

Abstract
A novel enzyme-linked DNA hybridization assay on an interdigitated array (IDA) microelectrode integrated into a microfluidic channel is demonstrated with sub-nM detection limit. To improve the detection limit as compared to conventional electrochemical biosensors, a recyclable redox product, 4-aminophenol (PAP) is used with an IDA microelectrode. The IDA has a modest and easily fabricated inter-digit spacing of 10 μm, yet we were able to demonstrate 97% recycling efficiency of PAP due to the integration in a microfluidic channel. With a 70 nL sample volume, the characterized detection limit for PAP of 1.0 × 10−10 M is achieved, with a linear dynamic range that extends from 1.0 × 10−9 to 1.0 × 10−5 M. This detection limit, which is the lowest reported detection limit for PAP, is due to the increased sensitivity provided by the sample confinement in the microfluidic channel, as well as the increased repeatability due to perfectly static flow in the microchannel and an additional anti-fouling step in the protocol. DNA sequence detection is achieved through a hybridization sandwich of an immobilized complementary probe, the target DNA sequence, and a second complementary probe labeled with β-galactosidase (β-GAL); the β-GAL converts its substrate, 4-aminophenyl-D-galactopyranoside (PAPG), into PAP. In this report we present the lowest reported observed detection limit (1.0 × 10−10 M) for an enzyme-linked DNA hybridization assay using an IDA microelectrode and a redox signaling paradigm. Thus, we have demonstrated highly sensitive detection of a targeted DNA sequence using a low-cost easily fabricated electrochemical biosensor integrated into a microfluidic channel.
Keywords: microfluidic, electrochemical, IDA, PAP, β-galactosidase, DNA hybridization
1. Introduction
Electrochemical biosensors are excellent candidates for miniaturization using microfabrication techniques, as well as for integration with other functions through microfluidics. Microelectrodes can be fabricated on-chip with higher yield and lower cost as compared to other biosensing modalities under investigation, including optical waveguide, surface plasmon resonance, nanowire, and field effect transistor sensors. Furthermore, microfluidics can easily be integrated onto surfaces containing microelectrodes without interfering with the signals. As a result, miniaturized and integrated electrochemical biosensors hold great potential for bio-analysis systems.
In particular, electrochemical immunoassays utilize the selectivity and sensitivity of immuno-recognition and can be applied to a number of critical applications, including biomolecular detection for disease diagnosis, protein quantification in cellular and molecular biology research, and protein-protein interaction studies. Similar to an immunoassay is a DNA hybridization assay, in which specific DNA sequences are detected by using complementary sequences as probes; the complementary probes are analogous to the antibodies in an immunoassay. For electrochemical immunoassays or DNA hybridization assays that employ enzyme labeling, the enzyme is conjugated to the biorecognition probe, and thus is immobilized onto the sensor surface upon biorecognition of the analyte. After a substrate is introduced, the enzyme catalyzes the conversion of the substrate to a product. The analytical signal gradually increases with time because of the continuous reaction between each enzyme and the substrate. The enzyme-generated product is quantified with an adequate electrochemical method, e.g., potentiometry, voltammetry, chronoamperometry, or impedance analysis, etc (Diaz-Gonzalez et al. 2005; Ronkainen-Matsuno et al., 2007; Skladal, 2005; Wang et al., 2001; Warsinke et al., 2000). More recent developments applied magnetic beads for both immobilization and bio-molecule carriers in electrochemical immunoassays for lower detection limit and higher throughput (Choi et al., 2002; Do and Ahn, 2008). Enzymes such as alkaline phosphatase (ALP), β-galactosidase (β-GAL) and horseradish peroxidase (HRP) are the most commonly used labeling enzymes in immunoassays (Skladal, 2005; Warsinke et al., 2000). Good candidates for enzymatic reactions for electrochemical immunoassays include 4-aminophenyl phosphate (PAPP) and 4-aminophenol β-d-galactopyranoside (Niwa et al. 1993; Purushothama et al., 2001). Various compounds, e.g. α-fetoprotein, thephylline, mouse IgG and avian influenza virus (AIV) have been electrochemically assayed via PAPP (Wang et al., 2001; Ohtsuka et al., 2008). Also, β-d-galactopyranoside (PAPG) has been used to assay human immunodeficiency virus (HIV) with microelectrodes (Laczka et al., 2009).
When oxidized on the anode, PAP goes through a 2-electron transfer process and loses 2 protons. Its oxidized form, para quenone imide (PQI), can either be reduced back to PAP, or further oxidized to quinone in acidic environments (Snead and Remick, 1957). The electrochemically reversible nature of PAP can be utilized for signal amplification when detected with an interdigitated array (IDA) microelectrode. Since the reversibility of the PAP redox reaction depends on the pH of the solvent and its electrochemical kinetics depend on electrode materials, different electrochemical techniques have been used to detect PAP.
