

Abstract
Background
Interleukin 13 (IL-13) is upregulated in ulcerative colitis (UC) and increases colon epithelial permeability by inducing apoptosis and expression of the pore-forming tight junction protein claudin-2. IL-13 induces activation of signal transducer and activator of transcription 6 (STAT6). However, the STAT6 phosphorylation status in patients with UC is unknown, as is the effect of STAT6 inhibition on colonic epithelium exposed to IL-13. The study aims were to determine if mucosal STAT6 phosphorylation is increased in patients with UC, and if STAT6 inhibition attenuates IL-13-induced colon epithelial cell dysfunction.
Methods
Immunohistochemical staining for phosphorylated (p) STAT6 was performed on colonic tissue from newly diagnosed pediatric subjects with UC (early UC) or Cohn’s disease (CD), colectomy tissue from adults with UC (advanced UC), and controls. Colon HT-29 and T84 cells were transfected with STAT6 small interfering RNA (siRNA), or treated with suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor that inhibits STAT6, prior to IL-13 treatment.
Results
Median score for epithelial pSTAT6 was 0 in control subjects, 2 in early UC (vs. control P=0.019), 4 in advanced UC (P=0.003), and 0 in CD (P=0.4). Cell transfection with STAT6 siRNA prevented IL-13-induced apoptosis and claudin-2 expression. SAHA inhibited IL-13-induced STAT6 phosphorylation, apoptosis, and claudin-2 expression, and mitigated IL-13-induced reductions in trans-epithelial resistance.
Conclusions
UC is associated with increased colonic epithelial STAT6 phosphorylation, and STAT6 inhibition prevents IL-13-induced apoptosis and barrier disruption. These data identify STAT6 as a novel target for UC treatment, and support further study of SAHA as a therapeutic agent.
Keywords: ulcerative colitis, STAT6 transcription factor, suberoylanilide hydroxamic acid, interleukin-13, cell membrane permeability
The inflammatory bowel diseases (IBD), ulcerative colitis (UC) and Cohn’s disease (CD), are associated with divergent patterns of injury and inflammation, yet current therapies for these conditions remain quite similar. The colonic mucosa in UC maintains a Th2 cytokine pattern distinct from the Th1 pattern seen in CD.(1–3) While this distinction has been recognized for many years, there are no therapies in use for UC that specifically target Th2 cytokines or signaling.
The Th2 cytokine interleukin 13 (IL-13) has been implicated as a primary cause of epithelial barrier disruption in UC. Lamina propriety lymphocytes isolated from patients with UC produce increased IL-13 compared with CD and controls.(4) Furthermore, in vitro, IL-13 causes increased permeability and delayed repair of model colon epithelial cell monolayers by stimulating apoptosis and increasing expression of the pore-forming tight junction protein claudin-2.(5) Thus, interfering with IL-13 cell signaling might be an effective strategy to treat UC.
IL-13 binding to its receptor, comprised of the IL-4 receptor alpha (IL-4Rα) and IL-13 receptor alpha 1 (IL-13Rα1) subunits, triggers a signaling cascade leading to the phosphorylation of signal transducer and activation of transcription 6 (STAT6). Phosphorylated (p) STAT6 dimerizes and translocates to the nucleus where it binds DNA promoter elements to regulate gene transcription.(6, 7) While IL-13 induces STAT6 activation in colonic epithelial cells in vitro,(5) the STAT6 activation status in the colonic mucosa of patients with UC has not been reported. Similarly, it is not known if inhibition of STAT6 has any effects on IL-13-induced colon epithelial cell dysfunction.
Constitutive activation of STAT6 is associated with certain cancer cells including cutaneous T-cell lymphoma (CTCL), and Hodgkin’s and Reed-Sternberg cells (HRS) of Hodgkin’s lymphoma.(8–11) Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase (HDAC) inhibitor FDA-approved for the treatment of cutaneous T-cell lymphoma, inhibits STAT6 in both CTCL and HRS cell lines.(12, 13) Furthermore, in HRS cells, SAHA inhibits chemokine and Th2 cytokine production.(12) However, It is not known if SAHA can inhibit IL-13 induced epithelial dysfunction.
The aims of this study were to determine whether 1) STAT6 phosphorylation is increased in the intestinal mucosa of pediatric subjects at diagnosis with ulcerative colitis, and 2) IL-13-induced colon epithelial cell dysfunction is STAT6-dependent and can be inhibited by SAHA. Understanding the role of STAT6 activation in UC and IL-13-induced colon epithelial dysfunction could inform the development of future disease-specific therapies targeted to the Th2-mediated inflammation seen in UC.
MATERIALS AND METHODS
Human Samples
Archived paraffin-embedded tissue sections of biopsies from pediatric patients at presenting colonoscopies for UC and Crohn’s colitis, non-inflammatory controls, and colectomy specimens from adults with active UC were accessed from the Vanderbilt Digestive Disease Research Center Human Tissue Acquisition and Pathology Shared Resources.
Immunohistochemistry
Immunostaining was performed as previously described.(14) Briefly, sections were deparaffinized, rehydrated, and antigen was unmasked in a citrate-containing buffer (Vector Labs, Burlingame, CA). Sections were stained with rabbit anti-phospho Y641-STAT6 (pSTAT6) antibodies (ab32520, Abcam, Cambridge, MA). An isotype rabbit polyclonal IgG negative control was used to test for the specificity of the involved antibody at matched concentration and incubation conditions (ab27478, Abcam, Cambridge, MA). The Envision Plus System-HRP labeled polymer detection system (Dao, Carpentaria, CA) was used to unmask the antibody binding sites with 3,3-diamino-benzidine as a chromogen. Sections were counterstained with Mayer’s hematoxylin. A pathologist (MKW) blinded to diagnosis then scored stained sections for both epithelial and lamina propria nuclear pSTAT6 on a semi-quantitative scale from 0 (no staining) to 4 (>100 nuclear stained cells per high-powered field). To be conservative, only cells with nuclear pSTAT6 staining were counted.
