

Abstract
MACS (Model-based Analysis of ChIP-Seq) is a command line tool designed by X. Shirley Liu and colleagues to analyze data generated by ChIP-Seq experiments in eukaryote, especially in mammal. Given the ChIP-Seq data with or without control samples, MACS can be used to identify transcription factor binding sites and histone modification enriched regions. This unit describes two basic protocols that provide detailed information of how to use MACS to identify either the binding sites of a transcription factor, or the enriched regions of a histone modification with broad peaks. Furthermore, the basic ideas of MACS algorithm and its appropriate usage are discussed.
Keywords: MACS, ChIP-Seq, peak-calling, cistrome, epigenome
INTRODUCTION
One type of special proteins, called transcription factors (TFs), performs important functions by regulating the transcription of genes via physically interaction with certain DNA sequence patterns, or called motifs. To uncover the regulation mechanisms, one promising approach is to identify all cis-acting targets, or called binding sites, for a given TF in the genome scale, which is defined as the TF’s cistrome (Carroll, et al., 2006; Lupien, et al., 2008), and the popular technology to study cistrome is Chromatin Immunoprecipitation coupled with sequencing (ChIP-Seq) (Johnson, et al., 2007). Briefly, in the ChIP step, DNA sequences are fragmentized into hundreds of base pairs, and fragments with certain TF binding are enriched through immunoprecipitation. The enriched DNA fragments are then sequenced using massively parallel DNA sequencing technology, with outputs called sequencing reads or tags. MACS was originally designed to give robust and high resolution peak identification for ChIP-Seq data with two main features (Zhang, et al., 2008). Firstly, MACS empirically models the shift size of ChIP-Seq reads, and uses it to improve the spatial resolution of inferred TF binding sites. Secondly, MACS estimates a dynamic background reads distribution to effectively capture local biases in the genome, allowing for more robust identifications.
Besides cistrome studies, ChIP-Seq technology is also widely used to generate epigenome profiles, especially histone modification status (Barski, et al., 2007; Mikkelsen, et al., 2007). As different histone modifications have distinct effects on chromatin environments by altering the binding to DNA or providing recognition sites for chromatin effector modules, genome-wide ChIP-Seq approaches can dramatically increase the understanding on the relationships between specific histone modifications and gene regulation outcomes. Although the procedure to generate histone modification ChIP-Seq data is quite similar to that for cistrome, the distributions of sequencing reads for both cases are usually different. For most TFs, sequencing reads enriched regions are generally discrete, and typically they form sharp peaks along the genome. However for many types of histone modifications, the distribution of reads obeys a continuous property, as the epigenetic status of nearby nucleosomes tend to be similar, usually resulting in quite broad peaks. With proper parameter setting, MACS performs well to detect histone modification enriched regions. Similarly, MACS can also be applied in affinity enrichment based DNA methylation studies, such as MeDIP-Seq data.
This unit firstly describes the basic protocol of analyzing FoxA1 ChIP-Seq data in human MCF7 cell line. FoxA1 is a typical TF, which regulates gene expression as a pioneer factor (Lupien, et al., 2008). This protocol contains a control sample. Another basic protocol analyzes H3K27me3 ChIP-Seq in mouse ES cell (Mikkelsen, et al., 2007). H3K27me3 is a widely studied histone modification with broad peaks. Each protocol includes the required data to run MACS, the exact parameters with explanation and the understanding of MACS results. After the two basic protocols, the basic idea behind MACS is presented‥ Besides, a complete parameter list of MACS software with description is followed for user’s reference. The software is available via http://liulab.dfci.harvard.edu/MACS/. It is written in Python and distributed under the terms of Artistic License. The latest version is 1.4.0beta. Specialized terms used in this unit are defined in Table 2.14.1.
Table 2.14.1.
Terminologies
| Term | Definition |
|---|---|
| ChIP-Seq | A biological experiment technology combines chromatin immunoprecipitation (ChIP) and massively parallel DNA sequencing technology (Seq) to identify the DNA binding sites of a transcription factor of interest or certain epigenomic status. |
| epigenome | Epigenome is a parallel to the word genome, which means the overall epigenetic state of a cell, including histone modification, DNA methylation, etc. |
| cistrome | Cistrome is the set of cis-acting targets of a trans-acting factor on a genome scale. With a transcription factor’s cistrome will help to understand its target genes and regulatory mechanism. |
BASIC PROTOCOL 1
RUNNING MACS PROGRAM TO IDENTIFY TRANSCRIPTION FACTOR BINDING SITES
As a non-interactive command line tool, MACS takes input by setting proper command line parameters and no input is needed during running. The input should be mapped reads from ChIP-Seq experiments, and several widely used formats are accepted, while the control data is optional. The minimum output of MACS contains the called peaks and their summits, together with an R script used to draw the shifting size model built by MACS. MACS can also generate wiggle format files, which can be loaded into Affymetrix Integrated Genome Browser (IGB) (Nicol, et al., 2009) or UCSC Genome Browser (Kent, et al., 2002) to visually present the ChIP-Seq signal.