Several groups have reported on the detection of PAP and its application with various methods of measurement, such as enzymatic recycling in the PAP/PQI redox system, colorimetry, rotation disk electrode method, adsorptive stripping voltammetry, differential pulse voltammetry, or amperometry. The utilized electrodes are typically bare Au electrodes, glassy carbon, or carbon nanotube modified carbon electrodes. Despite the high number of reported approaches, the best detection limit reported so far is 500 pM and the most impressive dynamic range achieved is between 5 nM and 2 μM (Niwa et al., 1993; Purushothama et al., 2001; Ohtsuka et al., 2008; Yamaguchi et al., 1992; Bocxlaer et al., 1997; Huang et al., 2003; Wyszecka-Kaszuba et al., 2003). A comparison of these various methods is shown in Table S1 in the Supplementary Material.
To improve upon the reported detection limit, there are two primary factors to be considered: increasing the sensitivity to the analyte concentration, and decreasing the variation in the measurement of the analyte. First, the sensitivity for detection of PAP with an IDA can be improved by increasing the recycling efficiency of PAP. This can be accomplished by decreasing the spacing of the digits in the IDA; however, doing so increases the fabrication costs and decreases the yield of the biosensor. Alternatively, the recycling efficiency can be increased by preventing the loss of available signaling species at the electrode surface. This can be accomplished by integrating the IDA into a microfluidic channel with a low height; because of the confinement, species that are recently oxidized will remain close to the electrode where they can be reduced, and vice versa. Furthermore, this integration step does not have a major impact on cost or yield; in fact it leads to better integration of the biosensor with other functions, such as sample preparation. Therefore, in this work, we have integrated the IDA into a microfluidic channel in order to improve the detection limit of our enzyme-linked electrochemical assay.
In addition, we investigated methods of minimizing the variation in the measured signal from PAP in order to further improve the detection limit. One important characteristic of electrochemical sensors using microband electrodes is that the signal is dependent upon the flow rate. In a microchannel, the limiting current measured on a microband electrode can be presented with a modified Levich equation (Girault, 2004):
| Eq. (1) |
where n is the number of electron transfers, F is the Faraday constant, c and D are the bulk concentration and the diffusion coefficient of the redox compound, l and L are the width and length of the band, Q̇ is the volumetric flow rate, and 2h and d are the height and width of the channel, respectively. The flow rate dependency for microelectrodes in a microfluidic channel has been verified previously (Amatore et al., 2004; Amatore et al., 2005; Amatore et al., 2006; Compton et al., 1993; Ordeig et al., 2008). Therefore, to reduce measurement variability while at the same time minimizing the consumed sample volume, it is advantageous to detect analytes at a perfectly static state after the microchannel is filled with sample solution.
In addition to measurement variations caused by convection flow in a channel, microelectrode-based assays are notorious for a lack of repeatability due to electrode fouling. While utilizing PAP with an IDA microelectrode in an electrochemical immunoassay can increase the detected signal, PAP polymerizes in acidic, neutral or basic solutions during electrochemical reactions or interactions with an enzyme such as HRP (Aizawa et al., 1996; Eddy et al., 1995; Reihmann and Ritter, 2002). The conductivity and other electrochemical properties of the electrode are then altered after the thin film is formed (Eddy et al., 1995; Vieira et al., 2006; Ferreira et al., 2008; Tsuchida et al., 1970). The poly-PAP film fouls the electrode surface and reduces the sensitivity and reproducibility. Therefore, an effective cleaning procedure between measurements must be introduced. Low molecular weight oxidative polymer of PAP is soluble in organic solvents, such as methanol and acetone, and can further reduce back to PAP (Tsuchida et al., 1970; Lewis et al., 2010). This technique can be adapted to an IDA microsystem to improve the repeatability between measurements, which leads to better detection limit.
In this work, we implement an IDA in a microfluidic channel under static flow with an electrode regeneration protocol to improve the detection limit for PAP as compared to previous reports. The flow rate dependence of the signal with our IDA microelectrode is experimentally verified. The improvement in measurement variability when implementing our electrode regeneration protocol is demonstrated. Utilizing all of these advances in our assay, our system achieves an excellent detection limit and dynamic range for PAP detection on a gold IDA electrode. Building upon this result, we first immobilize the enzyme directly onto the microchannel surface and perform high sensitivity enzyme activity detection. Further, we implement an enzyme-linked DNA hybridization assay using a β-GAL-labeled probe and a PAPG substrate. Our results show a measured detection limit of 100.0 pM of target DNA. To our knowledge, we are presenting the first redox/enzyme coupling assisted single stranded DNA detection with a sub-nM detection limit.
2. Materials and Methods
2.1 Fabrication of electrochemical detector
The electrochemical detector is integrated into a microfluidic system. Rectangular microchannels with the cross-section of 35 μm height, 200 μm width, and a 1-cm length are made with polydimethylsiloxane (PDMS) via soft lithography methods. PDMS (Sylgard 184) is purchased from Dow Corning, MO. A sample port and a waste port ( φ=1.0 mm) are punched at the two ends of the microchannel with a UniCore hole puncher (as shown in Fig. 1A). The PDMS slab is sonicated in 190-proof ethanol (111USP190, Pharmco-Aaper, Brookfield, CT) and blow-dried with air before being assembled with the electrode chips. The microchannel is connected to a syringe pump at the waste port. Unless specified otherwise, the PDMS is not modified and remains hydrophobic.