Cell Culture
HT-29 cells were grown in Dulbecco’s Modified Eagles medium (DMEM) containing 10% fetal calf serum. T84 cells were grown in 1:1 DMEM/Ham’s F-12 medium containing 5% fetal calf serum. All media contained 100 U/ml penicillin, and 100 μg/ml streptomycin. Cultures were incubated at 37°C in a 95% air/5% CO2 atmosphere. In some experiments, cells were pre-treated with 0.1% DMSO vehicle or SAHA (Chemie Tek, Indianapolis, IN) at 1 or 5 μM for 6 hours prior to exposure to IL-13 or IL-4 (R&D Systems, Minneapolis, MN) at 10 ng/ml for various time periods. In other experiments, cells were transfected with 50 nmol STAT6 small interfering RNA (siRNA) SMARTpool or nontargeting siRNA pool (Dharmacon, Layafette, CO) using Lipofectamine RNAi/MAX (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Western Blot Analysis
Western Blot analysis on cell lysates was performed as previously described.(14) Briefly, membranes were blocked with 5% nonfat dry milk in TBS-Tween (0.05%) for 1 hour and incubated overnight at 4°C with primary antibodies against total STAT6 (AB3165, Millipore, Billerica, MA), claudin-2, IL-13 receptor alpha 1 (ab7297, Abcam, Cambridge, MA), pSTAT6, cleaved caspase-3 (9361 and 9661, Cell Signaling, Danvers, MA), IL-4 receptor alpha (MAB230, R&D Systems, Minneapolis, MN), or β-actins (A5441, Sigma-Aldrich, St. Louis, MO). Membranes were incubated with secondary antibodies, anti-rabbit- or anti-mouse horseradish peroxidase (Cell Signaling, Danvers, MA), and protein bands were detected by chemiluminescence using Western Lightning (Perkin Elmer, Waltham, MA).
Apoptosis
Both detached and adherent cells were collected and washed once in ice-cold phosphate buffered saline containing 0.5% bovine serum albumin. Cells were resuspended in binding buffer and stained with Annexin V-PE and 7-aminoactinomycin D (7-AAD) according to the manufacturer’s instructions (BD Biosciences, San Jose, CA). Flow cytometry for Annexin V binding was determined by flow cytometric analysis using a BD LSRII instrument and FlowJo software (Tree Star, Ashland, OR). Apoptosis results were confirmed by immunoblot for cleaved caspase-3.
RNA Expression
RNA was isolated from cells using the RNeasy Mini Kit (Quiagen, Valecia, CA) per the manufacturer’s instructions. RNA (1 μg) was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Separate real-time PCR (RT-PCR) reactions were carried out in triplicate using TaqMan Gene Expression Assays (Applied Bio systems, Foster City, Ca) for STAT6 (Assay ID: Hs00598618_m1), claudin-2 (Assay ID: Hs00252666_s1), and the reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (4333764F) on the Light Cycler 480 II RT-PCR platform (Roche, Indianapolis, IN). Thermal cycling conditions included an initial denaturation step at 95°C for 10 min followed by 40 cycles at 95°C for 15s and 60°C for 1 min. Relative mRNA levels were determined using the 2−ΔΔCT method with GAPDH as the reference.
Transepithelial Resistance
For measurements of transepithelial resistance (TER), T84 cell monolayers were grown on standing mixed cellulose esters membrane inserts (Millipore, Billerica, MA). TER across each monolayer was assessed by using a voltmeter (WPI, Sarasota, FL). Measurements were calculated in Ω • cm2 and expressed as a percentage of baselines. For all experiments, baseline TER was > 1000 Ω • cm2.
Statistical Analyses
All data are representative of at least three independent experiments. For each set of experiments yielding continuous data, analysis of variance was applied as a global test for differences in the primary outcome variable. Predetermined pair-wise comparisons were made using Student’s t-test with Bonferroni correction only when an overall effect was detected. Dose dependent effects were detected by assessing for linear trend using linear regression. Differences in patient characteristics across diagnosis groups were analyzed with one-way analysis of variance or Fisher’s Exact Test as appropriate. Since pSTAT6 immunohistochemistry was scored semiquantitatively, groups were compared globally using the nonparametric Kruskal-Wallis test. If global differences were detected, pair-wise comparisons relative to control patients were made using the Wilcoxon Rank Sum test with Bonferroni correction. We determined that based on a two-tailed t-test with 10 patients in each group, and assuming a type 1 error rate of 0.05, we would have 80 percent power to detect a difference of 1.3 times the standard deviation in pSTAT6 staining scores between groups. Statistical analyses were performed using Prism 5.0b (GraphPad Software, La Jolla, CA) and PASW Statistics 18.0.1 (SPSS Inc., Chicago, IL).
ETHICAL CONSDIERATIONS
The study was reviewed by the Vanderbilt University Institutional Review Board and determined not to qualify as “human subject” research since the pathologic specimens were stripped of all protected health information.
