

Abstract
Mitochondrial and chloroplast ATP synthases are key enzymes in plant metabolism, providing cells with ATP, the universal energy currency. ATP synthases use a transmembrane electrochemical proton gradient to drive synthesis of ATP. The enzyme complexes function as miniature rotary engines, ensuring energy coupling with very high efficiency. Although our understanding of the structure and functioning of the synthase has made enormous progress in recent years, our understanding of regulatory mechanisms is still rather preliminary. Here we report a role for 14-3-3 proteins in the regulation of ATP synthases. These 14-3-3 proteins are highly conserved phosphoserine/phosphothreonine-binding proteins that regulate a wide range of enzymes in plants, animals, and yeast. Recently, the presence of 14-3-3 proteins in chloroplasts was illustrated, and we show here that plant mitochondria harbor 14-3-3s within the inner mitochondrial-membrane compartment. There, the 14-3-3 proteins were found to be associated with the ATP synthases, in a phosphorylation-dependent manner, through direct interaction with the F1 β-subunit. The activity of the ATP synthases in both organelles is drastically reduced by recombinant 14-3-3. The rapid reduction in chloroplast ATPase activity during dark adaptation was prevented by a phosphopeptide containing the 14-3-3 interaction motif, demonstrating a role for endogenous 14-3-3 in the down-regulation of the CFoF1 activity. We conclude that regulation of the ATP synthases by 14-3-3 represents a mechanism for plant adaptation to environmental changes such as light/dark transitions, anoxia in roots, and fluctuations in nutrient supply.
Adenosine triphosphate (ATP) is the cell's energy currency, and control over its production and hydrolysis is of vital importance for survival of all living organisms. ATP is synthesized by the F type ATPase (ATP synthase), which is present in mitochondria, chloroplasts, and (photosynthetic) bacteria (1–4). ATP synthases consist of an extrinsic catalytic F1 complex that contains five different subunits (α3β3γ1δ1ɛ1), and a proton pathway Fo that is formed from a variable number of subunits, the stoichiometry of which is still under investigation (5, 6). The crystal structure of the bovine α3β3γ1 complex indicates that the α- and β-subunits are arranged alternately around the NH2- and COOH-terminal α-helices of the γ-subunit. The isolated F1 hydrolyzes ATP, followed by γ-subunit rotation, which is driven by conformational changes of the catalytic subunits (7). The synthesis of ATP requires an electrochemical proton gradient across the inner mitochondrial or thylakoid membrane that is driven by the transport of protons back into the matrix through the Fo domain. When a mitochondrion is deprived of oxygen or chloroplasts are deprived of light, the electrochemical gradient across the membrane collapses, and the enzyme switches its catalytic state from ATP synthesis to ATP hydrolysis. To overcome a possible wasteful hydrolysis of cellular ATP, the organelles are equipped with mechanisms to down-regulate the hydrolytic capability of the ATP synthase complex. In mitochondria, the most thoroughly studied mechanism involves the inhibitor protein, IF1 (8). In bovine mitochondria, IF1 is a basic protein of 84 amino acids whose binding to ATP synthase is pH dependent. Although IF1 can be attributed to down-regulating the ATP synthase in some cell types, it is by no means the only mechanism and, in some instances (such as frog skeletal muscle; ref. 9), IF1 is argued not to be involved in the down-regulation of the ATP synthase under anoxia. Furthermore, in these mitochondria, there is no pH change associated with anoxia, which is a prerequisite for binding IF1 to the ATP synthase. This reaction to anoxia would suggest that organelles possess other mechanisms of down-regulating the complex.
Chloroplast ATP synthase shows a complex regulation of its catalytic activity. ΔpH, thiol modulation, and nucleotide binding are all mechanisms used to avoid the dissipative cleavage of ATP (5). However, despite knowledge of these mechanisms, there remains an unexplained paradox in chloroplast energetics; namely, within whole isolated chloroplasts, the ATP synthase is rapidly deactivated in the dark, although broken chloroplasts (thylakoid particles) remain active for several hours after washing. The implication is that a factor present in the chloroplast stroma responsible for the down-regulation of the complex is lost during the production of broken chloroplasts (10).