Fig. 1.
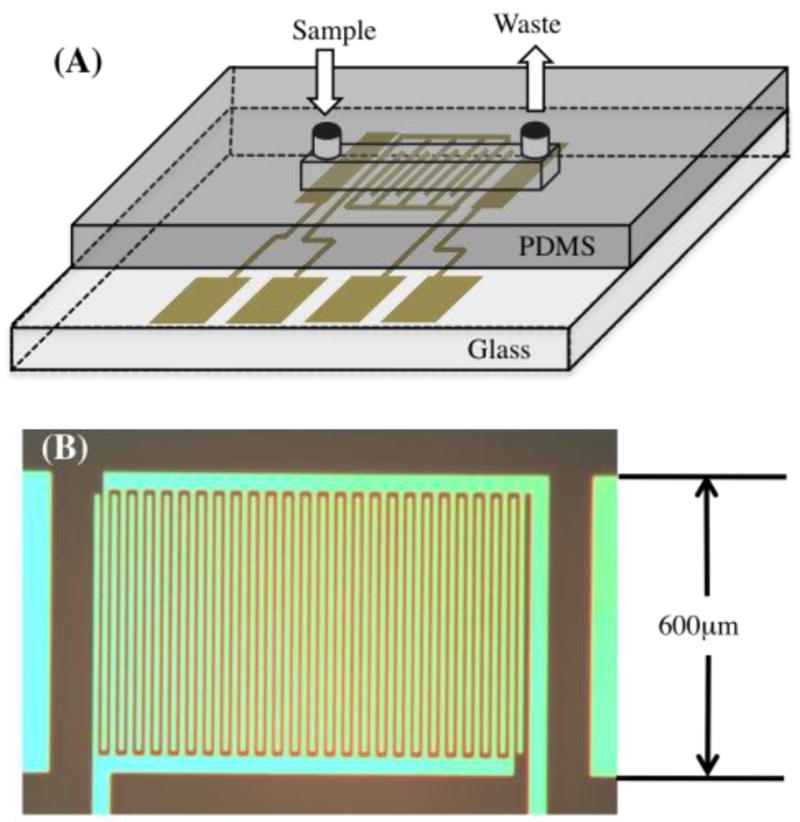
Setup of electrode chips. (A) The electrochemical detectors are composed of electrodes patterned on a glass chip and a PDMS microchannel. The electrode set includes Ag|AgCl reference electrode, IDA working electrode, and Au counter electrode. The microchannel of 35 μm height, 200 μm width and 1 cm length is connected to a syringe pump. (B) Micrograph of the 25-pair Au IDA with 10 μm-wide microbands and 10 μm gaps.
Electrode chips are produced in the FabLab at the University of Maryland. A Pyrex 7740 wafer (Corning, NY) is first cleaned with piranha solution (3:1 H2SO4:H2O2) for 5 minutes, followed by the sputtering of 100 Å chrome and 1000 Å gold layers with the AJA Sputtering System (Scituate, MA). Electrode patterns are made by photolithography using Shipley 1813 (Marlboro, MA) photoresist. A chrome-on-glass mask (PhotoplotStore, Denver, CO) with the resolution of 0.6 μm is used to pattern S1813. After developing S1813 with Shipley CD-30 for 30 seconds, TFA gold and chrome etchant solution (Danvers, MA) is used to dissolve the exposed Au and Cr. Then S1813 is stripped from the wafer and a 1100 Å thick silicon dioxide insulation layer is deposited on the electrode wafer with an Oxford plasma enhanced chemical vapor deposition system (Oxon, UK). The second photolithography step and etching of silicon dioxide with 10:1 HF (EMD, Brookfield, WI) are performed to expose the electrode areas. Electrode wafers are then cut into 1.5 cm × 2 cm chips with a dicing saw. The S1813 is removed in 1165 stripper (Rohm and Haas, Midland, MI) and 2-propanol (JT Baker cat #9079, Phillipsburg, NJ) after the chips are diced.
Interdigitated electrodes are composed of two sets of interconnected microband electrodes. Each microband is 10 μm wide and 540 μm long, with the gap of 10 μm between each band (as shown in Fig. 1B). Two rectangular electrodes with the dimension of 200 μm × 600 μm are used as reference and counter electrodes. The Ag|AgCl reference electrode is electroplated on the Au surface with Techni Silver Cyless II Ag plating solution (Cranston, RI) at the current density of 10 mA/cm2 for 10 minutes, followed by the oxidation of Ag in 0.1 M HCl (BDH 7647-01-0, West Chester, PA) at the current density of 10 mA/cm2 for 5 minutes.