RESULTS
Human Ulcerative Colitis is Associated with Increased Activated STAT6
To examine STAT6 activation status early in the clinical course of human IBD, we analyzed endoscopic biopsy tissue from pediatric patients at presenting colonoscopy for UC (early UC), CD, and controls (n=10 for each group). Patient characteristics are presented in Table 1. As another comparison, we also examined surgical sections from adults who underwent colectomy for refractory UC (advanced UC, n=5). Stained sections were scored as described in the methods. There was increased pSTAT6 in both newly diagnosed and advanced UC (Figure 1). Median pSTAT6 nuclear epithelial staining scores were 0 in control subjects, 2 in early UC (vs. control P=0.019), 4 in advanced UC (P=0.003), and 0 in CD (P=0.4).
Table 1.
Patient characteristics
| Normal (n=10) | UC (n=10) | CD (n=10) | P | |
|---|---|---|---|---|
| Age, y, mean±SD | 13.1±4.2 | 11.1±3.6 | 10.7±3.7 | 0.34 |
| Male sex, n (%) | 2 (20) | 5 (50) | 4(40) | 0.37 |
| Disease involvement | ||||
| Small intestine only | 0 (0) | 0 (0) | .003 | |
| Colon only | 10 (100) | 3 (30) | ||
| Small intestine and colon | 0 (0) | 7 (70) | ||
| Perianal disease | 0 (0) | 2 (20) | .474 | |
| Granuloma | 0 (0) | 5 (50) | .033 | |
Figure 1.

Increased nuclear pSTAT6 in the colonic epithelium of pediatric subjects at diagnosis with UC. (A) Representative endoscopic biopsy sections from pediatric subjects at diagnosis with UC and normal subjects were stained using anti-pSTAT6 antibody or immunoglobulin control. Magnified inserts (60x) show representative crypts with nuclear staining in the epithelium of the UC tissue. Representative sections stained with anti-pSTAT6 from (B) an adult subject at colostomy for advanced UC and (C) pediatric subjects at diagnosis with CD. (D) Results of pathologist scoring in a fashion blinded to diagnosis for nuclear pSTAT6 in the epithelium and lamina propriety. Bars = 100μm. Pathologist nuclear pSTAT6 scoring system: 0 – no staining, 1 – trace, 2 – <50 cells/hpf, 3 – 50–100 cells/hpf, 4 – >100 cells/hpf. *P ≤ 0.05, **P ≤ 0.01.
Interestingly, there was a dichotomy in pSTAT6 nuclear epithelial staining amongst the subjects with CD such that 6 had no nuclear staining (score of 0) and 4 had 50–100 positive epithelial cells per high-powered field (score of 3). Two of the four CD patients with positive staining had disease limited to the colon, without per anal involvement or granulomas.
Within the lamina propria, there was increased nuclear pSTAT6 in the advanced UC group and a non-statistically significant increase in the early UC group compared to the control group. Median pSTAT6 nuclear lamina propria staining scores were 0 in control subjects, 2 in early UC (vs. control P=0.3), 3.5 in advanced UC (P=0.012) and 1 in CD (P=1.0).
IL-13-induced apoptosis and claudin-2 expression are STAT6 dependent
To determine the role of STAT6 activity in IL-13-induced epithelial dysfunction, apoptosis and claudin-2 expression, human colon epithelial HT-29 cells were transfected with STAT6-specific or non-targeting siRNA. STAT6 siRNA transfection resulted in a 72% reduction in STAT6 protein expression (P<0.001) and a 95% reduction in IL-13-induced STAT6 phosphorylation (P<0.001) (Figure 2A and B). Transfected cells were exposed to IL-13 (10 ng/ml) for 48 hours and apoptosis was quantified by flow cytometry of Annexin V-stained cells. While IL-13 exposure increased the apoptosis 1.5-fold (P<0.01) in cells transfected with non-targeting siRNA, IL-13 exposure had no effect on apoptosis in cells transfected with STAT6 siRNA (P<0.01) (Figure 2C and D). The main effect of transfection with STAT6 siRNA was prevention of early apoptosis (annexinV+/7-AAD−). The results of Annexin V staining were confirmed by Western blot analysis for cleaved caspase 3 (Figure 2E). In addition, transfection with STAT6 siRNA completely eliminated IL-13-induced claudin-2 expression (Figure 2F).
Figure 2.

IL-13-induced apoptosis and claudin-2 expression in colon epithelial cells is STAT6-dependent. HT-29 cells were transfected with STAT6 siRNA (SI) or nontargeting (NT) siRNA multiplexes prior to exposure to IL-13 10 ng/ml. (A) STAT6 expression was determined by Western blot analysis. Densitometry was performed to quantify the effectiveness of STAT6 siRNA knockdown. (B) To determine whether STAT6 siRNA transfection reduced STAT6 signaling, cells were exposed to IL-13 for 45 minutes and the lysates were subjected to Western blot analysis for pSTAT6 and total STAT6. The percent of cells undergoing apoptosis was quantified by flow cytometry of Annexin V stained cells. Representative flow cytometry plots (C) and quantification of mean percent Annexin V+ cells from three experiments (D) are shown. Lysates from cells exposed to IL-13 were subjected to Western blot analysis for (E) cleaved caspase-3 and (F) claudin-2. Pooled results of densitometry showing the mean density relative to untreated cells transfected with NT siRNA are shown. Data presented are the results of at least 3 independent experiments. *P ≤ 0.05, **P ≤ 0.01, *** P ≤ 0.001.
