

Abstract
Pendrin is an anion transporter encoded by the PDS/Pds gene. In humans, mutations in PDS cause the genetic disorder Pendred syndrome, which is associated with deafness and goiter. Previous studies have shown that this gene has a relatively restricted pattern of expression, with PDS/Pds mRNA detected only in the thyroid, inner ear, and kidney. The present study examined the distribution and function of pendrin in the mammalian kidney. Immunolocalization studies were performed using anti-pendrin polyclonal and monoclonal antibodies. Labeling was detected on the apical surface of a subpopulation of cells within the cortical collecting ducts (CCDs) that also express the H+-ATPase but not aquaporin-2, indicating that pendrin is present in intercalated cells of the CCD. Furthermore, pendrin was detected exclusively within the subpopulation of intercalated cells that express the H+-ATPase but not the anion exchanger 1 (AE1) and that are thought to mediate bicarbonate secretion. The same distribution of pendrin was observed in mouse, rat, and human kidney. However, pendrin was not detected in kidneys from a Pds-knockout mouse. Perfused CCD tubules isolated from alkali-loaded wild-type mice secreted bicarbonate, whereas tubules from alkali-loaded Pds-knockout mice failed to secrete bicarbonate. Together, these studies indicate that pendrin is an apical anion transporter in intercalated cells of CCDs and has an essential role in renal bicarbonate secretion.
In 1997, the gene (PDS) defective in Pendred syndrome, a genetic disorder associated with deafness and goiter, was identified (1). Since that time, there have been numerous studies aiming to catalog mutations associated with Pendred syndrome (2, 3), to characterize the expression pattern of the gene (4), and to elucidate the function of the encoded protein, pendrin. Because of the hallmark clinical features of Pendred syndrome, initial studies focused on pendrin's role in the inner ear and the thyroid. Based on sequence similarity, pendrin was predicted to function as an anion transporter (1). Heterologous expression studies revealed that pendrin is capable of transporting several different anions; for example, Xenopus laevis oocytes and Sf9 insect cells expressing pendrin manifest sodium-independent iodide/chloride (5) and chloride/formate exchange (6). Immunolocalization studies of human thyroid demonstrated that pendrin is present in a limited subset of cells within the thyroid follicles, exclusively in the apical membrane of the follicular epithelium (7, 8); based on these results and evidence of pendrin's ability to transport iodide, we proposed that pendrin is an apical iodide transporter in the thyroid (7). RNA in situ hybridization studies revealed that Pds is expressed in discrete areas of the mouse inner ear, including regions thought to play a key role in fluid transport (4).
The likely distinct functions of pendrin in the thyroid and inner ear coupled with the protein's ability to transport more than one anion prompted us to investigate pendrin expression and function in other tissues. Here we have investigated the role of pendrin in the mammalian kidney, providing evidence for its role in bicarbonate secretion by the intercalated cells of the cortical collecting ducts (CCDs).
Materials and Methods
Northern Blot Analysis.
RNA was prepared by using the RNeasy Total RNA kit (Qiagen, Chatsworth, CA). Northern blots were prepared as described (9) and hybridized with a Pds-specific probe [see GenBank accession no. AF167412; consisting of a mixture of three PCR products generated as described (7)] or a Pax-8-specific probe (9).
Generation of Anti-Pendrin Antibodies.
We previously reported (7) the generation and characterization of rabbit polyclonal anti-pendrin antibodies (e.g., r630–643). By using the same methods, an additional antibody (h766–780) was raised to a peptide corresponding to amino acids 766–780 of human pendrin (GenBank accession no. AF030880). In addition, an anti-pendrin monoclonal antibody was generated as follows. Recombinant His-tagged pendrin produced in Sf9 cells was purified on Talon nickel affinity matrix (CLONTECH), and mice were injected four times (≈24 days apart) with 10 μg of this purified pendrin. Four days after the final immunization, spleens were harvested for cell fusion (10). Thirteen days after fusion, hybridoma supernatants were screened against 0.25 μg of the purified pendrin. Single-cell cloning was then performed by limiting dilution, and the final clones were cultured in serum-free medium.
Other Antibodies.
The following other antibodies were used for double-labeling studies: a rabbit anti-human aquaporin-2 antibody (11), a rabbit antibody against the B1 subunit of the H+-ATPase [prepared as described by Nelson et al. (12) and characterized by Yasui et al. (13)], a rabbit polyclonal antibody against mouse anion exchanger 1 (AE1; ref. 14), and the mouse monoclonal antibody E11 against the bovine kidney 31-kDa subunit of vacuolar H+-ATPase (15).