Until recently, 14-3-3s were thought to be present only in the cytoplasm and on the plasma membrane. It has now been shown that 14-3-3s are present in the nucleus (11), where they serve as transcriptional regulators (12), and within the stroma of the chloroplast (13). 14-3-3 has a role in protein import into mitochondria in animal cells (14) and has recently been shown to be important for guiding nuclear-encoded chloroplasts' proteins to this organelle (15). Furthermore, it has recently been shown that 14-3-3 may play a role in modulating the activity of uncoupling factor at the mitochondrial inner membrane (16).
In plants, consumers of energy and reducing power produced by the
chloroplasts and mitochondria—e.g., the plasma membrane
H+ ATPase (P type), nitrate reductase (NR),
sucrose-phosphate synthase (SPS), trehalose-6-phosphate synthase, and
plasma membrane K+ channels—are all regulated by
14-3-3 proteins (17–19). A picture is emerging wherein 14-3-3 proteins
play a central role in controlling the primary metabolism of plants,
and the question arises whether activities are coordinately regulated
by the different 14-3-3 isoforms present in plants. Thus, the
H+ ATPase, in combination with
K+ channels, determines the driving force for
H+-coupled nitrate uptake (supplying NR with
NO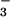 ); coordinate regulation of NR and SPS determines
the partitioning of fixed carbon between sugars and proteins (19).
); coordinate regulation of NR and SPS determines
the partitioning of fixed carbon between sugars and proteins (19).
In this study, we addressed the questions of whether 14-3-3 proteins play a role in the regulation of the mitochondrial and chloroplast ATP synthases, and what implications this regulation has for our knowledge of the regulation of metabolic processes in plants.
Materials and Methods
Plant Material.
Barley seeds (Hordeum vulgare cv. Scarlet) were imbibed for 2 h in running tap water. Plants used for mitochondria experiments were grown for 5 days in darkness at 17 ± 2°C on stainless steel screens over a 1 mM CaSO4 solution. Humidity was kept constant at 70%. Plants used to isolate chloroplasts were grown in soil under normal greenhouse conditions, and leaves were harvested from 7- to 8-day-old plants.
Isolation of Organelles.
Barley mitochondria were isolated according to the method outlined (20), with the exception that the final pellet was resuspended in 1 ml of wash buffer, and then layered on top of a Percoll buffer consisting of 28% (vol/vol) Percoll in wash buffer. Cantharidin (10 μM; ref. 21) was also included in buffers where ATP synthase was isolated in the presence of phosphatase inhibitor. Mitoplasts were prepared by diluting a mitochondrial preparation (protein: 5 mg/ml) in 5 vol of hypotonic buffer [10 mM Hepes/KOH (pH 7.2) and 1% (wt/vol) BSA]. Mitoplasts were centrifuged at 6,000 × g and resuspended in wash buffer (20). Broken barley chloroplasts were prepared according to ref. 22, with the exception that the chloroplasts were osmotically shocked for 5 min in 5 mM MgCl2, after which 4 vol of the following isolation medium was added: 300 mM sorbitol/10 mM KCl/3 mM MgCl2/30 mM Tricine–NaOH (pH 7.8). After centrifugation at 3,500 × g for 3 min, the broken chloroplasts were resuspended in isolation medium to a chlorophyll concentration of about 4.5 mg/ml. All steps were carried out at 4°C.
Marker Enzyme Assays.