Necessary Resources List
Hardware
A computer with proper versions of Python and R installed.
Software
Python version must be equal to 2.6 or 2.7 to run MACS, and the version 2.6.5 is preferred. R is also needed if user wants to generate a PDF image of the shifting size model. As MACS is a command line program, a terminal is also necessary, which is integrated into most popular operating systems, including Unix, Linux, Windows and Macintosh.
Files
To identify TF binding sites, user must have the file with the information of mapped genomic locations for sequencing reads in certain formats. Currently, eight types of formats are supported, including “ELAND”, “BED”, “ELANDMULTI”, “ELANDEXPORT”, “ELANDMULTIPET”, “SAM”, “BAM” and “BOWTIE”. The detailed description for each format can be found in the “00README” file distributed along MACS package. MACS could detect the format automatically in case it is not overridden by user specified parameters. MACS works on both cases of with or without control data. This protocol uses single-end reads in “BED” format with control data.
The example is FoxA1 ChIP-Seq data in human MCF7 cell line. The data contain two files in “BED” format: “Treatment_tag.bed” for ChIP-seq data and “Input_tag.bed” for control data. Example lines together with indicator for each column are shown as follows. Each line contains genomic location of a sequencing read.
#chrom chromStart chromEnd name score strand
chr1 233604 233639 0 2 −
chr1 559767 559802 0 3 +
chr1 742600 742635 0 2 +
chr1 742600 742635 0 0 +
chr1 744231 744266 0 0 +
chr1 744307 744342 0 2 −
Download and install MACS in local computer (see Support Protocol).
Download and decompress FoxA1 ChIP-Seq data from http://liulab.dfci.harvard.edu/MACS/Sample.html.
Open a terminal and change the working directory to that contains the sample data.
-
Execute MACS by the following command:
macs14 –t ./Treatment_tags.bed -c ./Input_tags.bed -g hs -n FoxA1 –w
In this command, -t specifies the path and file name of ChIP-Seq data and -c specifies the control data, which can be omitted if there is no control data. Parameter -g gives the genome size, while ‘hs’ is a shortcut for human (2.7e9). Prefix of output files is set via -n. Parameter -w indicates to store the pileup of shifted reads into wiggle files at 10 bp resolution. It worth noting that using -w parameter is time and space consuming. The list of MACS critical parameters is shown in Table 2.14.2.
-
Check the MACS critical and progress messages displayed in the terminal, shown as follows.
INFO @ Wed, 28 Jan 2011 16:29:05: # ARGUMENTS LIST: # format = AUTO # ChIP-seq file = ./Treatment_tags.bed # control file = ./Input_tags.bed # effective genome size = 2.70e+09 # band width = 300 # model fold = 10,30 # pvalue cutoff = 1.00e-05 # Range for calculating regional lambda is: 1000 bps and 10000 bps INFO @ Fri, 28 Jan 2011 16:29:05: #1 read tag files… INFO @ Fri, 28 Jan 2011 16:29:05: #1 read treatment tags… INFO @ Fri, 28 Jan 2011 16:29:05: Detected format is: BED INFO @ Fri, 28 Jan 2011 16:29:14: 1000000 <Some lines are deleted here> INFO @ Fri, 28 Jan 2011 16:30:36: #2 Build Peak Model… INFO @ Fri, 28 Jan 2011 16:30:52: #2 number of paired peaks: 11861 INFO @ Fri, 28 Jan 2011 16:30:52: #2 finished! INFO @ Fri, 28 Jan 2011 16:30:52: #2 predicted fragment length is 119 bps INFO @ Fri, 28 Jan 2011 16:30:52: #2.2 Generate R script for model: FoxA1_model.r INFO @ Fri, 28 Jan 2011 16:30:52: #3 Call peaks… <Some lines are deleted here> INFO @ Fri, 28 Jan 2011 16:50:50: #3 call negative peak candidates INFO @ Fri, 28 Jan 2011 16:51:04: #3 use control data to filter peak candidates… INFO @ Fri, 28 Jan 2011 16:51:10: #3 Finally, 13639 peaks are called! INFO @ Fri, 28 Jan 2011 16:51:10: #3 find negative peaks by swapping treat and control INFO @ Fri, 28 Jan 2011 16:51:16: #3 Finally, 2481 peaks are called! INFO @ Fri, 28 Jan 2011 16:51:16: #4 Write output xls file… FoxA1_peaks.xls INFO @ Fri, 28 Jan 2011 16:51:17: #4 Write peak bed file… FoxA1_peaks.bed INFO @ Fri, 28 Jan 2011 16:51:17: #4 Write summits bed file… FoxA1_summits.bed INFO @ Fri, 28 Jan 2011 16:51:17: #4 Write output xls file for negative peaks… FoxA1_negative_peaks.xls INFO @ Fri, 28 Jan 2011 16:51:17: #5 Done! Check the output files!