Immunolocalization Studies.
Mouse or rat kidneys were removed, longitudinally sliced, fixed in 4% paraformaldehyde in PBS (10 mM sodium phosphate buffer containing 0.9% NaCl, pH 7.4) overnight, and then embedded in paraffin. A paraffin-embedded piece of normal human kidney cortex was obtained from the Pathology Department of Suburban Hospital, Bethesda.
Kidney sections (2 or 3 μm thick) were derived from each sample, deparaffinized, and rehydrated with xylene and graded alcohol. After a 5-minute incubation in PBS, sections were blocked with 3% BSA/5% normal goat serum in PBS for 1 h and then subjected to a 5-min antigen-retrieval treatment with antigen unmasking solution (Vector Laboratories). Incubations with the primary antibodies, diluted in 1% BSA in PBS to the indicated concentrations, were performed for 2 h at room temperature or overnight in a humidified chamber at 4°C. After three rinses in PBS, binding sites of the primary antibodies were detected with a 1:250 dilution of fluorescein-conjugated goat anti-rabbit IgG (Vector Laboratories). Finally, the sections were rinsed with PBS, and coverslips were mounted by using Vectashield (Vector Laboratories) as a fading retardant. For double-labeling experiments, the rabbit polyclonal antibody was detected with fluorescein anti-rabbit and the anti-pendrin monoclonal antibody with Texas red-conjugated horse anti-mouse IgG (1:250; Vector Laboratories). The resulting sections were examined by fluorescence microscopy, with images captured by use of a Photometrics (Tucson, AZ) PXl cooled charge-coupled device camera controlled by iplab spectrum software.
Immunohistochemistry was performed by using the Vectastain Elite kit (Vector Laboratories) according to the manufacturer's instructions. Briefly, the primary antibodies were revealed with biotinylated goat anti-rabbit IgG (1:200), and the sites of peroxidase activity were visualized by using the Vector VIP substrate kit. Immunostaining was detected by light microscopy.
Isolated Perfused Tubule Studies with Pds+/+and Pds−/−Mice.
CCDs were dissected from male or female wild-type (Pds+/+) and pendrin-deficient (i.e., Pds-knockout; Pds−/−) mice (16) at 6–8 weeks of age. The mice received 5 mg/100 g body weight deoxycorticosterone pivalate (DOCP; CIBA–Geigy) by intramuscular injection 5–14 days before sacrifice and drank 50 mM NaHCO3 in drinking water ad libitum for 4 days before sacrifice. They also ate a balanced rodent diet (LabDiet 5001; PMI Nutrition International, Richmond, IN) and were pair fed for at least 4 days before sacrifice. Animals were injected with furosemide (5 mg/100 g body weight i.p.) 30 min before sacrifice by cervical dislocation. The kidneys were immediately removed, and coronal slices were placed in a dissection dish containing the chilled bath and perfusate solution (see below) at 11°C. CCDs were dissected from medullary rays. The juncture of the superficial distal tubule and the CCD was identified, and the CCDs were dissected distal to that site. Tubules were mounted on concentric glass pipettes and perfused in vitro at 37°C. The tubule end, which fell at the juncture of the superficial distal tubule and the collecting duct, was placed in the collection pipette.
Experiments were performed with symmetric solutions in the bath and
perfusate. The solution composition was as follows (in mM): 125 NaCl, 5
KCl, 24 NaHCO3/5% CO2, 1
Na2HPO4, 2
CaCl2, 1.2 MgSO4, and 5.5
glucose. Osmolality was measured in all solutions (17). To maintain the
desired CO2 concentration in the
HCO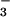 /CO2-buffered solutions,
the perfusate was passed through jacketed concentric tubing through
which 95% air/5% CO2 was blown in a
counter-current direction around the perfusate line (17, 18). To
maintain pH in the bicarbonate-containing solutions, the bath fluid was
constantly bubbled with 95% air/5% CO2. Bath
pH was measured continuously during all experiments as described (17,
19). All collections began 30 min before and terminated 75 min after
warming the tubule.
/CO2-buffered solutions,
the perfusate was passed through jacketed concentric tubing through
which 95% air/5% CO2 was blown in a
counter-current direction around the perfusate line (17, 18). To
maintain pH in the bicarbonate-containing solutions, the bath fluid was
constantly bubbled with 95% air/5% CO2. Bath
pH was measured continuously during all experiments as described (17,
19). All collections began 30 min before and terminated 75 min after
warming the tubule.