ATPase activities were measured as the release of inorganic phosphate from ATP, according to the method of Ames (23). Subcellular fractions (10 μg of protein) were made up to a final volume of 100 μl with water and added to 200 μl of a reaction mix [3 mM ATP/3 mM MgSO4/50 mM KCl/1 mM sodium molybdate/0.02% (wt/vol) Brij 58, buffered to pH 8.0 for F type, or to pH 6.0 for P type, with 1,3-bis[tris(hydroxymethyl)methylamino]propane (BTP)/Mes], and incubated for 20 min at 28°C. The reaction was stopped by the addition of 0.9 ml of Ames reagent [6 parts 0.4% (wt/vol) ammonium molybdate in 0.5 M H2SO4 to 1 part 10% (wt/vol) ascorbic acid], and allowed to stand for 30 min at room temperature. The absorbance at 820 nm was measured in 1-ml cuvettes against a reagent blank. Reaction mixtures included inhibitors of specific ATPases—mitochondrial F type (3 mM sodium azide) and plasma membrane P type (0.25 mM sodium vanadate). Subsequently, the F ATPase activity was calculated as the azide-sensitive activity (with respect to total ATPase activity), and the P ATPase activity as the vanadate-sensitive activity (with respect to total ATPase activity). NR activity was measured as the amount of nitrite produced per unit time. Subcellular fractions (10 μg of protein) were made up to a final volume of 100 μl with water and added to 400 μl of the NR-assay medium (50 mM Hepes–KOH/2 mM KNO3/0.5 mM NADH/10 μM FAD/1 mM EDTA/50 μM leupeptin, pH 7.5). Reactions were incubated for 30 min at 28°C; reactions were stopped by the addition of 100 μl of 500 mM zinc acetate. The mixture was clarified by centrifuging at 15,000 × g for 2 min; to the supernatant was added 0.75 ml of 1 part 1% (wt/vol) sulfanilamide (in 3 M HCl) to 1 part 0.02% (wt/vol) N-(1-naphthyl)ethylenediamine hydrochloride. The absorbance was measured at 540 nm, and the amount of nitrite formed was calculated by using a standard curve of known nitrite/A540 readings. NADH-cytochrome c reductase activity was assayed according to the method of Dignam and Strobel (24). This assay is based on the spectrophotometric measurement of the increase in absorbance at 550 nm caused by the reductase-catalyzed production of reduced cytochrome c by using an extinction coefficient of 21 × 103 cm−1⋅M−1.
Protease Protection Assays.
Whole barley mitochondria and mitoplasts (750 μg of protein) were incubated on ice for 30 min in the presence of 200 μg/ml proteinase K, with or without 0.5% Triton X-100. Subsequently, PMSF was added to a final concentration of 1 mM, and the samples were boiled in SDS/PAGE loading buffer. Mitochondrial proteins (35 μg per sample) were separated by SDS/PAGE and blotted onto nitrocellulose. Specific 14-3-3 isoforms were identified by using anti-14-3-3 antibodies.
Production of Recombinant Barley 14-3-3 Protein.
The cDNA for barley 14-3-3B isoform (accession number Q43470) was cloned as a BamHI fragment into the pPinpoint vector (Promega); the manufacturer's instructions were followed for expression and purification of the recombinant protein. For overlay blot experiments, a full-length biotin-tagged 14-3-3 protein was used. For the removal of the biotinylated tag, the Promega pPinpoint protocol book was followed, and a concentration of 0.1 mg/ml protease factor Xa was used. The full-length 14-3-3 was subsequently purified by using a mono Q column (Amersham Pharmacia), and the resulting protein was desalted by using a 5-ml HiTrap column (Amersham Pharmacia) followed by freeze drying. Lyophilized protein was stored at −20°C.
Purification of Mitochondrial (M)FoF1 and Chloroplast (C)FoF1 ATP Synthases.
The method of Pick and Racker (25) was used to purify synthase complexes from both chloroplasts and mitochondria. Ammonium sulfate pellets were stored at −20°C in a medium consisting of 5 mM MgCl2/200 mM sucrose/20 mM Tricine–NaOH (pH 8.0) at a protein concentration of 30 mg/ml. Pellets were dissolved in gel filtration buffer [50 mM Tris–Acetate/250 mM sucrose/3 mM EDTA/2 mM ATP/0.2% Triton X-100 (pH 8.0)] at a protein concentration of 5 mg/ml. Cantharidin was added to a final concentration of 10 μM as indicated in the figure legends. Dissolved complexes (1 mg of protein) were fractionated on a Superdex 200 (Amersham Pharmacia) gel filtration column, and fractions between the void volume and salt elution were further analyzed by using SDS/PAGE and Western blotting techniques. The gel-filtration fractions were subjected to 12% (wt/vol) SDS/PAGE according to Laemmli (26). Immunoblotting was performed following the method of Towbin et al. (27). Briefly, the proteins resolved in the gel were electrophoretically transferred to a 0.2-μm pore nitrocellulose membrane (Bio-Rad). The transferred membrane was treated with TBS (20 mM Tris⋅HCl/150 mM NaCl, pH 7.5) containing 1% (wt/vol) BSA and 0.1% Tween-20 for 1 h. The membrane was then incubated overnight at 4°C with the appropriate dilutions of the primary antibody (1:50,000 for anti-14-3-3A and 14-3-3B; 1:10,000 for anti-14-3-3C). This was followed by washing and incubation with the secondary antibody, conjugated to horseradish peroxidase (HRP) [anti-rabbit IgG-HRP: 1:2,000 dilution (DAKO)]; the membrane was developed by using the enhanced chemiluminescence (ECL; Amersham Pharmacia) system. Western blot analysis was performed by using specific antibodies directed against the C-terminal regions of the three barley 14-3-3 isoforms (28).