The messages starts with critical parameters, lines starting with ‘#’, used by MACS in the current run, which is useful to check whether MACS has adopted proper parameters. The followed messages show the progress information and also warning messages if exist.
Check the output files generated by MACS. MACS will generate five files and a directory in current working directory, as listed in Table 2.14.3.
-
Load the script “FoxA1_model.r” with R by:
R --vanilla < FoxA1_model.r
This command will produce a PDF image named “FoxA1_model.pdf” in current working directory, as shown in Figure 2.14.1. Red curve represents the distribution of locations relative to the midpoints for reads from forward strand, while the blue curve for reads from reverse strand. The d is determined as the distance between the summits of red and blue curves, and MACS shifts all reads by d/2 toward the 3’ends to improve the spatial resolution of inferred TF binding sites. Black curve is drawn based on the locations of shifted reads.
-
Understand “FoxA1_peaks.xls”. Besides the annotation lines starting with ‘#’, top 10 lines together with indicator for each column are shown as follows. Each line contains full information of a called peak.
chr start end length summit tags −10*log10(pvalue) fold_enrichment FDR(%) chr1 858357 858641 285 128 6 51.00 13.93 17.36 chr1 998955 999229 275 106 9 74.39 18.28 3.18 chr1 1050021 1050286 266 154 13 152.00 52.23 0.03 chr1 1684288 1684577 290 176 9 89.70 32.14 0.91 chr1 1775031 1775371 341 270 6 51.08 16.71 17.30 chr1 1780682 1780965 284 168 7 61.17 19.90 10.64 chr1 1923147 1923449 303 202 16 164.87 44.31 0.03 chr1 2110970 2111170 201 100 6 67.53 20.89 6.24 chr1 2111408 2111732 325 103 9 71.96 30.95 4.24 chr1 2232814 2233120 307 202 7 63.91 24.10 8.47
For each peak, the detailed information includes the chromosome name, start position, end position, length of peak region, summit location related to the peak start position, number of reads in peak region, −10*log10(p-value) for the peak region (i.e. a value 100 means p-value 1e-10), fold enrichment for this region (compared to the expectation from Poisson distribution with local lambda) and false discovery rate (FDR). “FoxA1_peaks.xls” is a tabular plain text file, and user can open it in Excel and sort or filter using Excel functions.
Understand “FoxA1_peak.bed” and “FoxA1_summits.bed”. The former contains the positions and −10*log10(pvalue) for called peaks, while the latter has the location and height information of peak summits, which is especially useful for DNA motif finding at TF binding sites. Both files are in BED format, which can be loaded directly to Affymetrix IGB or UCSC Genome Browser for visualization. It worth noting that coordinates in BED format starts from 0, therefore the peak locations in “FoxA1_peak.bed” is slightly different from that in “FoxA1_peaks.xls”.
Visualization of wiggle and BED files generated by MACS. Load “FoxA1_MACS_wiggle/treat/FoxA1_treat_afterfiting_chr1.wig.gz” into Affymetrix IGB to visually display the FoxA1 ChIP-Seq signal in chromosome1. Two BED files, “FoxA1_peaks.bed” and “FoxA1_summit.bed”, are also added to examine the peak calling results. A sample 1kb region is shown in Figure 2.14.2, with genome version NCBI36/hg18. In this region, the peak called by MACS is consistent well with the ChIP-Seq signal enriched region. Additionally, with the availability of FKHR motif locations, which dictate the precise FoxA1 binding sites, the reliability of peak summit calling in Figure 2.14.2 is confirmed.
Table 2.14.2.
