following expression of I4897T alone and wild-type RyR1 alone (Table 1). In addition, fluorescence microscopic analysis of dyspedic myotubes injected with cDNA constructs that fused a green fluorescent protein molecule to the N terminus of RyR1 and I4897T indicated that both proteins were expressed to similar levels (data not shown). Coexpression of I4897T and RyR1 in dyspedic myotubes (Fig. 3A, third column) also fully restored L-current density (Gmax = 152 ± 14 nS/nF, n = 19), but only partially restored voltage-gated SR Ca2+ release (ΔF/Fmax = 1.3 ± 0.3, n = 19). On average, coexpression of I4897T/RyR1 resulted in a 60% reduction in the amplitude of the maximal Ca2+ transient (Table 1). Nevertheless, the reduced transients observed in I4897T/RyR1-expressing myotubes represent a skeletal EC coupling mechanism because (i) they exhibit a sigmoidal dependence on voltage (Fig. 3 C and D) and (ii) electrically evoked transients persist in the absence of extracellular Ca2+ (Fig. 1C). Thus, these data demonstrate that the I4897T mutation exerts a potent and selective reduction in voltage-gated SR Ca2+ release.
Table 1.
Parameters of fitted I–V and ΔF/F–V curves
| n | I–V
data
|
ΔF/F–V data
|
|||||
|---|---|---|---|---|---|---|---|
| Gmax, nS/nF | VG1/2, mV | kG, mV | ΔF/Fmax | VF1/2, mV | kF, mV | ||
| Dyspedic | 14 | 38 ± 4* | 32.3 ± 1.8* | 9.4 ± 0.5* | |||
| RyR-1 | 22 | 133 ± 9 | 25.0 ± 1.1 | 7.0 ± 0.2 | 3.3 ± 0.5 | 12.8 ± 1.6 | 5.5 ± 0.4 |
| RyR-1/I4897T | 19 | 152 ± 14 | 24.3 ± 1.1 | 6.8 ± 0.2 | 1.3 ± 0.3* | 15.5 ± 1.9 | 6.4 ± 0.6 |
| I4897T | 14 | 156 ± 13 | 18.9 ± 1.2* | 6.0 ± 0.2* | |||
Compared to RyR-1, P < 0.001.
Discussion
The clinical and histologic abnormalities associated with CCD were originally hypothesized (24) to result from a defect in Ca2+ regulation in central regions of skeletal muscle fibers caused by the presence of overactive or “leaky” SR Ca2+ release channels. This “leaky channel hypothesis” is now the prevailing mechanism used to explain all of the pathophysiologic effects produced by each of the different CCD mutations in RyR1 (1, 8, 10, 11, 16–18, 24). Though this may be the case for certain CCD mutations in RyR1, our results indicate that I4897T-containing release channels operating within skeletal muscle are not excessively leaky to Ca2+, but on the contrary, release Ca2+ inefficiently even following activation by sarcolemmal DHPRs. It is not clear exactly what accounts for the difference between our data and those obtained by Lynch et al. (16). However, because RyR1 activity in skeletal muscle is strongly influenced by the presence of several regulatory proteins and factors, some of which are relatively muscle-specific (e.g., skeletal DHPR, triadin, calsequestrin, and FKBP12), it seems likely that the behavior of mutant release channels expressed in HEK293 cells may not precisely reflect their activity within a skeletal muscle environment. Clearly, it will be important for future studies to determine the effects of the other MH and CCD mutations in RyR1 on resting Ca2+ levels, SR Ca2+ content, and EC coupling in excised muscle fibers obtained from affected individuals and/or following expression of identified mutants in dyspedic myotubes.
Two affected individuals in the family possessing the I4897T mutation in RyR1 were diagnosed as malignant hyperthermia susceptible (MHS) by using the European MH Group in vitro contracture test protocol. However, no anaesthetic complications were recorded in the medical history of the family, despite the exposure of 19 affected members to MH-triggering agents. A similar lack of MH occurrence was also reported for another C-terminal CCD mutation in RyR1 (T4637A) in which no MH episodes were reported for ten affected individuals that underwent a total of 22 surgical procedures conducted under general anesthesia (13). Thus, CCD arising from certain mutations within MH/CCD region 3 may not correlate with MH susceptibility as strongly as some of the other mutations identified in MH/CCD regions 1 and 2. This observation supports the notion that at least two distinct cellular mechanisms may lead to the skeletal muscle abnormalities associated with CCD in humans.
In skeletal muscle, the release of Ca2+ from the SR is controlled by a unique, reciprocal gating interaction between sarcolemmal L-channels and SR Ca2+ release channels (9, 19, 23). The most plausible explanation for this bidirectional control of channel activity (19, 20) is that the channel activities of the DHPR and RyR1 are regulated by a mechanical, possibly direct, interaction between the two proteins (9, 23). Thus, our observation that homozygous and heterozygous expression of I4897T in dyspedic myotubes fully restored DHPR channel activity suggests that I4897T-containing release channels are not only targeted to junctions between the sarcolemma and the SR, but also interact with DHPRs that are present in the junction.