2.2 Characterization of Microelectrodes
Cyclic voltammetry is used to characterize the IDA integrated into the microfluidic channel (inside of the microchannels under static condition). The cyclic voltammograms of the IDA electrodes were recorded by scanning the anode array (generating electrode) in 10−3 M 4-aminophenol hydrochloride (PAP, Sigma, cat # A6161, St. Louis, MO) between -0.2 and 0.3 V against the on-chip Ag|AgCl reference electrode at the rate of 10, 20, 50, 100 and 200 mV/sec, while keeping the cathode array (collecting electrode) potential at −0.15 V. A CHI 820A (CH Instrument, Austin, TX) is used for electrochemical analysis. All aqueous solutions are prepared with deionized water at the resistivity of 18.2 MΩ freshly generated from a Purelab Ultra water purification system (ELGA, Woodridge, IL). All electrodes are electrochemically cleaned by cycling in 0.05 M H2SO4 followed by cycling in 0.1 M NaOH and conditioning in phosphate buffer for 3 minutes before use. All PAP solution is prepared freshly prior use in pH 7.0 background solution (phosphate buffer) containing 20 mM dibasic phosphate (JT Baker cat #3828).
2.3 Impact of flow velocity on microfluidic IDA performance
Two sets of experiments are performed for detecting the flow rate effect on current when integrating electrodes into a microchannel. In the first set of experiments, currents are recorded at a steady flow. The 4-aminophenol at the concentration of 1.0 × 10−5 M in phosphate buffer is primed through the chip by a syringe pump at the flow rates of 0.3, 0.6, 0.9, 1.2, 1.5 and 1.8 mL/hr for a 70 second period of time. Sixty seconds after the pumping starts, a potential is applied on the working electrodes for 10 seconds. In the second set of experiments, sample is primed at 1.2 mL/hr for a 30 second period of time. A potential is applied on the working electrodes for 10 seconds after 0, 5, 10, 20, 30, 45 and 60 seconds from when the pump is stopped. Based on the results of cyclic voltammograms (as shown in Fig. S1 in the Supplementary materials), the oxidation and reduction potential are kept at 0.15 and −0.1 V respectively. The current signals acquired in between 9.75 and 10 seconds are averaged to represent a single point of measurement. Each flow rate is repeated 3 times.
2.4 Impact of electrode regeneration on microfluidic IDA performance
PAP solution in the concentration range of 1.0 × 10−10 M to 1.0 × 10−7 M is primed into the microchannel at the rate of 1.2 mL/hr for a 30 second time period and then idles for 30 seconds before a potential is applied on the working electrodes. Two sets of experiments are used to demonstrate the effect of the cleanliness of the electrode surface. On the electrode where the surface is not regenerated, 15 μL of buffer is primed through the channel after each measurement. On the electrode where the surface is regenerated, 7.5 μL of ethanol and 7.5 μL of buffer are primed through the channel after each measurement.
2.5 Electrochemical detection
Amperometry is used to characterize the detection limit and dynamic range of the IDA inside of the microchannel. The IDA is first conditioned with amperometry at 0.15 V for 30 seconds in background solution before measurements. 10 μL of sample solution is delivered into the channel by pumping at the flow rate of 1.2 mL/hr for 30 seconds. The pump is then stopped for 30 seconds to allow the fluid to reach stationary status before the currents are collected by applying 0.15 V and −0.1 V potential on the generating and collecting electrodes respectively. To regenerate the electrode surface to its initial status and remove residual sample solution from the channel, 7.5 μL ethanol and 7.5 μL background solution are primed through the microchannel after each measurement of PAP.
2.6 Immobilization of beta-galactosidase on the microchannel surface
β-GAL of different concentrations is prepared in phosphate buffered saline (PBS, Sigma P4417) solution. The PDMS channel is first functionalized with amine groups via 3-aminopropyl-trimethoxysilane (3-APS, Aldrich 28177-8) followed by the activation by 2% glutaraldehyde. β-GAL solution is then introduced into the channel for immobilization. The activity of the immobilized β-GAL is assessed by introducing 1.0 × 10−3 M PAPG (Sigma A9545) in 2.0 × 10−2 M phosphate buffer and allowing it to react for up to 120 seconds. Amperometric detection is then conducted at the potential of 0.15 and −0.1V on the anode and cathode respectively. This protocol is described in detail in the Supplementary Material.
2.7 Enzyme-linked DNA hybridization assay
DNA hybridization is performed on the PDMS microchannel surface, which is integrated with the electrode chip to form a microfluidic system. The target DNA sequence is 41 bases in length (see Supplementary Material for sequence). A 20 nt probe (primary probe) with an amine 5’ end that is complementary to a portion of the target is immobilized onto the amino-silanized microchannel surface with a glutaraldehyde cross-linker. Ten μL of target DNA solution with the concentration of zero, 1.0 × 10−10 M, 1.0 × 10−9 M or 1.0 × 10−8 M are then primed into the channel where the primary probe DNA is immobilized. A second ssDNA (secondary DNA probe), which is 20 nt in length and is complementary to another region of the target, is conjugated with β-Galactosidase via a thiol group on the 3’ end and an SPDP cross-linker. The hybridization of these three elements results in the immobilization of β-GAL onto the channel surface, as illustrated in Fig. S2 in the Supplementary Material. The entire assay time takes less than 30 minutes. The complete protocol and DNA sequences are described in detail in the Supplementary Material.