Tubule fluid samples were collected under oil in calibrated constriction pipettes. Flow rate was determined as described (18). Total CO2 concentration was measured in the collected fluid (CL) and perfusate (Co) by using a continuous flow fluorimeter (17, 20). The CO2 reagent was purchased as a kit (132-A from Sigma) and diluted to 50% strength with water. By using this method, bicarbonate (total CO2) concentration differences of less than 1 mM can be detected with a pipette of 8 nl (17, 20). Bicarbonate flux, JtCO2, was calculated according to the equation
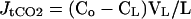 |
where Co and CL are the perfusate and collected fluid total CO2 concentration, respectively. VL is the flow rate in nl/min, and L is the tubule length. In the CCD tubules, net water absorption has not been observed in tubules perfused in vitro with symmetric solutions (21, 22). Thus, net fluid transport was taken to be zero in the absence of an imposed osmolality gradient.
Statistics.
One to three replicate measurements of tCO2 were performed and averaged to obtain a single value for each tubule. When only one measurement was made, this value was reported for that tubule. The “n” reported equals the number of mice studied (with one tubule isolated from each mouse). Statistical significance was determined by using an unpaired two-tailed Student's t test. Statistical significance was achieved with a P < 0.05. Data are displayed as mean ± SE.
Results
Pds Expression in Various Tissues.
Whereas the expression of Pds in the inner ear (4) and thyroid (7) is well established, we sought to identify other tissues expressing significant levels of the gene. Northern blots containing mRNA derived from various rat tissues were hybridized with a Pds-specific probe. Interestingly, Pds expression was most pronounced in the kidney (Fig. 1), with much lower amounts of Pds mRNA detected in the thyroid. The strong expression of Pds in rat kidney, which is much greater than that encountered in human adult and fetal kidney (1), prompted us to investigate further pendrin's localization and function in this tissue.
Figure 1.

Northern blot analysis of Pds mRNA in various rat tissues. A Northern blot containing 10 μg of total RNA per lane from the indicated rat tissues was prepared and hybridized with a Pds-, Pax-8-, or Gapdh-specific probe, as indicated. The Pds-specific probe hybridized only to the ≈5-kb mRNA shown. The Pax-8-specific probe was used as a positive control for the thyroid and kidney mRNA.
Immunolocalization of Pendrin in the Kidney.
To establish the presence and distribution of pendrin in the kidney, we performed immunolocalization studies of mouse, rat, and human kidney samples. Anti-pendrin antibodies label the apical and subapical regions of a subpopulation of mouse (Fig. 2 A and B) and rat (Fig. 2 C and D) kidney cortical cells. No labeling is seen in the basolateral region of cortical cells or any cells within the inner or outer medulla. There was also no evidence of pendrin staining in the proximal tubules. Immunoperoxidase-based staining gave the same general pattern of staining as immunofluorescence (Fig. 3A). Prior incubation of the antibody with an excess of immunizing peptide abolishes the labeling (compare Fig. 3 B with A). Importantly, there is no labeling of kidney sections derived from Pds-knockout mice (Fig. 3C).
Figure 2.

Immunofluorescent staining of pendrin in mouse and rat kidney. Paraformaldehyde-fixed, paraffin-embedded kidney sections were incubated with rabbit anti-pendrin antibodies followed by fluorescein-labeled anti- rabbit secondary antibody (1:250). (A) A low-power composite view (original magnification ×100) of mouse kidney showing cortical cells staining with an anti-pendrin antibody (h766–780; 1:1000). Strong pendrin-specific staining (in green) is seen in the cortex; no staining is detected in the outer or inner medulla. (B) A higher-power view (original magnification ×630) of the section shown in A illustrating the apical staining of pendrin-positive cells. Note that pendrin is only detected in a subset of cells within a given tubule. Similar studies were performed with rat kidney stained with the same antibody as A and B (C) or with a published (7) anti-pendrin antibody (D; r612–625; 1:1000), revealing an identical pattern of apical staining in the cortex. All photomicrographs represent merged images captured with three distinct excitation lights (345, 490, and 540 nm); this approach allows the cellular structure to be visualized in conjunction with the FITC-associated staining (in green). Note that the white areas in A reflect fluorescence of the proximal tubules; the same background is also seen in the presence of immunizing peptide as well as in kidney sections derived from Pds-knockout mice.