A reduction in voltage-gated SR Ca2+ release following expression of I4897T mutant RyR1 proteins could arise from a selective disruption of orthograde coupling or via alterations in Ca2+ flux through activated release channels (25). With regard to the latter possibility, the isoleucine at position 4897 (corresponding to 4898 within the human RyR1 protein) has been shown to contribute to a conserved RyR pore-forming sequence (25–27). In fact, conservative hydrophobic mutations (A, L, or V) of I4897 in RyR1 result in marked reductions in the single channel conductance, activation by Ca2+, and Ca2+ permeation of the resulting mutant release channels (27). Although the single channel properties of homomeric and heteromeric release channels comprised of the I4897T CCD mutant RyR1 protein are currently unknown, it seems likely that a nonconservative, hydrophilic threonine substitution for I4897 would also result in dire alterations in release channel permeation and/or gating.
Whatever the effects on release channel permeation properties, our results demonstrate that the I4897T mutation imparts a strong loss-of-function alteration in SR Ca2+ release channel activity and suggests that compromised voltage-gated SR Ca2+ release contributes to the muscle weakness and atrophy experienced by individuals heterozygous for this form of CCD. In addition, our results are to our knowledge the first to support the hypothesis originally proposed by Quane et al. (28) that some forms of CCD might result from an uncoupling of muscle excitation from SR Ca2+ release within the core of the muscle fiber. Interestingly, the presence of central cores may in fact not contribute significantly to muscle weakness because there is not a clear relationship between the extent of the central cores and the clinical severity of the disease (29, 30). Thus, alterations in EC coupling, rather than the formation of central cores per se, may underlie muscle weakness associated with certain forms of CCD. In any event, the precise mechanism(s) by which “leaky” or “uncoupled” SR Ca2+ release channels ultimately lead to the development of central cores remains elusive and will require further investigation.
Acknowledgments
We thank Drs. Kurt G. Beam and Paul D. Allen for providing access to the dyspedic mice used in this study, as well as for their advice and continued support. We also thank Drs. T. Begenisich, D. Giovannucci, and S.-S. Sheu and Ms. K.M.S. O'Connell for helpful discussions and comments on the manuscript, and Linda Groom for excellent technical assistance. This work was supported by the National Institute of Health (AR44657 to R.T.D.), a Neuromuscular Disease Research grant from the Muscular Dystrophy Association (to R.T.D.), and a Consejo Nacional de Ciencia y Tecnologia postdoctoral fellowship (to G.A.).
Abbreviations
- CCD
central core disease
- DHPR
dihydropyridine receptor
- L-currents
L-type Ca2+ currents
- RyRs
ryanodine receptors
- SR
sarcoplasmic reticulum
- EC coupling
excitation–contraction coupling
- MH
malignant hyperthermia
- CPA
cyclopiazonic acid
Footnotes
SR Ca2+ stores were evaluated by using CPA instead of an RyR1 agonist because (i) mutations in RyR1 could alter agonist binding and/or channel activation and (ii) the degree of SR Ca2+ leak would be reflected in the rate of CPA-induced elevations in cytosolic Ca2+. Moreover, mutations in RyR1 that abolish or greatly reduce Ca2+ permeation through activated release channels (see Discussion) could lead to an underestimation of SR Ca2+ content when the release channel is activated by an agonist.