MACS critical parameters
| Parameters (short format) |
Parameters (long format) |
Description |
|---|---|---|
| -h | --help | Show help message and exit |
| -t TFILE | --treatment=TFILE | ChIP-Seq data files. REQUIRED. When ELANDMULTIPET is selected, user must provide two files separated by comma, e.g. s_1_1_eland_multi.txt,s_1_2_eland_multi.txt |
| -c CFILE | --control=CFILE | Control files. When ELANDMULTIPET is selected, user must provide two files separated by comma, e.g. s_2_1_eland_multi.txt,s_2_2_eland_multi.txt |
| -n NAME | --name=NAME | Experiment name, which will be used as prefix of output files. DEFAULT: “NA” |
| -f FORMAT | --format=FORMAT | Format of reads file, “AUTO”, “BED” or “ELAND” or “ELANDMULTI” or “ELANDMULTIPET” or “ELANDEXPORT” or “SAM” or “BAM” or “BOWTIE”. The AUTO option will let MACS estimate the format type. DEFAULT: “AUTO” |
| -g GSIZE | --gsize=GSIZE | Effective genome size. The value can be 1.0e+9 or 1000000000, or shortcuts: ‘hs’ for human (2.7e9), ‘mm’ for mouse (1.87e9), ‘ce’ for C. elegans (9e7) and ‘dm’ for fruitfly (1.2e8), Default: hs |
| -s TSIZE | --tsize=TSIZE | Read size. This parameter will override the auto detected read size. DEFAULT: 25 |
| -p PVALUE | --pvalue=PVALUE | P-value cutoff for peak detection. DEFAULT: 1e-5 |
| -m MFOLD | --mfold=MFOLD | Select regions with read enrichment ratio (compared to background) within MFOLD interval to build shifting size model. The ratio must be higher than the lower limit, and lower than the upper limit. DEFAULT: 10,30 |
| -w | --wig | Whether or not to store the pileup of shifted reads into wiggle files. Turn on the parameter results in time and space consuming. |
Table 2.14.3.
MACS output files
| File name | Description |
|---|---|
| FoxA1_model.r | An R script to produce a PDF image about the shifting size model based on ChIP-Seq data. |
| FoxA1_peaks.xls | A tabular file containing information about called peaks. |
| FoxA1_peaks.bed | A BED format file containing the peak locations |
| FoxA1_summits.bed | A BED format file containing the summits locations for called peaks. |
| FoxA1_negative_peaks.xls | A tabular file containing information about negative peaks, which are called by swapping the ChIP-Seq and control channel. The file will only be generated when control data are available. |
| FoxA1_MACS_wiggle | A directory containing compressed wiggle format files, which are generated through the pileup of shifted reads for each chromosome. |
Figure 2.14.1.
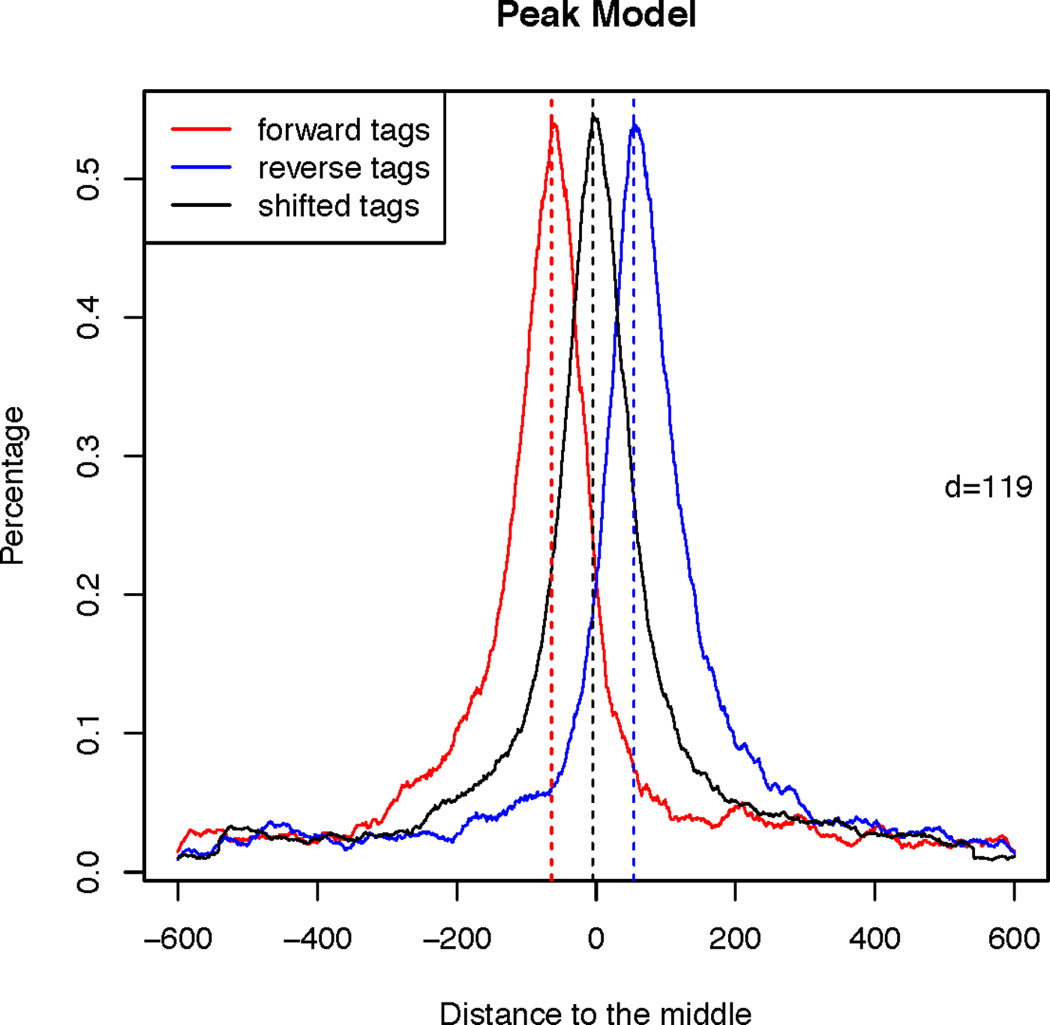
Shifting size model for FoxA1 ChIP-Seq data.