Figure 3.

Immunohistochemical staining of pendrin in mouse kidney. Paraformaldehyde-fixed, paraffin-embedded kidney sections from wild-type (Pds+/+; A and B) or pendrin-deficient (Pds−/−; C) mice were incubated with a rabbit anti-pendrin antibody (h766–780; 1:2000). In B, the antibody was preincubated with the pendrin-specific h766–780 peptide. (Original magnification ×100.)
The pattern of immunostaining, with pendrin-positive cells interspersed among unlabeled cells, appears to reflect the heterogeneous nature of the CCD epithelium. To investigate this further, double-labeling studies were performed by using rabbit polyclonal antibodies and an anti-pendrin monoclonal antibody. The CCD protein aquaporin-2 (vasopressin regulated water channel) represents a principal cell-specific marker (23). An anti-aquaporin-2 antibody labels a different set of cells than the anti-pendrin monoclonal antibody on a human kidney section (Fig. 4A), although both sets of cells reside within the same renal tubules. These data indicate that pendrin indeed resides within the intercalated cells of the CCD. Classically, different subtypes of CCD intercalated cells can be distinguished based on their staining for H+-ATPase and the AE1 isoform of the anion exchanger family. For example, in the case of the rat, H+-ATPase is present in the apical membrane of α-intercalated cells (acid-secreting cells) and in the basolateral membrane of β-intercalated cells (bicarbonate-secreting cells) (24), whereas AE1 is restricted to the basolateral membrane of α-intercalated cells (25, 26). Human CCD cells that stain for pendrin in their apical membrane also stain for H+-ATPase in their basolateral and apical membranes (Fig. 4B). In mouse, pendrin-positive cells also stain for H+-ATPase in both the basolateral and apical membranes (Fig. 5A); however, there is no overlap between cells staining for pendrin and AE1 (compare Fig. 5 A and B). Together, our immunolocalization studies demonstrate that pendrin resides in an intercalated cell(s) in the CCD that is distinct from α-cells.
Figure 4.

Localization of pendrin relative to other proteins in human kidney. Paraformaldehyde-fixed, paraffin-embedded human kidney sections were double-labeled with a polyclonal antibody to aquaporin-2 (green; 1:1000) and a monoclonal antibody to pendrin (red; 1:50), with the staining seen in the CCDs shown in A. In B, similar sections were stained with a polyclonal antibody to H+-ATPase (green; 1:1000) and the anti-pendrin monoclonal antibody (red; 1:50). In the merged image, note the α-intercalated cell with apical staining for H+-ATPase (arrowhead) and the two other intercalated cells staining for both H+-ATPase and pendrin (arrows), with the resulting yellow areas corresponding to the pendrin-containing apical regions. Also note the other cells in the same tubule that are negative for both proteins; these are the principal cells. (Original magnification ×1000.)
Figure 5.

Localization of pendrin relative to other proteins in mouse kidney. Paraformaldehyde-fixed, paraffin-embedded mouse kidney sections were double-labeled with the monoclonal antibody E11 to H+-ATPase (red; undiluted) and either the anti-pendrin polyclonal antibody h766–780 (green; 1:1000) or an anti-AE1 antibody (green, 1:500). The micrographs shown in A and B reflect identical areas of two serial sections (2 μm each). When comparing the merged images in A and B, note the different subpopulations of cells staining for pendrin (arrows) and AE1 (arrowheads). Also note that some intercalated cells are negative for both pendrin and AE1. The asterisk indicates red blood cells staining only for AE1. (Original magnification ×1000).
Role of Pendrin in Bicarbonate Transport by the Collecting Duct.
To stimulate bicarbonate secretion in the CCD, mice received deoxycorticosterone pivalate parenterally in addition to NaHCO3 in their drinking water, mimicking protocols previously developed in rats (21). Bicarbonate flux was then measured in perfused CCD segments isolated from these animals. CCD tubules from wild-type mice (Pds+/+) secreted bicarbonate. The collected tCO2 concentration in CCD tubules from these animals was 26.2 ± 0.6 mM, which was above the perfusate tCO2 concentration of 24.8 ± 0.4 mM. In contrast, CCD tubules from pendrin-deficient mice (Pds−/−) failed to secrete bicarbonate and, in fact, manifested a significant rate of bicarbonate absorption. The collected tCO2 concentration was 23.6 ± 0.8 mM, which was below the perfusate tCO2 concentration of 24.9 ± 1.0 mM. The bicarbonate flux, JtCO2, with Pds−/− mice was thus +2.7 ± 0.6 pmol/mm/min (n = 4) vs. −2.6 ± 1.4 pmol/mm/min with Pds+/+ mice (n = 4; P < 0.05). These data are summarized in Fig. 6 and Table 1.