References
- 1.MacLennan D H. Eur J Biochem. 2000;267:5291–5297. doi: 10.1046/j.1432-1327.2000.01566.x. [DOI] [PubMed] [Google Scholar]
- 2.Berchtold M W, Brinkmeier H, Muntener M. Physiol Rev. 2000;80:1215–1265. doi: 10.1152/physrev.2000.80.3.1215. [DOI] [PubMed] [Google Scholar]
- 3.Isaacs H, Heffron J J, Badenhorst M. J Neurol Neurosurg Psychiatry. 1975;38:1177–1186. doi: 10.1136/jnnp.38.12.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shy G M, Magee K R. Brain. 1956;79:610–621. doi: 10.1093/brain/79.4.610. [DOI] [PubMed] [Google Scholar]
- 5.Shuaib A, Paasuke R T, Brownwell K W. J Comp Pathol. 1987;97:597–600. [Google Scholar]
- 6.Dubowitz, V. & Pearse, A. G. E. (1960) Lancet 23–24. [DOI] [PubMed]
- 7.Hayashi K, Miller R G, Brownell K W. Muscle Nerve. 1989;12:95–102. doi: 10.1002/mus.880120203. [DOI] [PubMed] [Google Scholar]
- 8.MacLennan D H, Phillips M S. Science. 1992;256:789–794. doi: 10.1126/science.1589759. [DOI] [PubMed] [Google Scholar]
- 9.Franzini-Armstrong C, Protasi F. Physiol Rev. 1997;77:699–729. doi: 10.1152/physrev.1997.77.3.699. [DOI] [PubMed] [Google Scholar]
- 10.McCarthy T V, Quane K, Lynch L P. Hum Mutat. 2000;15:410–417. doi: 10.1002/(SICI)1098-1004(200005)15:5<410::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 11.Monnier N, Romero N B, Lerale J, Nivoche Y, Qi D, MacLennan D H, Fardeau M, Lunardi J. Hum Mol Genet. 2000;9:2599–2608. doi: 10.1093/hmg/9.18.2599. [DOI] [PubMed] [Google Scholar]
- 12.Brown R L, Pollock A N, Couchman K G, Hodges M, Hutchinson D O, Waaka R, Lynch P, McCarthy T V, Stowell K M. Hum Mol Genet. 2000;9:1515–1524. doi: 10.1093/hmg/9.10.1515. [DOI] [PubMed] [Google Scholar]
- 13.Scacheri P C, Hoffman E P, Fratkin J D, Semino-Mora C, Senchak A, Davis M R, Laing N G, Vedanarayanan V, Subramony S H. Neurology. 2000;55:1689–1696. doi: 10.1212/wnl.55.11.1689. [DOI] [PubMed] [Google Scholar]
- 14.Monnier N, Procaccio V, Stieglitz P, Lunardi J. Am J Hum Genet. 1997;60:1316–1325. doi: 10.1086/515454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto T, El-Hayek R, Ikemoto N. J Biol Chem. 2000;275:11618–11625. doi: 10.1074/jbc.275.16.11618. [DOI] [PubMed] [Google Scholar]
- 16.Lynch P J, Tong J, Lehane M, Mallet A, Giblin L, Heffron J J, Vaughan P, Zafra G, MacLennan D H, McCarthy T V. Proc Natl Acad Sci USA. 1999;96:4164–4169. doi: 10.1073/pnas.96.7.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong J, Oyamada H, Demaurex N, Grinstein S, McCarthy T V, MacLennan D H. J Biol Chem. 1997;272:26332–26339. doi: 10.1074/jbc.272.42.26332. [DOI] [PubMed] [Google Scholar]
- 18.Tong J, McCarthy T V, MacLennan D H. J Biol Chem. 1999;272:693–702. doi: 10.1074/jbc.274.2.693. [DOI] [PubMed] [Google Scholar]
- 19.Nakai J, Dirksen R T, Nguyen H T, Pessah I N, Beam K G, Allen P D. Nature (London) 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- 20.Avila G, Dirksen R T. J Gen Physiol. 2000;115:467–479. doi: 10.1085/jgp.115.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia J, Beam K G. J Gen Physiol. 1994;103:107–123. doi: 10.1085/jgp.103.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krause E, Schmid A, Gonzalez A, Schulz I. J Biol Chem. 1999;274:36957–36962. doi: 10.1074/jbc.274.52.36957. [DOI] [PubMed] [Google Scholar]
- 23.Melzer W, Herrmann-Frank A, Luttgau H C. Biochim Biophys Acta. 1995;1241:59–116. doi: 10.1016/0304-4157(94)00014-5. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Chen H S, Khanna V K, de Leon S, Phillips M S, Schappert K, Britt B A, Browell A K, MacLennan D H. Nat Genet. 1993;5:46–50. doi: 10.1038/ng0993-46. [DOI] [PubMed] [Google Scholar]
- 25.Balshaw D, Gao L, Meissner G. Proc Natl Acad Sci USA. 1999;96:3345–3347. doi: 10.1073/pnas.96.7.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao M, Li P, Zhang L, Winkfein R J, Chen S R W. J Biol Chem. 1999;274:25971–25974. doi: 10.1074/jbc.274.37.25971. [DOI] [PubMed] [Google Scholar]
- 27.Gao L, Balshaw D, Xu L, Tripathy A, Xin C, Meissner G. Biophys J. 2000;79:828–840. doi: 10.1016/S0006-3495(00)76339-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quane K A, Healy J M, Keating K E, Manning B M, Couch F J, Palmucci L M, Doriguzzi C, Fagerlund T H, Berg K, Ording H, et al. Nat Genet. 1993;5:51–55. doi: 10.1038/ng0993-51. [DOI] [PubMed] [Google Scholar]
- 29.Morgan-Hughes J A, Brett E M, Lake B D, Tome F M S. Brain. 1973;96:527–535. doi: 10.1093/brain/96.3.527. [DOI] [PubMed] [Google Scholar]
- 30.Palmucci L, Schiffer D, Monga G, Mollo F, de Marchi M. J Neurol. 1978;218:55–61. doi: 10.1007/BF00314719. [DOI] [PubMed] [Google Scholar]