Figure 2.14.2.
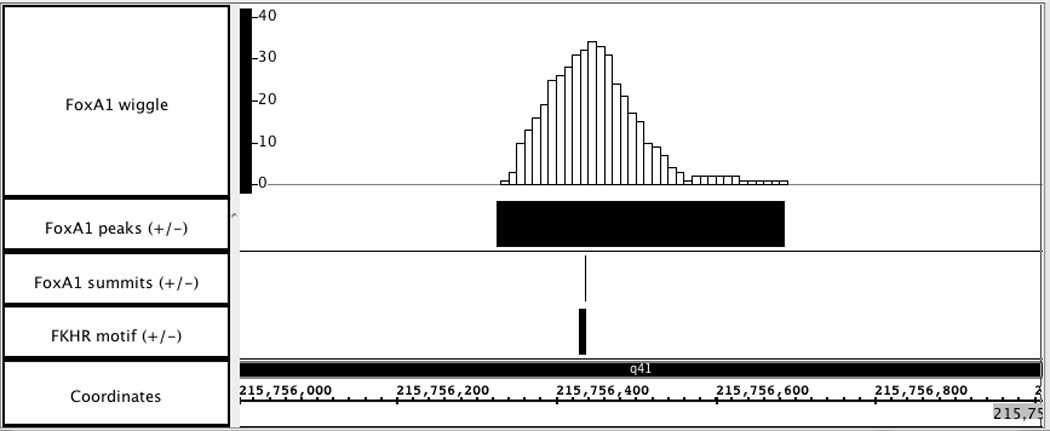
Visualization of wiggle and BED files for FoxA1 ChIP-Seq data in Affymetrix IGB. Genome region: chr1: 215,756,000 – 215,757,000.
BASIC PROTOCOL 2
RUNNING MACS PROGRAM TO PROFILE HISTONE MODIFICATION STATUS
The reads distribution of histone modification ChIP-Seq data usually obeys a continuous property, which is different from that of most TF ChIP-Seq data. When applying MACS to ChIP-Seq data with broad peaks, two advanced parameters should be set properly. First, such data increase the difficulty to build robust shifting size model, therefore it is recommended to skip the model building step by setting --nomodel in the command line. Second, the estimation of dynamic background works well for histone modification ChIP-Seq data with control. However, if no control data is available for ChIP-Seq data with broad peaks, the local background estimation should be skipped via setting --nolambda in the command line.
Necessary Resources List
Hardware
A computer with proper versions of Python and R installed.
Software
Python version must be equal to 2.6 or 2.7 to run MACS, and the version 2.6.5 is preferred. R is also needed if user wants to generate a PDF image of the shifting size model. As MACS is a command line program, a terminal is also necessary, which is integrated into most popular operating systems, including Unix, Linux, Windows and Macintosh.
Files
This basic protocol uses H3K27me3 ChIP-Seq data in mouse ES cells data as example. No proper control is available. Data can be downloaded from GEO with accession number GSM307619 (file name “GSM307619_ES.H3K27me3.aligned.txt.gz”). The file format is custom defined, which cannot be directly recognized by MACS. A simple script is needed to convert it to BED format, and the following command is a solution in shell environment.
awk '{print $1"\t"$2"\t"$3"\t0\t"$6"\t"$4}' GSM307619_ES.H3K27me3.aligned.txt > mES.H3K27me3.bed
Download and install MACS in local computer (see Support Protocol).
Download and decompress H3K27me3 ChIP-Seq data from GEO (GSM307619), and convert the file to BED format.
Open a terminal and change the working directory to that contains the sample data.