3. Results and Discussion
3.1 Microfluidic IDA characterization and collection efficiency
The combination of an IDA microelectrode and the recyclable PAP signaling molecule was chosen because of the potential signal gain due to multiple redox cycles of a single PAP molecule at adjacent digits of the microelectrode. To characterize the collection efficiency of the IDA, cyclic voltammograms (CVs) were recorded by scanning the potential on one of the electrode arrays while keeping the potential of the other at −0.15 V. Both the generating and collecting electrodes showed relatively steady state voltammograms at the scan rate lower than 50 mV/sec. The collection efficiency of the redox-recycling process on the IDA electrode is given by the ratio of the steady state currents measured on the constant potential (collection) and scanned (generation) electrodes. The calculated collection efficiency increases (i.e., higher recycling occurs) as scan rates get lower. As shown by the results summarized in Table S2 in the Supplementary Material, 97% collection efficiency is observed at the scan rate of 10 mV/sec. This high recycling efficiency implies an improved sensitivity to a particular concentration of PAP as compared to conventional electrochemical constructs, which ultimately leads to a better detection limit. The detailed CVs are depicted in Fig. S1 in the Supplementary Material.
3.2 Impact of flow velocity on microfluidic IDA performance
The detection limit can also be improved by reducing the measurement variation. In an IDA microelectrode system, convective sample movement can affect the measurement variation because the compound generated on the anodic microband can be flushed down to the cathodic microband for reduction and further flushed down to another anodic microband for oxidation. As the flow rate increases, the convection has a larger effect and therefore the signal increases. The Levich equation (Eq. 1) shows that the limiting current of an IDA in a microchannel is dependent on flow velocity; we found that the amperometric current also follows a similar relation. The current between 9.75 and 10 seconds of each measurement is averaged and used to represent each single point of concentration. As Fig. 2A shows, the anodic current is linearly proportional to the cubic root of flow rate between 0.3 to 1.8 mL/hr. This dependence of current on the flow rate illustrates the importance of applying consistent flow rate for reproducible analysis when integrating IDAs in microchannels. Similarly, the oxidation current measured under the stationary status is found to be dependent on the gap between the time when the pump is stopped and the measurement. As the flow rate decreases gradually after the pump is stopped due to the inertia of the fluid, the detected current decreases to a steady state accordingly. Fig. 2B shows that the current drops rapidly after the pumping is stopped and it does not change after the fluid is idle for 20 seconds. To avoid the fluctuation of the signal caused by flow while minimizing the consumed sample volume, we adopt the stationary status for more consistent measurements to pursue a better detection limit and dynamic range. A minimum idling time is desired to achieve a high turnover rate; therefore we chose the idling time of 30 seconds for further measurements.
Fig. 2.

(A) Flow rate effect on the measured current. Sample containing 1.0 × 10−3 M PAP is purged into the microchannel at different flow rates between 0.3 and 1.8 mL/hr by a syringe pump. The correlation follows the cubic root of the flow rate with correlation coefficient of 0.984 (mean ± σ, n=3). (B) Impact of idle time on measured current after pump is stopped. Sample is pumped at 1.2 mL/hr for 30 seconds. After 0, 5, 10, 20, 30, 45 or 60 seconds, oxidation current is collected for 10 seconds (mean ± σ, n=3). (C) Comparison of measured current at different PAP concentration with (solid bars) or without (open bars) regeneration of electrode by flushing ethanol in between measurements (mean ± σ, n=3).
3.3 Impact of electrode regeneration on microfluidic IDA performance
Maintaining a clean electrode surface is critical for preserving low measurement variability, and thus also contributes to improved detection limit. To maintain a clean electrode surface, we introduce a regeneration step into our protocol that has not been reported in previous PAP detection work. A comparison of PAP detection with and without electrode regeneration between measurements is shown in Fig. 2C. In general, the oxidation currents of PAP show higher signal magnitude when the chips are conditioned with both buffer and ethanol than only with buffer after each measurement. Also, the standard deviation of the measured current is much higher on the chips when the electrode surface is not flushed with ethanol. This result implies that the oxidation of PAP fouls the electrode surface and reduces the reproducibility of the system. Therefore, we use this cleaning protocol for our further experiments on the detection on PAP and its application of enzyme-linked biomolecule detection.
3.4 Detection limit of PAP on the microfluidic IDA
Given the improvements in sensitivity and measurement variation due to our microfluidic design and measurement protocol, we can expect an improved detection limit for PAP as compared to previously reported results. To determine the detection limit, we perform repeated amperometric detection of PAP, with fresh sample flushed into the channel after each measurement. After the potential is applied on the IDAs, the currents decrease gradually until reaching steady state. The current between 9.75 and 10 seconds of each measurement is averaged and used to represent each single point of concentration. When performing the detection in a microchannel, between each measurement ethanol is purged into the channel to flush away polymerized PAP and to regenerate the Au surface. The concentration dependence of the amperometric current is shown in Fig. 3 (mean + standard deviation, n=3). The background signal from buffer solution appears as low as 54.6 ± 0.482 pA (mean ± standard deviation). The current of PAP at the concentration of 1.0 × 10−10 M is 600 pA, which is significantly higher than the detection limit signal (IUPAC defines the detection limit as background signal plus 3 times the background standard deviation, i.e., 56.046 pA). A linear response between 1.0 × 10−9 M to 1.0 × 10−5 M with a correlation coefficient of 0.994 is measured, as Fig. 3 shows. Results in Fig. 3 are collected from the same electrode chip. Similar data of detection limit and dynamic range are collected from different electrode chips as well.