SAHA Inhibits IL-13-induced STAT6 Phosphorylation in Human Colon Epithelial Cells
The HDAC inhibitor SAHA inhibits STAT6 activation in lymphoma cell lines. To test the effects of SAHA on IL-13 signal transduction in human colon epithelial cells, HT-29 cell cultures were pre-treated with SAHA (1 or 5 μM) for 6 hours and then exposed to IL-13 at 10 ng/ml for 45 minutes. Whole cell lysates were prepared and subjected to Western blot analysis for pSTAT6. As expected, IL-13 induced STAT6 phosphorylation in these cells. SAHA pre-treatment reduced IL-13-induced STAT6 phosphorylation in a dose-dependent fashion (P<0.001) (Figure 3A). Expression of total STAT6 and the two components of the IL-13 receptor, IL-4Rα and IL-13Rα1, were unaltered by IL-13 or SAHA (Figure 3A and 3B).
Figure 3.

SAHA inhibits IL-13-induced STAT6 phosphorylation in colon epithelial cells. HT-29 cells were pre-treated with SAHA at the indicated concentrations for 6 hours followed by exposure to IL-13 (10 ng/ml) for 45 minutes. (A) Whole cell lysates were subjected to Western blot analysis for pSTAT6 and total STAT6. β-actin was assessed as a loading control. Effect of SAHA on IL-13-induced STAT6 phosphorylation was assessed by densitometry. (B) Western blot analysis was performed for IL-13Rα1 and IL-4Rα expression and (C) SOCS1 and SOCS3 expression. (D) HT-29 cells pre-treated with SAHA were exposed to either IL-4 or IL-13 (10 ng/ml) for 45 minutes and cell lysates were subjected to Western blot analysis for pSTAT6. (E) RNA was isolated from cells pre-treated with SAHA followed by exposure to IL-13 for 48 hours and STAT6 expression as measured by real-time PCR. Results are expressed relative to cells treated with DMSO alone. Data presented are the results of at least 3 independent experiments. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Members of the suppressor of cytokine signaling (SOCS) protein family are important regulators of JAK/STAT signal transduction, and both SOCS1 and SOCS3 are known inhibitors of STAT6 phosphorylation.(15, 16) Therefore, we speculated that SAHA may inhibit STAT6 by increasing the expression of these proteins. While IL-13 slightly augmented SOCS1 and SOCS3 expression, SAHA did not further induce expression of these negative regulators of STAT signaling (Figure 3C).
IL-4 also signals through STAT6 and its receptor shares the IL-4Rα protein with the IL-13 receptor. Therefore, we tested the effect of SAHA on IL-4 signaling and found that SAHA similarly inhibited IL-4-induced STAT6 activation in a dose-dependent manner (Figure 3D).
To further confirm that SAHA did not alter STAT6 expression, RNA was isolated from cells pre-treated with SAHA and exposed to IL-13 for 48 hours and STAT6 expression as measured by RT-PCR. STAT6 mRNA expression was not effected by either SAHA or IL-13 exposure (Figure 3E).
SAHA Inhibits IL-13-Induced Apoptosis in Human Colonic Epithelial Cells
To examine the effects of SAHA on IL-13-induced epithelial cell dysfunction, HT-29 cells were pre-treated with SAHA prior to exposure to IL-13 for 48 hours. Cells were stained with Annexin V and apoptosis quantified by flow cytometric analysis. IL-13 induced a 2.3-fold increase in apoptosis (12.8±1.9% Annexin V+ cellsvs. 5.7±2.1% in controls, P<0.001), which was inhibited by SAHA in a dose-dependent manner (IL-13 + SAHA 1 μM 10.3±1.4%; + SAHA 5 μM 6.9±0.8%, P<0.01) (Figure 4A and B). The dose-dependent effect of SAHA on IL-13-induced apoptosis was verified by testing for linear trend (P<0.001). Importantly, SAHA alone had no effect on baseline apoptosis, supporting the notion that inhibition of apoptosis was specific to IL-13-induced signaling. Suppression of IL-13-induced apoptosis by SAHA was confirmed with immunoblot analysis for cleaved caspase-3 (Figure 4C).
Figure 4.

SAHA inhibits IL-13-induced apoptosis in colon epithelial cells. HT-29 cells were pre-treated with SAHA at the indicated concentrations for 6 hours followed by exposure to IL-13 (10 ng/ml) for 48 hours. The percent of cells undergoing apoptosis was quantified by flow cytometry of Annexin V stained cells. Representative flow cytometry plots (A) and quantification of mean percent Annexin V+ cells from three experiments (B) are shown. (C) Cell lysates were subjected to Western blot analysis for cleaved caspase-3. The results of at least three independent experiments were analyzed by densitometry. *P ≤ 0.05, **P ≤ 0.01, *** P ≤ 0.001.
SAHA Inhibits IL-13-induced Claudin-2 Expression in Human Colon Epithelial Cells
IL-13 induced colon epithelial barrier dysfunction, a key factor in UC pathogenesis, is at least in part the result of increased expression of the pore-forming tight junction protein claudin-2.(5) To examine whether SAHA could inhibit IL-13 induction of claudin-2 expression, cultured HT-29 cells were pre-treated with SAHA prior to exposure to IL-13 for 48 hours and claudin-2 was detected by Western blot analysis of whole cell lysates. A small amount of claudin-2 expression was detected at baseline, which appeared to be decreased by SAHA. This effect of SAHA on baseline claudin-2 expression, however, did not reach statistical significance (P>0.05) after correcting for multiple comparisons. IL-13 stimulated claudin-2 expression 2-fold (P<0.05). This induction was inhibited by SAHA in a dose-dependent manner (P<0.01) (Figure 5A). RT-PCR for claudin-2 mRNA verified that both IL-13 and SAHA regulate claudin-2 protein levels by reducing gene expression (Figure 5B).