-
Execute MACS by the following command:
macs14 -t ./mES.H3K27me3.bed -g mm --nomodel --nolambda -w -n H3K27me3 --space=30
No control data is indicated here. The shortcut ‘mm’ is for mouse (1.87e9). This command introduces three new parameters: --nomodel, --nolambda and --space. The parameter --nomodel is set to skip the model building step. Similarly, --nolambda indicates MACS will use fixed background lambda, instead of dynamics local lambda, for peak calling. Parameter --space is used together with -w to define the resolution for generated wiggle files. The default resolution is 10 bp; by setting a lower resolution, 30 bp in this basic protocol, the size of generated wiggle files will be reduced by approximately 3 times.
-
Check the MACS critical and progress messages displayed in the terminal, shown as follows
INFO @ Sat, 29 Jan 2011 12:17:39: # ARGUMENTS LIST: # name = H3K27me3 # format = AUTO # ChIP-seq file = ./mES.H3K27me3.bed # control file = None # effective genome size = 1.87e+09 # band width = 300 # model fold = 10,30 # pvalue cutoff = 1.00e-05 # Range for calculating regional lambda is: 10000 bps INFO @ Sat, 29 Jan 2011 12:17:39: #1 read tag files… INFO @ Sat, 29 Jan 2011 12:17:39: #1 read treatment tags… INFO @ Sat, 29 Jan 2011 12:17:39: Detected format is: BED INFO @ Sat, 29 Jan 2011 12:17:48: 1000000 <Some lines are deleted here> INFO @ Sat, 29 Jan 2011 12:18:54: #2 Build Peak Model… INFO @ Sat, 29 Jan 2011 12:18:54: #2 Skipped… INFO @ Sat, 29 Jan 2011 12:18:54: #2 Use 100 as shiftsize, 200 as fragment length INFO @ Sat, 29 Jan 2011 12:18:54: #3 Call peaks… INFO @ Sat, 29 Jan 2011 12:18:54: # local lambda is disabled! INFO @ Sat, 29 Jan 2011 12:18:54: #3 !!!! DYNAMIC LAMBDA IS DISABLED !!!! <Some lines are deleted here> INFO @ Sat, 29 Jan 2011 12:31:15: #3 call peak candidates INFO @ Sat, 29 Jan 2011 12:32:04: #3 use self to calculate local lambda and filter peak candidates… INFO @ Sat, 29 Jan 2011 12:32:06: #3 Finally, 23866 peaks are called! INFO @ Sat, 29 Jan 2011 12:32:06: #4 Write output xls file… H3K27me3_peaks.xls INFO @ Sat, 29 Jan 2011 12:32:06: #4 Write peak bed file… H3K27me3_peaks.bed INFO @ Sat, 29 Jan 2011 12:32:07: #4 Write summits bed file… H3K27me3_summits.bed INFO @ Sat, 29 Jan 2011 12:32:07: #5 Done! Check the output files!
As expected, progress information indicates that MACS skips the model building step and disables the estimation of dynamic lambda.
Check the output files generated by MACS. MACS will generate three files (“H3K27me3_peaks.xls”, “H3K27me3_peaks.bed” and “H3K27me3_summits.bed”) and a directory (“H3K27me3_MACS_wiggle”) in current working directory.
Visualization of wiggle and BED files generated by MACS. Load “H3K27me3_MACS_wiggle/treat/H3K27me3_treat_afterfiting_chr1.wig.gz” into Affymetrix IGB to visually display the H3K27me3 ChIP-Seq signal in chromosome1. Two BED files, “H3K27me3_peaks.bed” and “H3K27me3_summit.bed”, are also added to examine the peak calling results. A sample 10 kb region is shown in Figure 2.14.3, with genome version NCBI36/mm8. In this region, the peak called by MACS is consistent well with the ChIP-Seq signal enriched region around the promoter of Twist2 gene.
Figure 2.14.3.
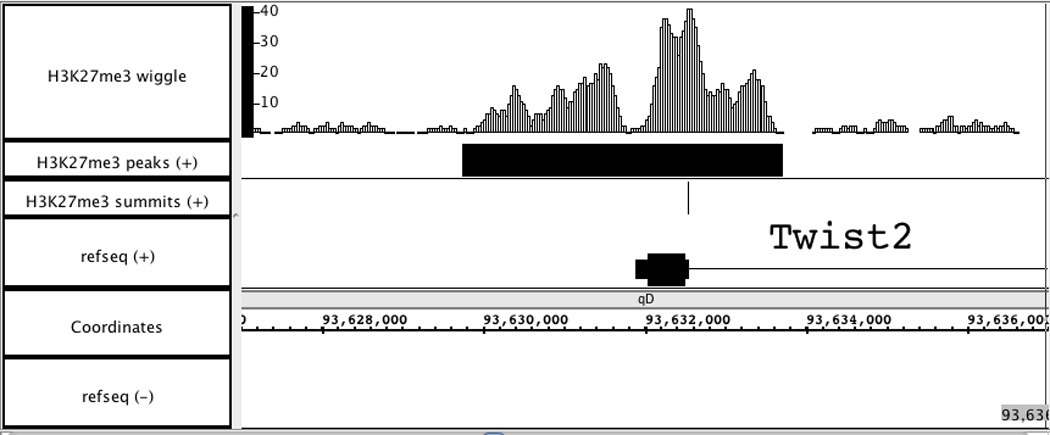
Visualization of wiggle and BED files for H3K27me3 ChIP-Seq data in Affymetrix IGB. Genome region: chr1: 93,627,000 – 93,637,000.