Fig. 3.

Oxidation current vs. PAP concentration on log-log scale. A 15 μL sample was delivered into the microchannel with a syringe pump. The detection limit is 1.0 × 10−10 M. The dynamic range is between 1.0 × 10−9 M and 1.0 × 10−5 M with a correlation coefficient of 0.994. The points represent the average value of 3 parallel measurements of the average current recorded between 9.75 and 10 second (mean + σ, n=3).
We believe that the improved detection limit is the result of several factors that relate either to increased sensitivity or decreased signal variation. First, since the electrodes are integrated into the microchannel and form a flow injection system, as opposed to sensors in beakers, the analyte is directed to the electrode surface with higher efficiency and better control; the reproducibility is therefore increased (Niwa et al., 1990). Also, the microchannel limits the diffusion of the redox compound, which leads to efficient amplification of the IDA and thus contributes to the lower detection limit (Ruzicka and Hansen, 2000). In addition, the regeneration of the Au electrode surface by flushing ethanol through the microchannel offers a consistent electrode condition for detection. We also carefully control the fluid at a static state for measurement, and therefore the signal fluctuation due to flow velocity is avoided. The result of all of these improvements is the lowest reported detection limit for PAP, despite the modest and easily fabricated inter-digit spacing of the microelectrode.
3.5 Detection of immobilized β –GAL in the microfluidic channel
In order to demonstrate the potential detection performance using β-GAL for enzyme-linked electrochemical immunoassays or DNA hybridization assays in our microfluidic IDA, the detection of PAP produced by surface confined β-GAL in a microchannel was investigated. 1.0 nM β-GAL is flushed into the microchannel and is immobilized via amine groups onto a 3-APS functionalized microchannel through a glutaraldehyde crosslinker. After the immobilization of β-GAL, the microfluidic channel is exposed to 1.0 × 10−6 M PAPG for the enzymatic reaction for 30, 60 or 120 seconds. The current responses of chips to (i) buffer solution, (ii) 1.0 × 10−3 M PAPG and (iii) 1.0 × 10−6 M PAP standard solution are shown in Fig. 4. Solid bars represent the current responses of chips with 1.0 × 10−9 M β-GAL used for immobilization, while empty bars represent no immobilized β-GAL in the channel (negative control). For the chip with no immobilized β-GAL, a small increment in current is observed when replacing buffer solution with PAPG solution. However, no significant difference is observed when increasing the exposure time of the chip to PAPG solutions. We can therefore conclude that the change in current between buffer and PAPG is caused by the oxidation of PAPG in a very limited level. However, PAP is not generated in this system since the oxidation currents did not increase as the reaction time increased. On the chip in which β-GAL is immobilized, the current shows a significant and monotonic increase as the enzymatic reaction time increases. This implies the successful immobilization of β-GAL on the surface and the high availability of its activity on the IDA. The detection of PAP standard solution is used to calibrate the measured current of PAPG. The immobilized β-GAL (from a solution of 1.0 × 10−9 M β-GAL) generates about 3.0 × 10−9 M of PAP, which diffuses to the electrode surface after 2 minutes of reaction. Based on these preliminary results, we have demonstrated the feasibility and high sensitivity of an electrochemical assay via β-GAL on the IDA biosensor integrated into a microfluidic system.
Fig. 4.

Oxidation current recorded on IDA after exposing to buffer, 1.0 × 10−3 M PAPG or 1.0 × 10−6 M PAP. 0 M (open bar) and 1.0 × 10−9 M (solid bar) β-GAL had been applied for immobilization on the microchannel surface (mean ± σ, n=3).
3.6 Enzyme-linked DNA hybridization assay
Target DNA solution with the concentration of 0, 1.0 × 10−10, 1.0 × 10−9 and 1.0 × 10−8 M are used as the samples for the hybridization assay. To detect the hybridization of the target DNA sequence with the two probes, 1.0 × 10−3 M PAPG in 1.0 × 10−2 M phosphate buffer is introduced into the channel for 90 seconds to allow the hydrolyzation reaction. The dual working electrode mode is used for detection with the same parameters as the PAP measurement described in the Electrochemical detection subsection within the Experimental section. In between each measurement, 7.5 μL of ethanol and DI water are flushed through the channel to refresh the electrode surface. On each chip, oxidation currents of 1.0 × 10−2 M phosphate buffer and 1.0 × 10−6 M PAP solution are used as calibration points and all the oxidation currents collected from the PAPG hydrolyzation are converted to the equivalent PAP concentrations. As Fig. 5 presents, the equivalent PAP concentration shows a monotonic increase as target DNA concentration increases from zero to 1.0 × 10−8 M. The current generated on the negative control chip is caused by the oxidation of the 1.0 × 10−3 M PAPG and is considered as background signal. A significant difference between chips with zero and 1.0 × 10−10 M target DNA immobilized is observed (P value = 0.05); therefore, for 90 seconds of enzymatic reaction time, the detection limit of the enzyme-linked DNA hybridization in our system is 1.0 × 10−10 M. The inset in Fig. 5 shows the data points plotted on a log-log scale. A linear dependence of the equivalent PAP and the target DNA concentration is found in the range of 1.0 × 10−10 to 1.0 × 10−8 M with the correlation coefficient of 0.999. This implies that the PAP generated in the microchannel is proportional to the target DNA. To our knowledge, the previously lowest reported detection limit for an enzyme-amplified DNA hybridization assay on microelectrodes in a microfluidic system is 1.0 × 10−9 M, with the assay time over 170 minutes (Liu et al., 2004). In this report, we demonstrate a much shorter assay time (30 minutes) with lower observed detection limit (1.0 × 10−10 M) using our microfluidic system and an optimized protocol.