Measurement of Mitoplast Respiration.
Mitoplast oxygen consumption rates were measured with a Rank oxygen electrode at 25°C in an air-equilibrated assay medium (0.4 M mannitol/1 mM potassium phosphate/10 mM Hepes–KOH/5 mM MgCl2, pH 6.8). The signals from the oxygen electrode were recorded on a computer-driven data acquisition system (MacLab, AD Instruments). Mitoplasts were first added to the 1-ml chamber at a final protein concentration of 1 mg of mitoplastal protein per milliliter. Subsequently, NADH (2.8 mM) was added to spark the Krebs cycle. The respiration rate of mitoplasts under these conditions is called state 2. The state 3 respiration rate was then obtained by the addition of 0.15 mM ADP. The rate of oxygen consumption was also measured in the presence of the respiratory uncoupler S13, which was applied at a final concentration of 4 μM. 14-3-3 was used at a final concentration of 100 nM. For competitive inhibition, phosphorylated or unphosphorylated NR-peptide (18) was used at a final concentration of 1 μM.
Protein and Chlorophyll Determination.
Protein was determined by using the method of Bradford (29) with BSA as the standard. Chlorophyll concentrations were determined according to the method of Bruinsma (30).
Measurement of Chloroplast ATP Hydrolysis.
ATP hydrolysis activities were determined by scalar proton release measured with a sensitive pH electrode, as described in Krab et al. (31). The reaction medium (2 ml) contained 2 mM Tricine–NaOH, 5 mM MgCl2, 50 mM KCl, 50 mM NaCl and 2.5 mM K2HPO4 at pH 8.0. We added 25 μM phenazine methosulfate and chloroplasts equivalent to 60 μg of chlorophyll per ml. ATP hydrolysis was started by the addition of 2.5 mM ATP after illumination at saturating light intensity for 30 sec. All measurements were made at a constant temperature of 25°C. 14-3-3 was used at a final concentration of 100 nM. For competitive inhibition, phosphorylated or unphosphorylated NR-peptide was used at a final concentration of 1 μM.
Overlay Blotting.
Proteins blotted onto nitrocellulose filters were blocked with 1% (wt/vol) BSA in phosphate buffered saline plus 0.05% Tween 20 (PBST) for 1 h. Blots were subsequently incubated with PBST containing 10 μg/ml 14-3-3B biotin, tagged and incubated overnight. Blots were washed three times for 10 min each in PBST and incubated for 1 h in PBST containing 10 μg/ml streptavidin-HRP. The blot was again washed three times for 10 min each in PBST, and then bands were detected by using ECL (Amersham Pharmacia).
Results
14-3-3 Isoforms Are Present Within Plant Mitochondria.
Although it has been reported that in animal cells 14-3-3 proteins are involved in the targeting of nuclear encoded mitochondrial proteins to the mitochondria (14), there are no reports so far of an intramitochondrial localization for 14-3-3 proteins in mammalian or plant cells. To examine the presence of 14-3-3 proteins in mitochondria, we isolated and purified mitochondria from 5-day-old barley roots (20). To illustrate the purity of the mitochondria, we performed marker-enzyme assays as shown in Table 1. Mitoplasts (i.e., mitochondria lacking an outer membrane; ref. 32) were prepared from the mitochondria either in the presence or absence of cantharidin, a phosphatase 1 and 2A inhibitor (21). To verify the removal of the outer membrane in the mitoplasts, an NADH-cytochrome c reductase determination was carried out. This assay showed that mitoplasts possess less than 4% outer membrane when compared with mitochondria. Total mitochondrial and mitoplastal proteins were separated by SDS/PAGE, and blotted and probed by using antibodies specific for the three barley 14-3-3 isoforms (28). All three isoforms were shown to be associated with the mitochondria (Fig. 1). The presence of 14-3-3B only in cantharidin-treated mitoplasts illustrates that this isoform is likely to be associated with phosphorylated complexes in the mitochondria. The data shown in Fig. 1 and Table 1 demonstrate that 14-3-3 proteins are present inside plant mitochondria and are not simply attached to the mitochondrial outer membrane. To conclusively show that 14-3-3s are localized inside the mitochondrion, we carried out a series of protease-protection assays. In these experiments, the organelles were incubated with a general protease that removes all peripheral proteins and protruding external membrane proteins. As can be seen from Fig. 1B, the 14-3-3 bands are protected from degradation in both the mitochondria and mitoplasts. In the control experiment where the membranes were permeabilized with detergent, all three 14-3-3 isoforms were fully degraded (Fig. 1B). This assay showed that the 14-3-3 proteins must be present in the matrix or the inner face of the inner membrane where they are protease protected. In further studies, we focused on the 14-3-3B isoform in view of its phosphorylation-dependent association with mitoplasts.