SUPPORT PROTOCOL
OBTAINING AND INSTALLING MACS PROGRAM
Necessary Resources List
Hardware
A computer with proper versions of Python and R installed.
Software
Python version must be equal to 2.6 or 2.7 to run MACS. The version 2.6.5 is preferred.
Download MACS from http://liulab.dfci.harvard.edu/MACS/.
Open up a command terminal and change the working directory to that contains the source package “MACS-1.4.0beta.tar.gz”.
-
Unpack the package by the command:
tar xvzf MACS-1.4.0beta.tar.gz
-
Change the working directory to “MACS-1.4.0beta” and simply run the install script:
python setup.py install
By default, the script will install python library and executable codes globally, which means user should be root or administrator of the machine to complete the installation. Please contact the administrator of the machine when necessary.
For user who prefers a nonstandard install prefix, for example to install everything under user’s own HOME directory, run the command:
python setup.py install --prefix /home/userhome/
-
Configure PYTHONPATH. After installation, user might need to add the install path to the PYTHONPATH environment variable. The detailed process varies across different platforms, however the general concept is identical. Using Linux with bash environment as example, add the following new line to the user’s shell configuration file, usually “~/.bashrc”:
export PYTHONPATH=/home/userhome/lib/python2.6/site-packages:$PYTHONPATH
In this example command, /home/userhome/ is the install prefix. If user doesn’t specify a prefix in the installation step, the value can be found via typing sys.prefix in a Python environment. The value 2.6 is the Python major-minor version used (2.6 or 2.7), which equals to Python’s sys.version[:3].
-
Configure PATH. Similar to PYTHONPATH configuration, user might also need to configure the PATH environment variable to use MACS command directly. The process for updating PATH variable is the same as that for PYTHONPATH variable. Using Linux with bash environment as example, add the following new line to “~/.bashrc”:
export PATH=/home/userhome/bin:$PATH
COMMENTARY
Background Information
ChIP-Seq is a popular technology combining Chromatin immunoprecipitation (ChIP) and high throughput sequencing (Seq) to study either the cistrome of TFs (Johnson, et al., 2007; Robertson, et al., 2007) or the epigenome status (Barski, et al., 2007; Mikkelsen, et al., 2007). ChIP-Seq provides several advantages over ChIP-chip, including less starting material, lower cost, and higher peak resolution, but also poses challenges in the data analysis. First, ChIP-Seq reads do not present the precise location of protein-DNA binding sites but the ends of ChIP fragments, and the fragment length is usually unknown to the user. Second, regional bias along the genome widely exists, due to sequencing and mapping biases, chromatin structure and genome copy number variations (Redon, et al., 2006). Although such biases would be corrected when matching control samples are sequenced deeply enough, it is still quite challenging if no control data is available or the sequence depth of the control data is low. MACS is designed to address these issues and give robust and high resolution ChIP-Seq peak identifications.
Based on the reads distribution, MACS empirically models the shift size of ChIP-Seq reads. Since ChIP-DNA fragments are equally likely to be sequenced from both ends, the reads density around a true TF binding site should show a bimodal enrichment pattern, with forward strand reads enriched upstream of binding and reverse strand reads enriched downstream. Given two parameters, bandwidth and mfold (a high-confidence fold-enrichment interval), MACS slides 2bandwidth windows across the genome to find regions with certain reads enrichment relative to the expectation (larger than 10 fold and smaller than 30 fold as default). MACS selects these high-quality peaks, separates their forward and reverse reads, and aligns them by the midpoint. The distance between the modes of the forward and reverse peaks in the alignment is defined as ‘d’, and MACS shifts all the reads by d/2 toward the 3’ ends to better locate the precise binding sites.
With the genome coverage of most ChIP-Seq experiments, reads distribution along the genome could be modeled by a Poisson distribution (Mikkelsen, et al., 2007). Instead of using a uniform λBG estimated from the whole genome, MACS uses a dynamic parameter, λlocal, defined for each read enriched region as: λlocal = max(λBG, [λ1k,] λ5k, λ10k). In this formula, λ1k, λ5k and λ10k are λ estimated from the 1 kb, 5 kb and 10 kb window centered at the peak location in the control sample, or in the ChIP-Seq sample when a control sample is not available (in which case λ1k is not used). Using λlocal could capture influence of local biases, and the value is robust against occasional low read counts at small local regions. MACS applies λlocal to calculate the p-value for each read enriched region, and only those with p-values below a user-defined threshold (default 10−5) are reported as identified peaks, with the ratio between the ChIP-Seq read count and λlocal as ‘fold_enrichment’.