Figure 6.

Influence of pendrin on JtCO2. Isolated CCD
tubules from wild-type (Pds+/+) or
pendrin-deficient (Pds−/−) mice were
perfused in symmetric,
HCO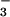 /CO2-buffered solutions.
tCO2 concentration was measured in collected perfusate
samples in CCD tubules. Values measured for each tubules are given (see
Table 1 for additional details). In wild-type mice, the
JtCO2 was −2.6 ± 1.4 pmol/mm/min
(n = 4); in pendrin-deficient mice, the
JtCO2 was +2.7 ± 0.6 pmol/mm/min
(n = 4, P < 0.05).
/CO2-buffered solutions.
tCO2 concentration was measured in collected perfusate
samples in CCD tubules. Values measured for each tubules are given (see
Table 1 for additional details). In wild-type mice, the
JtCO2 was −2.6 ± 1.4 pmol/mm/min
(n = 4); in pendrin-deficient mice, the
JtCO2 was +2.7 ± 0.6 pmol/mm/min
(n = 4, P < 0.05).
Table 1.
Contribution of pendrin to bicarbonate secretion in the mouse CCD
| Mice | Number of tubules | Tubule length, mm | Collection rate, nl/mm/min | Bath pH | Total CO2
|
||
|---|---|---|---|---|---|---|---|
| Perfusate concentration, mM | Collected concentration, mM | Total CO2 Flux, pmol/mm/min | |||||
| Pds+/+ | 4 | .40 ± .03 | 1.90 ± .12 | 7.41 ± .03 | 24.8 ± 0.4 | 26.2 ± 0.6 | −2.6 ± 1.4 |
| Pds−/− | 4 | .37 ± .04 | 2.04 ± .14 | 7.45 ± .02 | 24.9 ± 1.0 | 23.6 ± 0.8 | +2.7 ± 0.6 |
Discussion
To extend our previous work on the cellular distribution and function of pendrin in the inner ear (4) and thyroid (7), we have localized the protein in the mammalian kidney and carried out functional measurements in Pds-knockout mice. Immunolocalization studies revealed that pendrin has a highly restricted distribution within the kidney, being present in the apical membrane of a subpopulation of intercalated cells in the cortical part of the renal collecting duct.
The CCD contains a heterogeneous epithelium consisting of two main cell types: principal cells and intercalated cells (25, 27). The intercalated cells carry out fine regulation of acid-base excretion through regulated bicarbonate-transport processes (28). In general, the CCD can either absorb or secrete bicarbonate depending on the systemic acid-base status of the animal (29). Bicarbonate absorption is carried out by an intercalated cell subtype with H+-ATPase on the apical membrane (α or type A intercalated cells). Bicarbonate secretion is mediated by an intercalated cell subtype with H+-ATPase on the basolateral membrane (β or type B intercalated cells). There is also evidence for other types of intercalated cells that do not have any AE1 immunoreactivity and have the H+-ATPase either in the apical membrane (the so-called non A–non B cells) (26, 30, 31) or in both the apical and basolateral membranes (31).
Identification of the proteins responsible for bicarbonate transport in the various intercalated cell types remains an area of active investigation. In α-intercalated cells, the predominant bicarbonate transporter is the chloride/bicarbonate exchanger AE1, a splice variant product of the erythrocyte band 3 gene (32). AE1 resides in the basolateral membrane and functions in series with the H+-ATPase in the apical membrane to mediate net bicarbonate absorption (33, 34). In contrast, the transport protein(s) responsible for bicarbonate secretion across the apical membrane of other intercalated cells (35) has not yet been defined. One possibility is that AE1 mediates apical chloride/bicarbonate exchange in these cells. In fact, it has been proposed that β-intercalated cells and α-intercalated cells may interconvert by reversal of their apical-basolateral polarity (36). However, the presence of AE1 in the apical membrane of intercalated cells has never been directly demonstrated. Moreover, the inhibitor sensitivities and kinetic properties of the apical chloride/bicarbonate exchanger differ dramatically from those of the basolateral chloride/bicarbonate exchanger or AE1. Thus, the identification of the transporter(s) responsible for apical chloride/bicarbonate exchange in intercalated cells has been challenging (26, 37–42). Our data indicate that pendrin resides in the apical membrane of a non-α-intercalated cell. The precise classification of the pendrin-positive cells requires further investigation.