Table 1.
Marker enzyme assays illustrating the presence of cytoplasmic and plasma membrane contamination in the various stages of the purification of barley root mitochondria and mitoplasts
| Fraction | Cytoplasm marker: NR
specific activity, nmol NO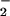 per mg protein per
h per mg protein per
h |
Plasma membrane marker: P-type ATPase specific activity, μmol Pi per mg protein per h | Mitochondrial marker: F-type ATPase specific activity, μmol Pi per mg protein−1 per min |
|---|---|---|---|
| Whole homogenate of barley roots | 155 ± 14 | 3.0 ± 0.4 | 0.03 ± 0.002 |
| Supernatant post 1,000 × g spin | 134 ± 18 | 2.9 ± 0.5 | 0.05 ± 0.002 |
| Pellet post 9,000 × g spin | 12 ± 3 | 1.1 ± 0.02 | 3.4 ± 0.5 |
| Pellet post Percoll gradient | 0 | 0.5 ± 0.01 | 3.6 ± 0.6 |
| Mitochondria after three washings | 0 | 0 | 4.1 ± 0.4 |
| Mitoplasts | 0 | 0 | 5.6 ± 0.7 |
Data presented are for three independent isolation and purification experiments with values for the various enzyme activities and their standard errors.
Figure 1.

Three barley 14-3-3 isoforms are present within the mitochondria. (A) Total mitochondrial (mc) or mitoplasts' (mp) (prepared in the presence or absence of cantharidin) protein (10 μg) was separated by SDS/PAGE, and proteins were blotted onto nitrocellulose. Identical blots were probed with antibodies specific for the A, B, or C barley 14-3-3 isoforms, with anti-rabbit IgG-HRP conjugates as secondary antibody. Blots were developed by using the ECL system. (B) Protease protection assay carried out on mitochondria and mitoplasts to identify the subcompartmental location of the 14-3-3 proteins.
14-3-3 Down-Regulates the Activity of the MFoF1 ATP Synthase.
To establish whether 14-3-3 proteins affect the respiration of mitoplasts and/or the photosynthesis in chloroplasts, we used recombinant 14-3-3B. Trace A in Fig. 2 illustrates a typical measurement of mitoplast respiration with NADH as substrate. Addition of excess ADP results in an increase in oxygen uptake (state 3 respiration) because ATP synthesis by the FoF1 synthases is coupled to electron transfer via complexes III and IV to oxygen. The stimulatory effect of the uncoupler S13 shows that the mitoplasts are well coupled. However, in the presence of recombinant 14-3-3, addition of ADP no longer stimulates oxygen uptake (Fig. 2, trace B). The fact that the uncoupler S13 still doubled the rate of oxygen uptake indicates that the effect of 14-3-3 is caused by blocking the ATP synthase rather than affecting complexes III and IV in the inner mitochondrial membrane. Phosphoserine peptides that match the binding sequences in 14-3-3 target proteins have been used successfully to competitively inhibit the binding of 14-3-3 proteins (18, 33, 34). We synthesized a phosphopeptide matching the 14-3-3 interaction domain in barley NR (18) containing a phosphorylated (*) motif (RSTS*TP) identical to the one found in Raf-kinase (34). This NR-peptide effectively removed the inhibitory effect of 14-3-3 and returned the mitoplasts to state 3 respiration (Fig. 2, trace C). Unphosphorylated NR-peptide was not able to alleviate the 14-3-3 inhibition (data not shown).
Figure 2.