Critical Parameters and Troubleshooting
MACS uses a two-step strategy to perform ChIP-Seq data analysis: modeling the read shift size, and then peak calling. It worth noting that the parameter mfold is used only in the first step, where a suitable mfold parameter will lead to several thousand paired peaks from ChIP-Seq data for model building. Some users might receive a warning message about “too few paired peaks” when running MACS. Although to decrease mfold may dramatically increase the number of paired peaks, it is not recommended to set mfold lower that 10, as many noises will also be introduced in model building.
When applying MACS to ChIP-Seq data with broad peaks, for example epigenome data, it is recommended to skip the model building step by setting --nomodel in the command line. If no control data is available for such ChIP-Seq data, the local background estimation should be skipped via setting --nolambda in the command line.
MACS empirically calculates FDR based on the number of peaks from control over ChIP that are called at the same p-value cutoff. Therefore if no control data is available, the FDR column does not exist in the output tabular file. Technically, MACS can also be applied to identify differential peaks between two conditions by treating one of the samples as the control. However, calculated FDR value should be ignored, as peaks from either sample are likely to be biologically meaningful in this case.
Advanced Parameters
The advanced parameters of MACS program together with descriptions are shown in Table 2.14.4.
Table 2.14.4.
MACS advanced parameters
| Parameters | Description |
|---|---|
| --bw=BW | Bandwidth. This value is used in model building. If --nomodel is set, 2 time of this value will be used as a scanning window size. DEFAULT: 300 |
| --version | Display MACS version and exit |
| --single-wig | When set, a single wiggle file will be stored for treatment and input separately. Default: False |
| --wigextend=WIGEXTEND | Integer type value. When MACS generates wiggle files, each shifted read will be extended from its middle point to a certain size, with modeled d as default. The value does not affect peak calling. |
| --space=SPACE | The resolution for stored wiggle files, 10 bps as default. Usable only with ‘--wig’ option. |
| --nolambda | If set, MACS will use fixed background lambda, instead of dynamics local lambda, for peak calling. |
| --slocal=SMALLLOCAL | The small region size in bp to calculate dynamic lambda, which is used to capture the bias near peak summit. Invalid if no control data is available. DEFAULT: 1000 |
| --llocal=LARGELOCAL | The large region size in bp to calculate dynamic lambda, which is used to capture the surrounding bias. DEFAULT: 10000 |
| --off-auto | If not set, when MACS fails to build model, it automatically continue the analysis using --nomodel settings. DEFAULT: False |
| --nomodel | Whether or not to build the shifting size model. If set, MACS will not build model. DEFAULT: False |
| --shiftsize=SHIFTSIZE | The predefined shift size in bp. When --nomodel is set, MACS will treat this value as d/2. DEFAULT: 100 |
| --call-subpeaks | If set, MACS will invoke Mali Salmon’s PeakSplitter soft through system call. If PeakSplitter cannot be found, an instruction will be shown for downloading and installing the PeakSplitter package. DEFAULT: False |
| --keep-dup | When set, MACS will keep all duplicate reads (same coordination and strand). Otherwise, MACS calculates the maximum reads at the exact same location based on binomial distribution, and only keep at most this number of reads at each location. Default: False |
| --petdist=PETDIST | Best distance between paired-end reads. Only available when format is ‘ELANDMULTIPET’. DEFAULT: 200 |
| --verbose=VERBOSE | Set verbose level. 0: only show critical message, 1: show additional warning message, 2: show process information, 3: show debug messages. DEFAULT: 2 |
| --diag | Whether or not to produce a diagnosis report. It is up to 9X time consuming. DEFAULT: False |
| --fe-min=FEMIN | For diagnostics, min fold enrichment to consider. DEFAULT: 0 |
| --fe-max=FEMAX | For diagnostics, max fold enrichment to consider. DEFAULT: maximum fold enrichment |
| --fe-step=FESTEP | For diagnostics, fold enrichment step. DEFAULT: 20 |
Acknowledgements
This article is based on research published in Genome Biology with title "Model-Based Analysis of ChIP-Seq (MACS)", which was funded partially by NIH grants HG004069, HG004270 and DK074967.
Literature Cited
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. FoxA1 translates epigenetic signatures into enhancer driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol JW, Helt GA, Blanchard SG, Jr, Raja A, Loraine AE. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25:2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]