The localization of pendrin to intercalated cells prompted us to investigate the possibility that this protein represents the transporter responsible for bicarbonate secretion in the CCD. When treated with deoxycorticosterone pivalate and bicarbonate in their drinking water to stimulate bicarbonate secretion, isolated CCDs from wild-type mice secrete bicarbonate. However, bicarbonate secretion does not occur in CCDs isolated from pendrin-deficient mice; indeed, these CCDs absorb bicarbonate, presumably because of the sustained function of the bicarbonate-absorbing α-intercalated cells. These results indicate that pendrin plays a critical role in the process of renal bicarbonate secretion. One possibility is that pendrin directly mediates the transport of bicarbonate across the apical membrane of intercalated cells, a conclusion congruent with the recent demonstration that heterologously expressed pendrin (in HEK-293 cells) is capable of chloride/bicarbonate exchange (43).
An alternate explanation is that pendrin might play an indirect role in apical bicarbonate transport in intercalated cells. Studies with pendrin-expressing Xenopus oocytes demonstrated that the protein can function as a chloride/base exchanger (5), including as a chloride/formate exchanger (6). Chloride/formate exchange has been demonstrated to occur across the apical membrane of the renal proximal tubule (44), thereby playing a critical role in net NaCl reabsorption. In this process, the formate is believed to be recycled across the plasma membrane in the form of nonionic formic acid after being titrated by luminal protons, resulting in net transport of base equivalents into the lumen. It is possible that an analogous process could be responsible for bicarbonate secretion in the CCD. Specifically, pendrin-mediated secretion of base equivalents, such as formate, could titrate protons in the luminal fluid. This process would generate bicarbonate in the lumen via dissociation of carbonic acid. For this latter scenario, pendrin-dependent bicarbonate secretion need not involve the direct transport of bicarbonate by pendrin. Although a role for chloride/formate exchange in bicarbonate secretion seems to be excluded by the demonstration of bicarbonate secretion in isolated perfused tubules in the absence of added formate, it is possible that endogenously produced formate could catalyze a low rate of transport.
Neither Pendred syndrome patients nor pendrin-deficient mice have been reported to develop overt acid-base disturbances, such as a metabolic alkalosis. This likely reflects the fact that the kidney has other means of regulating bicarbonate excretion in the absence of pendrin [e.g., by regulation provided by the Na+/H+ exchanger NHE3 in the proximal tubule and the loop of Henle (45)]. Alternatively, there may be compensatory regulation of other transporters in the more distal portions of the nephron. Overt abnormalities in acid-base balance in pendrin-deficient humans or mice may be induced under conditions of extensive alkali loading or severe metabolic alkalosis.
The studies reported here further illustrate the interesting and diverse roles of pendrin within the restricted set of tissues in which it is expressed. It is intriguing that such a nonubiquitous protein has evolved to serve discrete functions in highly limited subsets of cells within tissues as distinct as the inner ear, the thyroid, and the kidney. The characterization of pendrin performed to date in many ways reflects its established role in a human disease (Pendred syndrome); however, it seems inevitable that as we catalogue and characterize the large set of proteins encoded by the mammalian genome, other such examples will come to the forefront.
Acknowledgments
We thank Seth Alper for providing the rabbit polyclonal anti-AE1 antibody and Steven Gluck for providing the mouse monoclonal anti-H+-ATPase antibody. We also thank Kirsten Madsen for comments on the immunolocalization data. This work was supported in part by National Institutes of Health Grant DK52935 (to S.M.W.), a March of Dimes Birth Defects Foundation grant (to L.P.K.), and funds from the Office of Research and Development in the Department of Veterans Affairs (to L.P.K.).
Abbreviations
- CCD
cortical collecting duct
- AE1
anion exchanger 1
Note Added in Proof.