14-3-3 protein inhibits the mitoplastal ATP synthase. The respiration of isolated mitoplasts was measured in the presence of NADH as an electron donor. Trace A, the application of ADP converts the respiration of the mitoplasts from state 2 to state 3. Application of the uncoupler S13 leads to maximum oxygen uptake. Trace B, the application of 14-3-3 in combination with ADP inhibits the state 3 respiration. Trace C, the inhibition of the state 3 respiration by 14-3-3 can be overcome by the addition of excess phosphorylated NR-peptide, which competitively inhibits the binding of 14-3-3 to target proteins.
14-3-3 Rapidly Deactivates the CFoF1 in Broken Chloroplasts.
In contrast to the mitochondrial enzyme, the chloroplast ATP synthase shows a complex regulation of catalytic activity (5, 31). A high value of the proton motive force (ΔμH+) across the thylakoid membrane favors a high rate of ATP synthesis. Under low light conditions, when ΔμH+ collapses and thermodynamic conditions favor ATP hydrolysis, inactivation of the enzyme prevents dissipative cleavage of ATP. Although inactivation in intact chloroplasts takes place rapidly (within minutes), inactivation in broken chloroplasts is slow. It has been suggested that in the preparation of broken chloroplasts, a factor necessary for deactivation of the chloroplast ATP synthase is washed away (10). Immediately upon isolation, broken chloroplasts from barley show an ATP synthase with ATP hydrolysis activity, in contrast to spinach chloroplasts where the ATP synthase is not active (31). In isolated broken chloroplasts from barley, the ATP hydrolysis rate slowly decreased when chloroplasts were stored on ice in the dark (Fig. 3). In the presence of recombinant 14-3-3B, the reduction in hydrolytic activity was much more rapid, whereas the addition of phosphorylated NR-peptide stabilized the hydrolytic activity of the ATP synthase for up to 4 h when stored on ice. These results indicated a role for endogenous 14-3-3 in deactivation of the synthase, and this idea was corroborated by the observation that addition of phosphorylated NR-peptide to buffer control or 14-3-3 treated broken chloroplasts returned the rate of ATP hydrolysis to the initial maximum rate (see arrows in Fig. 3). In experiments where unphosphorylated NR-peptide was added, the results were indistinguishable from the buffer-only control (results not shown). On the basis of these experiments, we hypothesized that during dark adaptation, endogenous 14-3-3 down-regulates the CFoF1 activity. To investigate the role of phosphorylation in the regulation of activity, we isolated broken chloroplasts in the presence and absence of cantharidin. Broken chloroplasts isolated in the presence of cantharidin had consistently lower ATP hydrolysis rates than those isolated in the absence of the inhibitor (35% lower activity). Addition of phosphorylated NR-peptide to the cantharidin-treated chloroplasts resulted in an almost 50% increase in the hydrolysis rate. We also found 14-3-3 to have an inhibitory effect on ATP synthesis (data not shown). This effect was restricted to measurements of synthesis under low illumination. All these data are consistent with a model where 14-3-3 binds to and inhibits phosphorylated ATP synthase.
Figure 3.

The effect of 14-3-3 and phosphorylated NR-peptide on the rate of ATP hydrolysis of isolated broken chloroplasts. A batch of broken chloroplasts was split into three and treated with 14-3-3, phosphorylated NR-peptide, or buffer only. Aliquots were removed at the time intervals shown, and the rate of ATP hydrolysis was measured. ●, buffer control; ▴, 14-3-3; ♦, phosphorylated NR-peptide; ○, buffer control + phosphorylated NR-peptide; ▵, 14-3-3 + phosphorylated NR-peptide. The arrows illustrate the effect of adding phosphorylated NR-peptide to the buffer only and 14-3-3 batches. The experiment was repeated several times and a single representative experiment is shown.
14-3-3 Is Associated with ATP Synthase Complexes in a Phosphorylation-Dependant Manner.
The above-described functional effects of 14-3-3 on the organelles indicate that endogenous 14-3-3 is associated with phosphorylated FoF1 ATP synthase complexes. This is indeed the case, as is shown in Fig. 4A, which shows that MFoF1 and CFoF1 complexes, purified in the presence of cantharidin and separated on a Superdex 200 size exclusion column, were associated with 14-3-3B protein. By contrast, complexes isolated in the absence of cantharidin had a much lower amount of associated 14-3-3 (Fig. 4A). The peak of the ATPase complex in the latter purification eluted later from the column than the peak of the complex isolated in the presence of cantharidin, which supports our conclusion that 14-3-3 is associated with this complex (data not shown). These findings strengthen the argument that phosphorylation of the complex is necessary for 14-3-3 binding and offer an explanation for the absence of 14-3-3B in mitoplasts that were isolated in the absence of cantharidin (Fig. 1). Moreover, these data corroborate the conclusion that 14-3-3 proteins are present in the same compartment as the ATP synthase, namely, the mitochondrial matrix.