Figure 5.
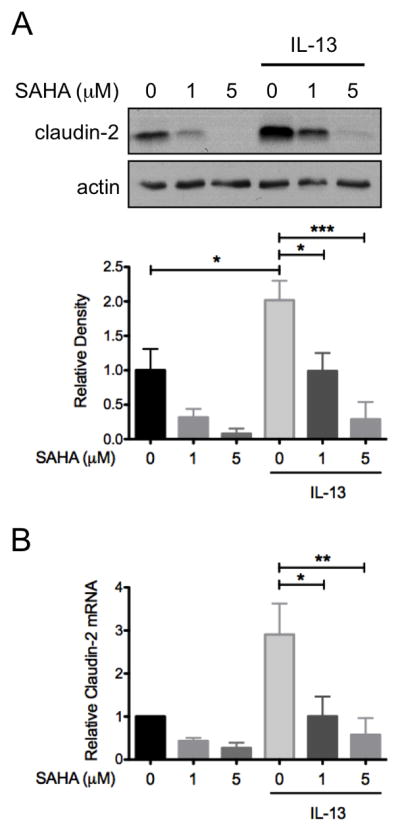
SAHA inhibits IL-13-induced apoptosis claudin-2 expression in colon epithelial cells. HT-29 cells were pre-treated with SAHA at the indicated concentrations for 6 hours followed by exposure to IL-13 (10 ng/ml) for 48 hours. (A) Cell lysates were subjected to Western blot analysis for claudin-2. The results of three independent experiments were analyzed by densitometry. (B) RNA was isolated from cells and STAT6 expression as measured by real-time PCR. Results of three independent experiments are expressed relative to cells treated with DMSO alone. *P ≤ 0.05, **P ≤ 0.01, *** P ≤ 0.001.
SAHA Protects from IL-13-induced Colon Epithelial Barrier Dysfunction
Since SAHA inhibits IL-13-induced apoptosis and claudin-2 expression, we questioned whether it would also protect colon epithelial cells from IL-13-induced epithelial barrier dysfunction. Our HT-29 cells do not spontaneously generate tight epithelial monolayers as measured by TER. Therefore, we used the human colon carcinoma T84 cell line, which generates high TER, to test the effect of SAHA on alterations in TER induced by IL-13. Western blot analysis performed on T84 cells pre-treated with SAHA and exposed to IL-13 for 45 minutes confirmed, as others have shown(17), that SAHA also inhibits IL-13-induced pSTAT6 in T84 cells (Figure 6A). T84 cells were pre-treated with SAHA and exposed to IL-13 for 48 hours. IL-13 reduced TER to 57±4%, 54±7%, and 35±4% at 12, 24, and 48 hours after treatment, respectively, compared to 95±5%, 81±6%, and 43±4%, respectively, in the presence of SAHA (P<0.001, P<0.01, and P<0.05 respectively for IL-13 + SAHA vs. IL-13 alone) (Figure 6B).
Figure 6.
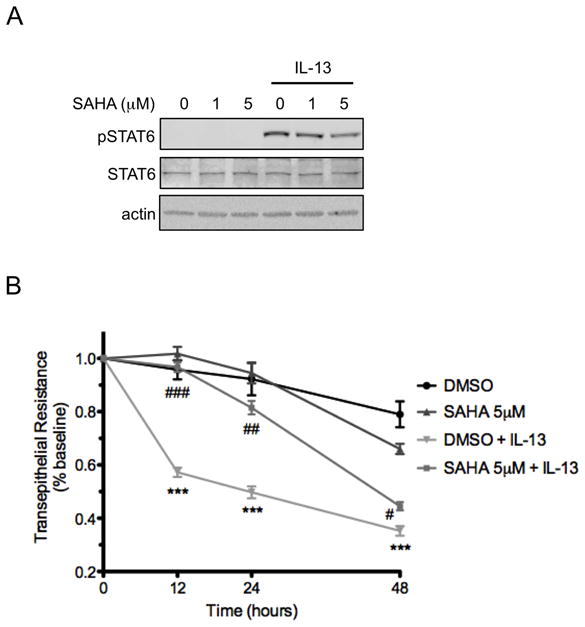
SAHA mitigates IL-13-induced epithelial barrier dysfunction. (A) T84 cells were pre-treated with SAHA at the indicated concentrations for 6 hours followed by exposure to IL-13 10 ng/ml for 45 minutes. Whole cell lysates were subjected to Western blot analysis for pSTAT6 and total STAT6. β-actin was assessed as a loading control. (B) T84 monolayers were cultured on standing membrane inserts, pre-treated with DMSO or SAHA 5 μM for 6 hours followed by exposure to IL-13 (10 ng/ml). TER was measured at the indicated time points and results of four independent experiments are expressed relative to baseline. ***P ≤ 0.001 compared to DMSO; #P ≤ 0.05, ##P ≤ 0.01, ###P ≤ 0.01 compared to DMSO + IL-13.
DISCUSSION
While Th2-driven inflammation is a distinguishing feature of UC (3–5, 18), no currently approved therapies for its treatment specifically target Th2 lymphocytes, cytokines, or transcription factors associated with a Th2 immune response. Since IL-13 is a key Th2 cytokine in the pathogenesis of UC(4, 5, 19), we sought to evaluate whether activation of STAT6, a transcription factor downstream of IL-13 signaling, is altered in UC, and whether STAT6 inhibition limits the effects of IL-13 on colon epithelial cells. To our knowledge, this study is the first demonstration of increased pSTAT6 in the epithelium of subjects with new-onset ulcerative colitis. Furthermore, we demonstrate that SAHA, a compound that inhibits constitutive STAT6 activation in lymphoma cell lines,(12, 13) inhibits IL-13-induced apoptosis, claudin-2 expression, and barrier dysfunction in colon epithelial cells.