Since submission of this paper, Soleimani et al. (43) reported the presence of rat pendrin in the proximal tubule, a finding not confirmed by our studies. Further work is required to determine whether the reported proximal tubule localization is due to a very low level of pendrin expression at that site or possibly the presence of a pendrin-like protein in proximal tubule cells.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Everett L A, Glaser B, Beck J C, Idol J R, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis A D, et al. Nat Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 2.Van Hauwe P, Everett L A, Coucke P, Scott D A, Kraft M L, Ris-Stalpers C, Bolder C, Otten B, de Vijlder J J, Dietrich N L, et al. Hum Mol Genet. 1998;7:1099–1104. doi: 10.1093/hmg/7.7.1099. [DOI] [PubMed] [Google Scholar]
- 3.Coyle B, Reardon W, Herbrick J A, Tsui L C, Gausden E, Lee J, Coffey R, Grueters A, Grossman A, Phelps P D, et al. Hum Mol Genet. 1998;7:1105–1112. doi: 10.1093/hmg/7.7.1105. [DOI] [PubMed] [Google Scholar]
- 4.Everett L A, Morsli H, Wu D K, Green E D. Proc Natl Acad Sci USA. 1999;96:9727–9732. doi: 10.1073/pnas.96.17.9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott D A, Wang R, Kreman T M, Sheffield V C, Karniski L P. Nat Genet. 1999;21:440–443. doi: 10.1038/7783. [DOI] [PubMed] [Google Scholar]
- 6.Scott D A, Karniski L P. Am J Physiol. 2000;278:C207–C211. doi: 10.1152/ajpcell.2000.278.1.C207. [DOI] [PubMed] [Google Scholar]
- 7.Royaux I E, Suzuki K, Mori A, Katoh R, Everett L A, Kohn L D, Green E D. Endocrinology. 2000;141:839–845. doi: 10.1210/endo.141.2.7303. [DOI] [PubMed] [Google Scholar]
- 8.Bidart J M, Mian C, Lazar V, Russo D, Filetti S, Caillou B, Schlumberger M. J Clin Endocrinol Metab. 2000;85:2028–2033. doi: 10.1210/jcem.85.5.6519. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki K, Lavaroni S, Mori A, Ohta M, Saito J, Pietrarelli M, Singer D S, Kimura S, Katoh R, Kawaoi A, et al. Proc Natl Acad Sci USA. 1998;95:8251–8256. doi: 10.1073/pnas.95.14.8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galfre G, Milstein C. Methods Enzymol. 1981;73:3–46. doi: 10.1016/0076-6879(81)73054-4. [DOI] [PubMed] [Google Scholar]
- 11.DiGiovanni S R, Nielsen S, Christensen E I, Knepper M A. Proc Natl Acad Sci USA. 1994;91:8984–8988. doi: 10.1073/pnas.91.19.8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson R D, Guo X L, Masood K, Brown D, Kalkbrenner M, Gluck S. Proc Natl Acad Sci USA. 1992;89:3541–3545. doi: 10.1073/pnas.89.8.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasui M, Kwon T H, Knepper M A, Nielsen S, Agre P. Proc Natl Acad Sci USA. 1999;96:5808–5813. doi: 10.1073/pnas.96.10.5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chernova M N, Humphreys B D, Robinson D H, Garcia A-M, Brosius F C, Alper S L. Biochim Biophys Acta. 1997;1329:111–123. doi: 10.1016/s0005-2736(97)00090-4. [DOI] [PubMed] [Google Scholar]
- 15.Hemken P, Guo X L, Wang Z Q, Zhang K, Gluck S. J Biol Chem. 1992;267:9948–9957. [PubMed] [Google Scholar]
- 16.Everett L A, Belyantseva I A, Noben-Trauth K, Cantos R, Chen A, Thakkar S I, Hoogstraten-Miller S L, Kachar B, Wu D K, Green E D. Hum Mol Genet. 2001;10:153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- 17.Wall S M. Am J Physiol. 1996;270:F432–F439. doi: 10.1152/ajprenal.1996.270.3.F432. [DOI] [PubMed] [Google Scholar]
- 18.Wall S M, Knepper M A. Semin Nephrol. 1990;10:148–158. [PubMed] [Google Scholar]
- 19.Wall S M, Sands J M, Flessner M F, Nonoguchi H, Spring K R, Knepper M A. Am J Physiol. 1990;258:F75–F84. doi: 10.1152/ajprenal.1990.258.1.F75. [DOI] [PubMed] [Google Scholar]
- 20.Star R A. Am J Physiol. 1990;258:F429–F432. doi: 10.1152/ajprenal.1990.258.2.F429. [DOI] [PubMed] [Google Scholar]