Fig. 5.
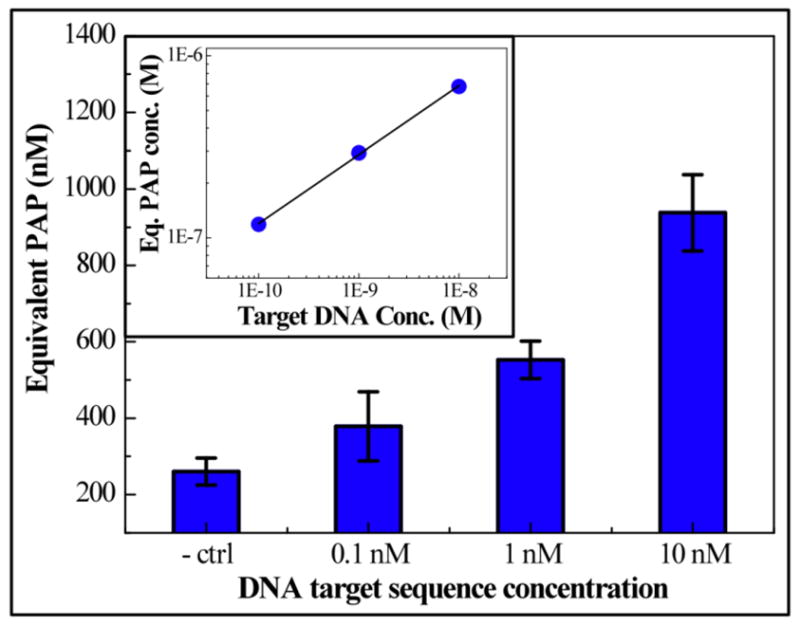
A monotonic increase of generated PAP is found as target DNA concentration increases from zero to 1.0 × 10−8 M. A significant difference is found between control and 1.0 × 10−10 M chips (P=0.05). Error bards are the standard deviation of 3 parallel measurements. Oxidation current is converted into equivalent PAP concentration using zero and 1.0 × 10−6 M PAP as calibration points. Inset: On log-log scale (axes are same as the big plot), equivalent PAP concentration shows a linear dependence on target DNA for concentrations between 1.0 × 10−10 and 1.0 × 10−8 M.
4. Conclusion
In this work, we presented the detection of 4-aminophenol (PAP) in a microfluidic system with a turnover time of 2 minutes using a gold IDA with 10 μm inter-digit spacing, which is simple to fabricate with high yield and low cost. With well-designed buffer conditions and proper sampling and cleaning procedures, we achieve the lowest reported observed detection limit for PAP of 1.0 × 10−10 M with broad linear dynamic range of 1.0 × 10−9 to 1.0 × 10−5 M. The recorded current shows a strong dependence on the flow rate of the sample, which emphasizes the advantage of the precise flow control afforded by the microfluidic channel when applying IDAs for sensing. The regeneration of the electrode surface using ethanol prevents fouling and thus preserves the high sensitivity of the electrode and improves repeatability. The enzyme activity is not hindered after flushing the ethanol through the microchannel. The surface confined β-GAL activity detection confirms the feasibility of using an IDA biosensor integrated into a microfluidic system for enzyme-labeled electrochemical assays. Finally, we demonstrate detection of a 41 nucleotide DNA sequence in an enzyme-linked DNA hybridization assay with the observed detection limit of 1.0 × 10−10 M with an assay time of less than 30 minutes.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the funding from: the Middle Atlantic RCE Program, NIAID/NIH 2 U54 AI057168-06, The United States Army, grant number W81XWH0920109, and NIBIB, grant number 5K25EB006011. The authors also thank Drs. David Mosser and Debra Weinstein for their commitment to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizawa M, Wang L. In: Polymeric Materials Encyclopedia: D-E. 1. Salamone JC, editor. Vol. 3. John Wiley; New York: 1996. pp. 2107–2115. [Google Scholar]
- Amatore C, Belotti M, Chen Y, Roy E, Sella C, Thouin L. J Eletroanal Chem. 2004;573:333–343. [Google Scholar]
- Amatore C, Oleinick A, Klymenko OV, Svir I. Chem Phys Phys Chem. 2005;6:1581–1589. doi: 10.1002/cphc.200500129. [DOI] [PubMed] [Google Scholar]
- Amatore C, Klymenko OV, Svir I. Chem Phys Phys Chem. 2006;7:482 – 487. doi: 10.1002/cphc.200500400. [DOI] [PubMed] [Google Scholar]
- Bocxlaer JFV, Clauwaert KM, Lambert WE, Leenheer APD. Clin Chem. 1997;43:627–634. [PubMed] [Google Scholar]
- Compton RG, Fisher AC, Wellington RG. J Phys Chem. 1993;97:10410–10415. [Google Scholar]
- Choi JW, Oh KW, Thomas JH, Heineman WR, Halsall HB, Nevin JH, Helmicki AJ, Henderson HT, Ahn CH. Lab Chip. 2002;2:27–30. doi: 10.1039/b107540n. [DOI] [PubMed] [Google Scholar]