Fuss and colleagues were the first to report the importance of IL-13 in UC by demonstrating that this cytokine is abundantly secreted by lamina propria lymphocytes from patients with advanced disease.(4) The potential importance of IL-13 in the pathogenesis of UC is underscored by the finding that neutralization of IL-13 prevents oxazalone-induced colitis, a mouse model with similar features to human UC.(19) These seminal studies, however, do not provide in situ evidence that the colon epithelium is actually exposed to IL-13 in patients with UC. In fact, other groups examining cytokine levels from tissue homogenates or supernatants from organ culture have reported down regulation of IL-13 in UC.(20, 21) Our finding of increased pSTAT6 in the colonic epithelium of pediatric subjects with UC is evidence for IL-13-induced signaling and consistent with the notion that the colonic epithelium in UC is exposed to increased IL-13. Although IL-4 is known to also signal through STAT6, many investigators using various methods have demonstrated low or normal levels of IL-4 in patients with both UC and CD. (3, 4, 22–26) While prior studies investigating IL-13 in UC used colectomy tissue from patients with severe or established UC, our findings are from tissues of pediatric patients at their diagnostic colonoscopies, which suggests a role for Th2 cytokine signaling in the early pathogenesis of UC.
We found that a subset of 4 patients with CD had increased epithelial pSTAT6 staining. Interestingly, 2 of these patients had strictly colonic involvement (without perianal disease or granulomas). The remainder of CD patients, with none to minimal epithelial pSTAT6, had both small bowel and colonic involvement. One possible explanation is the CD patients with only colonic involvement were misdiagnosed and truly had ulcerative colitis. Since the tissue specimens were obtained from a pathology repository, we did not have access to the entire detailed medical record to determine the clinical criteria on which each patient was diagnosed. However, in our practice, in the absence of granulomas, small bowel involvement, or perianal disease, patients would have to display clearly distinguishing signs of Crohn’s disease such as discrete apthous or linear ulceration, or skip lesions to be diagnosed with CD (as opposed to IBD-unspecified or UC). Alternatively, since CD is a phenotypically heterogeneous disorder, we can speculate that this finding could represent overlap in the pathogenesis of UC and a specific colonic subtype of CD. Interestingly, perinuclear antineutrophil cytoplasmic antibodies (pANCA) are another biomarker generally more specific for ulcerative colitis which, when present in patients with CD, are associated with a colonic phenotype and UC-like features.(27, 28) A larger prospective study of STAT6 signaling in the mucosa of patients with Crohn’s disease is needed to test this hypothesis.
Given our finding of increased pSTAT6 in UC and the established role of IL-13 in the disease, we hypothesize that STAT6 is a potential target against which to develop future UC therapies. We show that the two known mechanisms by which IL-13 directly increases colon epithelial permeability, induction of apoptosis and induction of claudin-2 expression(5), are STAT6 dependent. Our results support the findings of Madden et al who, using a STAT6 knockout mouse, demonstrated that IL-13-induced increases in mucosal permeability are STAT6-dependent.(29) In contrast, Capons et al found that in T84 cells, IL-13 regulation of epithelial permeability was not STAT6-dependent, but rather mediated by phosphoinositide 3-kinase signaling.(30) There are several explanations for these conflicting results including the use of different cell lines and model systems, and different approaches to reduce or eliminate STAT6 signaling; in the current report we used siRNA, whereas Madden et al used a knockout mouse, and Capons et al used transcription factor decoys. Recently, STAT proteins have been shown to have cellular roles other than as transcription factors.(31, 32) Therefore, transcription factor decoys might not mitigate all the relevant cellular effects of STAT6 regulation of epithelial permeability.
The HDAC inhibitor SAHA has been shown to inhibit constitutive STAT6 activation in several lymphoma cell lines(12, 13), and we demonstrate that SAHA also inhibits IL-13-induced STAT6 activation in HT-29 colon epithelial cells. Furthermore, SAHA prevented IL-13-induced apoptosis without altering baseline apoptosis. SAHA and other HDAC inhibitors induce cell cycle arrest and apoptosis in many transformed cell lines while sparing normal cells.(33–35) While HT29 cells are a transformed cell line, they are resistant to HDAC-inhibitor-induced apoptosis(36), which more closely models the response of non-transformed intestinal epithelial cells in this regard.(37)
Our finding that SAHA inhibits IL-13-induced claudin-2 expression in HT-29 cells is in line with those of Weber et al who demonstrated the same results in T84 cells.(17) Weber et al also demonstrated that IL-13-induced reductions in TER are dependent on the induction of claudin-2. Here we add to these findings by demonstrating that SAHA alone is capable of abrogating the detrimental effects of IL-13 on TER.
While the mechanism underlying blockade of STAT6 activation by SAHA remains an important area for further investigation, the findings presented here suggest avenues for future investigation. SAHA inhibited both IL-13- and IL-4-induced STAT6 activation, suggesting that SAHA acts on elements common to the signal transduction cascade of both cytokines. SAHA did not affect expression of either the IL-4Rα subunit, which is a component of both the IL-4 receptor and IL-13Rα1 heterodimers, or the IL-13Rα1 subunit, which is only a part of the IL-13Rα1 heterodimer. Therefore, SAHA may act on IL-4Rα activation (as opposed to expression) or on components of both the IL-13 and IL-4 signaling cascade downstream of the receptors.