- 21.Knepper M A, Good D W, Burg M B. Am J Physiol. 1985;249:F870–F877. doi: 10.1152/ajprenal.1985.249.6.F870. [DOI] [PubMed] [Google Scholar]
- 22.Knepper M A, Good D W, Burg M B. Contrib Nephrol. 1985;47:116–124. doi: 10.1159/000411217. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen S, DiGiovanni S R, Christensen E I, Knepper M A, Harris H W. Proc Natl Acad Sci USA. 1993;90:11663–11667. doi: 10.1073/pnas.90.24.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown D, Hirsch S, Gluck S L. J Clin Invest. 1988;82:2114–2126. doi: 10.1172/JCI113833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuster V L, Bonsib S M, Jennings M L. Am J Physiol. 1986;251:C347–C355. doi: 10.1152/ajpcell.1986.251.3.C347. [DOI] [PubMed] [Google Scholar]
- 26.Alper S L, Natale J, Gluck S, Lodish H F, Brown D. Proc Natl Acad Sci USA. 1989;86:5429–5433. doi: 10.1073/pnas.86.14.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madsen K M, Tisher C C. Am J Physiol. 1986;250:F1–F15. doi: 10.1152/ajprenal.1986.250.1.F1. [DOI] [PubMed] [Google Scholar]
- 28.Schuster V L. Annu Rev Physiol. 1993;55:267–288. doi: 10.1146/annurev.ph.55.030193.001411. [DOI] [PubMed] [Google Scholar]
- 29.McKinney T D, Burg M B. J Clin Invest. 1977;60:766–768. doi: 10.1172/JCI108830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teng-umnuay P, Verlander J W, Yuan W, Tisher C C, Madsen K M. J Am Soc Nephrol. 1996;7:260–274. doi: 10.1681/ASN.V72260. [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Kim Y H, Cha J H, Tisher C C, Madsen K M. J Am Soc Nephrol. 1999;10:1–12. doi: 10.1681/ASN.V1011. [DOI] [PubMed] [Google Scholar]
- 32.Alper S L. Annu Rev Physiol. 1991;53:549–564. doi: 10.1146/annurev.ph.53.030191.003001. [DOI] [PubMed] [Google Scholar]
- 33.Furuya H, Breyer M D, Jacobson H R. Am J Physiol. 1991;261:F377–F385. doi: 10.1152/ajprenal.1991.261.3.F377. [DOI] [PubMed] [Google Scholar]
- 34.Muto S, Yasoshima K, Yoshitomi K, Imai M, Asano Y. J Clin Invest. 1990;86:1829–1839. doi: 10.1172/JCI114913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Star R A, Burg M B, Knepper M A. J Clin Invest. 1985;76:1123–1130. doi: 10.1172/JCI112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.al-Awqati Q. Am J Physiol. 1996;270:C1571–C1580. doi: 10.1152/ajpcell.1996.270.6.C1571. [DOI] [PubMed] [Google Scholar]
- 37.Ridderstrale Y, Kashgarian M, Koeppen B, Giebisch G, Stetson D, Ardito T, Stanton B. Kidney Int. 1988;34:655–670. doi: 10.1038/ki.1988.230. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz G J, Satlin L M, Bergmann J E. Am J Physiol. 1988;255:F1003–F1014. doi: 10.1152/ajprenal.1988.255.5.F1003. [DOI] [PubMed] [Google Scholar]
- 39.Schuster V L. Kidney Int Suppl. 1991;33:S47–S50. [PubMed] [Google Scholar]
- 40.van Adelsberg J S, Edwards J C, al-Awqati Q. J Biol Chem. 1993;268:11283–11289. [PubMed] [Google Scholar]
- 41.Fejes-Toth G, Chen W R, Rusvai E, Moser T, Naray-Fejes-Toth A. J Biol Chem. 1994;269:26717–26721. [PubMed] [Google Scholar]
- 42.Tsuganezawa, H., Kobayashi, K., Iyori, M., Araki, T., Koizumi, A., Watanabe, S. I., Kaneko, A., Fukao, T., Monkawa, T., Yoshida, T., et al. (2001) J. Biol. Chem., in press. [DOI] [PubMed]
- 43.Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, Burham C E. Am J Physiol Renal Physiol. 2001;280:F356–F364. doi: 10.1152/ajprenal.2001.280.2.F356. [DOI] [PubMed] [Google Scholar]
- 44.Karniski L P, Aronson P S. Proc Natl Acad Sci USA. 1985;82:6362–6365. doi: 10.1073/pnas.82.18.6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schultheis P J, Clarke L L, Meneton P, Miller M L, Soleimani M, Gawenis L R, Riddle T M, Duffy J J, Doetschman T, Wang T, et al. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]