- Diaz-Gonzalez M, Gonzalez-Garcia MB, Costa-Garcia A. Electroanalysis. 2005;17:1901–1918. [Google Scholar]
- Do J, Ahn CH. Lab Chip. 2008;8:542–549. doi: 10.1039/b715569g. [DOI] [PubMed] [Google Scholar]
- Eddy S, Warriner K, Christie I, Ashworth D, Purkiss C, Vadgama P. Biosens Bioelectron. 1995;10:831–839. [Google Scholar]
- Ferreira LF, Boodts JFC, Brito-Madurro AG, Madurro JM. Polym Int. 2008;57:644–650. [Google Scholar]
- Girault HH. Analytical and Physical Electrochemistry. 1. EPFL Press; Lausanne, Switzerland: 2004. [Google Scholar]
- Huang W, Hu W, Song J. Talanta. 2003;61:411–416. doi: 10.1016/S0039-9140(03)00302-3. [DOI] [PubMed] [Google Scholar]
- Laczka O, Ferraz RM, Ferrer-Miralles N, Villaverde A, Munoz FX, Campo FJd. Anal Chim Acta. 2009;641:1–6. doi: 10.1016/j.aca.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Lewis PM, Sheridan LB, Gawley RE, Fritsch I. Anal Chem. 2010;82:1659–1668. doi: 10.1021/ac901066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Perdue RK, Sun L, Crooks RM. Langmuir. 2004;20:5905–5910. doi: 10.1021/la049605p. [DOI] [PubMed] [Google Scholar]
- Niwa O, Morita M, Tabei H. Anal Chem. 1990;62:447–452. doi: 10.1021/ac951232x. [DOI] [PubMed] [Google Scholar]
- Niwa O, Xu Y, Halsall HB, Heineman WR. Anal Chem. 1993;85:1559–1563. doi: 10.1021/ac00059a013. [DOI] [PubMed] [Google Scholar]
- Ohtsuka K, Endo H, Morimoto K, Vuong BN, Ogawa H, Imai K, Takenaka S. Anal Sci. 2008;24:1619–1622. doi: 10.2116/analsci.24.1619. [DOI] [PubMed] [Google Scholar]
- Ordeig O, Godino N, Campo Jd, Munoz FX, Nikolajeff F, Nyholm L. Anal Chem. 2008;80:3622–3632. doi: 10.1021/ac702570p. [DOI] [PubMed] [Google Scholar]
- Purushothama S, Kradtap S, Wijayawardhana CA, Halsall HB, Heineman WR. Analyst. 2001;126:337–341. doi: 10.1039/b006798i. [DOI] [PubMed] [Google Scholar]
- Reihmann MH, Ritter H. J Macromol Sci, Part A- Pure Appl Chem. 2002;39:1369–1382. [Google Scholar]
- Ronkainen-Matsuno NJ, Halsall HB, Heineman WR. Electrochemical Immunoassays and Immunosensors. CRC Press; 2007. [Google Scholar]
- Ruzicka J, Hansen EH. Anal Chem. 2000;72:212A–217A. doi: 10.1021/ac002756m. [DOI] [PubMed] [Google Scholar]
- Skladal P. Electroanalysis. 2005;9:737–745. [Google Scholar]
- Snead WK, Remick AE. J Am Chem Soc. 1957;6121–6127 [Google Scholar]
- Tsuchida E, Kaneko M, Kurimura Y. Die Makromolekulare Chemie. 1970;132:209–213. [Google Scholar]
- Vieira SN, Ferreira LF, Franco Diego Leoni, Afonso AS, Goncalves RA, Brito-Madurro AG, Madurro JM. Macromol Symp. 2006;245–246:236–242. [Google Scholar]
- Wang J, Ibanez A, Chatrathi MP, Escarpa A. Anal Chem. 2001;73:5323–5327. doi: 10.1021/ac010808h. [DOI] [PubMed] [Google Scholar]
- Warsinke A, Benkert A, Scheller FW. Fresenius J Anal Chem. 2000;366:622–634. doi: 10.1007/s002160051557. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Ozawa S, Ikeda T, Senda M. Anal Sci. 1992;8:87–88. [Google Scholar]
- Wyszecka-Kaszuba E, Warowna-Grzeskiewicz M, Fijalek Z. J Pharm Biomed Anal. 2003;32:1081–1086. doi: 10.1016/s0731-7085(03)00212-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.