Both STAT1 and STAT3 are directly acetylated, and thus regulated by HDACs.(38, 39) In the case of STAT1, acetylating leads to recruitment of tyrosine phosphotases, rendering the protein resistant to persistent phosphorylation. Furthermore, treatment of human embryonic kidney cells with HDAC inhibitors prevents interferon alpha-induced STAT1 phosphorylation.(39) Future studies will be needed to determine if direct acetylating of STAT6 regulates phosphorylation by a similar mechanism.
Our results suggest that the use of SAHA or other drugs that inhibit STAT6 activation may be a potential treatment strategy for ulcerative colitis. HDAC inhibitors have already shown potential in mouse models of Th1-mediated colitis, ostensibly due to their effects on T-cell function. (40–43) SAHA and another HDAC inhibitor, trichostatin A (TSA), ameliorated dextran sodium sulfate-induced colitis and a T-cell transfer model of colitis in association with decreases in pro-inflammatory cytokine production and augmentation of the number of regulatory T-cells, and their function. Here we add to these findings by demonstrating a protective effect of SAHA and inhibition of STAT6 on a cell culture model of colon epithelium exposed to IL-13, a key cytokine involved in Th2-mediated colitis.
In conclusion, we demonstrate increased epithelial pSTAT6 in pediatric subjects newly diagnosed with ulcerative colitis. We further find that IL-13-induced human intestinal epithelial cell apoptosis and claudin-2 expression are STAT6 dependent. Lastly, SAHA protects colon epithelial cells from IL-13-induced apoptosis and epithelial barrier dysfunction, likely through inhibition of STAT6 activation. Thus, the role of SAHA, or other means of STAT6 inhibition as a novel Th2-inflammation-targeted therapy for UC warrants further investigation.
Acknowledgments
Support: Supported by National Institutes of Health grants 2T32DK007673-16, R01DK056008 (to DBP), 5R01AT004821-03 and 3R01AT004821-02S1 (to KTW), K01DK077956 (to MRF), American College of Gastroenterology Clinical Research Award (to MJR), NASPGHAN Fellow to Faculty Transition Award and George Ferry Young Investigator Award (to MJR), a Vanderbilt Physician Scientist Development Award (to MJR), and the Vanderbilt Digestive Diseases Research Center (NIH P30DK058404) including the Center’s Pilot and Feasibility Program, Flow Cytometry and Cellular and Animal Modeling Shared Resources.
The authors would like to thank Dr. Louis J. Muglia for his critical review of the manuscript and thoughtful feedback.
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Kugathasan S, Saubermann LJ, Smith L, et al. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut. 2007;56:1696–1705. doi: 10.1136/gut.2006.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 4.Fuss IJ, Heller F, Boirivant M, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. The Journal of clinical investigation. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Hershey GK. IL-13 receptors and signaling pathways: an evolving web. The Journal of allergy and clinical immunology. 2003;111:677–690. doi: 10.1067/mai.2003.1333. [DOI] [PubMed] [Google Scholar]
- 7.Takeda K, Kamanaka M, Tanaka T, et al. Impaired IL-13-mediated functions of macrophages in STAT6-deficient mice. J Immunol. 1996;157:3220–3222. [PubMed] [Google Scholar]
- 8.Guiter C, Dusanter-Fourt I, Copie-Bergman C, et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood. 2004;104:543–549. doi: 10.1182/blood-2003-10-3545. [DOI] [PubMed] [Google Scholar]
- 9.Skinnider BF, Elia AJ, Gascoyne RD, et al. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2002;99:618–626. doi: 10.1182/blood.v99.2.618. [DOI] [PubMed] [Google Scholar]
- 10.Takemoto S, Mulloy JC, Cereseto A, et al. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13897–13902. doi: 10.1073/pnas.94.25.13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapp U, Yeh WC, Patterson B, et al. Interleukin 13 is secreted by and stimulates the growth of Hodgkin and Reed-Sternberg cells. The Journal of experimental medicine. 1999;189:1939–1946. doi: 10.1084/jem.189.12.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buglio D, Georgakis GV, Hanabuchi S, et al. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood. 2008;112:1424–1433. doi: 10.1182/blood-2008-01-133769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Richon V, Ni X, et al. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. The Journal of investigative dermatology. 2005;125:1045–1052. doi: 10.1111/j.0022-202X.2005.23925.x. [DOI] [PubMed] [Google Scholar]
- 14.Edelblum KL, Washington MK, Koyama T, et al. Raf protects against colitis by promoting mouse colon epithelial cell survival through NF-kappaB. Gastroenterology. 2008;135:539–551. doi: 10.1053/j.gastro.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu CR, Mahdi RM, Ebong S, et al. Cell proliferation and STAT6 pathways are negatively regulated in T cells by STAT1 and suppressors of cytokine signaling. J Immunol. 2004;173:737–746. doi: 10.4049/jimmunol.173.2.737. [DOI] [PubMed] [Google Scholar]
- 16.Hebenstreit D, Luft P, Schmiedlechner A, et al. SOCS-1 and SOCS-3 inhibit IL-4 and IL-13 induced activation of Eotaxin-3/CCL26 gene expression in HEK293 cells. Mol Immunol. 2005;42:295–303. doi: 10.1016/j.molimm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Weber CR, Raleigh DR, Su L, et al. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. The Journal of biological chemistry. 2010;285:12037–12046. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue S, Matsumoto T, Iida M, et al. Characterization of cytokine expression in the rectal mucosa of ulcerative colitis: correlation with disease activity. The American journal of gastroenterology. 1999;94:2441–2446. doi: 10.1111/j.1572-0241.1999.01372.x. [DOI] [PubMed] [Google Scholar]
- 19.Heller F, Fuss IJ, Nieuwenhuis EE, et al. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17:629–638. doi: 10.1016/s1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- 20.Kadivar K, Ruchelli ED, Markowitz JE, et al. Intestinal interleukin-13 in pediatric inflammatory bowel disease patients. Inflammatory bowel diseases. 2004;10:593–598. doi: 10.1097/00054725-200409000-00014. [DOI] [PubMed] [Google Scholar]
- 21.Vainer B, Nielsen OH, Hendel J, et al. Colonic expression and synthesis of interleukin 13 and interleukin 15 in inflammatory bowel disease. Cytokine. 2000;12:1531–1536. doi: 10.1006/cyto.2000.0744. [DOI] [PubMed] [Google Scholar]
- 22.Camoglio L, Te Velde AA, Tigges AJ, et al. Altered expression of interferon-gamma and interleukin-4 in inflammatory bowel disease. Inflammatory bowel diseases. 1998;4:285–290. doi: 10.1002/ibd.3780040406. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen OH, Koppen T, Rudiger N, et al. Involvement of interleukin-4 and -10 in inflammatory bowel disease. Digestive diseases and sciences. 1996;41:1786–1793. doi: 10.1007/BF02088746. [DOI] [PubMed] [Google Scholar]
- 24.West GA, Matsuura T, Levine AD, et al. Interleukin 4 in inflammatory bowel disease and mucosal immune reactivity. Gastroenterology. 1996;110:1683–1695. doi: 10.1053/gast.1996.v110.pm8964392. [DOI] [PubMed] [Google Scholar]
- 25.Niessner M, Volk BA. Altered Th1/Th2 cytokine profiles in the intestinal mucosa of patients with inflammatory bowel disease as assessed by quantitative reversed transcribed polymerase chain reaction (RT-PCR) Clin Exp Immunol. 1995;101:428–435. doi: 10.1111/j.1365-2249.1995.tb03130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karttunnen R, Breese EJ, Walker-Smith JA, et al. Decreased mucosal interleukin-4 (IL-4) production in gut inflammation. J Clin Pathol. 1994;47:1015–1018. doi: 10.1136/jcp.47.11.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasiliauskas EA, Plevy SE, Landers CJ, et al. Perinuclear antineutrophil cytoplasmic antibodies in patients with Crohn's disease define a clinical subgroup. Gastroenterology. 1996;110:1810–1819. doi: 10.1053/gast.1996.v110.pm8964407. [DOI] [PubMed] [Google Scholar]
- 28.Vasiliauskas EA, Kam LY, Karp LC, et al. Marker antibody expression stratifies Crohn's disease into immunologically homogeneous subgroups with distinct clinical characteristics. Gut. 2000;47:487–496. doi: 10.1136/gut.47.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madden KB, Whitman L, Sullivan C, et al. Role of STAT6 and mast cells in IL-4- and IL-13-induced alterations in murine intestinal epithelial cell function. J Immunol. 2002;169:4417–4422. doi: 10.4049/jimmunol.169.8.4417. [DOI] [PubMed] [Google Scholar]
- 30.Ceponis PJ, Botelho F, Richards CD, et al. Interleukins 4 and 13 increase intestinal epithelial permeability by a phosphatidylinositol 3-kinase pathway. Lack of evidence for STAT 6 involvement. The Journal of biological chemistry. 2000;275:29132–29137. doi: 10.1074/jbc.M003516200. [DOI] [PubMed] [Google Scholar]
- 31.Lee H, Herrmann A, Deng JH, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–293. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gough DJ, Corlett A, Schlessinger K, et al. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science (New York, NY) 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marks P, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: causes and therapies. Nature reviews. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 34.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature reviews. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 35.Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer letters. 2008;269:7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 36.Huang X, Guo B. Adenomatous polyposis coli determines sensitivity to histone deacetylase inhibitor-induced apoptosis in colon cancer cells. Cancer research. 2006;66:9245–9251. doi: 10.1158/0008-5472.CAN-06-0887. [DOI] [PubMed] [Google Scholar]
- 37.Klampfer L, Huang J, Shirasawa S, et al. Histone deacetylase inhibitors induce cell death selectively in cells that harbor activated kRasV12: The role of signal transducers and activators of transcription 1 and p21. Cancer Res. 2007;67:8477–8485. doi: 10.1158/0008-5472.CAN-07-0210. [DOI] [PubMed] [Google Scholar]
- 38.Yuan ZL, Guan YJ, Chatterjee D, et al. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science (New York, NY) 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 39.Kramer OH, Knauer SK, Greiner G, et al. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes & development. 2009;23:223–235. doi: 10.1101/gad.479209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glauben R, Batra A, Fedke I, et al. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol. 2006;176:5015–5022. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]
- 41.Glauben R, Batra A, Stroh T, et al. Histone deacetylases: novel targets for prevention of colitis-associated cancer in mice. Gut. 2008;57:613–622. doi: 10.1136/gut.2007.134650. [DOI] [PubMed] [Google Scholar]
- 42.Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nature medicine. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 43.de Zoeten EF, Wang L, Sai H, et al. Expression of HDAC9 by T Regulatory Cells Prevents Colitis in Mice. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]