Figure 4.

14-3-3B protein copurifies with FoF1 and interacts with the β-subunit. (A) Partially purified FoF1 [isolated in the presence or absence of cantharidin (can)] was further purified by gel filtration; proteins within each fraction (1 to 9) were separated by SDS/PAGE and blotted onto nitrocellulose. Blots were probed with antibody specific for 14-3-3B. In the presence of cantharidin, a single 14-3-3 band was seen in fraction 4, which is the fraction containing the peak of FoF1 (as determined by hydrolytic activity and silver staining; see B). When FoF1 was isolated in the absence of cantharidin, the amount of 14-3-3 in fraction 4 was strongly reduced (CFoF1) or zero (MFoF1). A small amount of 14-3-3 was found in fraction 7, which represents proteins with an apparent molecular mass between 45 and 70 kDa (the mass of uncomplexed 14-3-3 dimer). (B) Fraction 4 of the gel filtration experiments shown above was run on SDS/PAGE and silver stained to show the position of the α- and β-subunits. A second gel was blotted and probed with biotinylated recombinant 14-3-3B. The bound 14-3-3B was detected with streptavidin-HRP. The overlay assay yields a band of 55 kDa in the case of mitochondrial extract, and 57 kDa in the case of the chloroplast. The comparison with the silver-stained gel shows that these molecular masses correspond to the estimated molecular mass of chloroplast and mitochondrial ATP synthase β-subunit, respectively.
The blots, as shown in Fig. 4A, also allowed us to investigate which subunit(s) of the synthases interact(s) with 14-3-3. Blots were probed with biotin-tagged 14-3-3B protein in a so-called overlay assay (Fig. 4B). The overlays show that the biotinylated 14-3-3B protein recognizes a protein of 55 kDa and 57 kDa in the MFoF1 and CFoF1 complexes, respectively, when purified in the presence of cantharidin; no cross-reaction was detected in complexes isolated without cantharidin. When compared with the silver-stained gel, these bands match the calculated size of the ATP synthase β-subunits (Fig. 4B). The barley FoF1 α-subunits can be identified because they run as a doublet (35).
Discussion
Our results show that the activity of evolutionarily related FoF1 synthases from plant mitochondria and chloroplasts is controlled by interaction with 14-3-3 proteins. In view of the conserved structure of the synthases, it may be a more general type of regulation that is operational in other organisms as well. Interaction between 14-3-3 and target proteins relies on reversible protein (serine/threonine) phosphorylation (34), and all our data are consistent with this characteristic of 14-3-3 action. The fact that in most FoF1 isolation procedures no precautions are taken to inhibit phosphatase activity (4, 25, 35) may offer an explanation why, thus far, 14-3-3 was not identified as a regulatory protein of the ATP synthases. Our findings are in line with a recent report that the chloroplast F1 β-subunit from cauliflower binds to a yeast 14-3-3 affinity column (33). We also demonstrated that purified MFoF1 and CFoF1 complexes bind to 14-3-3B affinity columns and are specifically eluted with phosphorylated NR-peptide (data not shown). The β-subunits are attractive targets for 14-3-3 interaction, because they undergo large conformational changes during catalysis. The chloroplast β-subunit is phosphorylated by casein kinase II located in the chloroplast stroma (36), although it is not yet clear whether the putative 14-3-3 recognition motif (429RFLSQP434 in the barley mitochondria β-subunit) conserved in the C-terminal domain of both chloroplast and mitochondrial ATP synthases is involved. Interestingly, this putative 14-3-3 binding motif lies in close proximity to the so-called DELSEED sequence (1), which contains a casein kinase II phosphorylation motif (37). This sequence in the C terminus of the β-subunits is thought to play a crucial role in coupling transport and catalysis (38). Furthermore, the DELSEED sequence is important for the binding of the inhibitor protein IF1 (39), where it locks the β-subunit in a conformation where the complex cannot undergo cooperative catalysis. The IF1 protein will only bind to the ATP synthase under specific conditions of acidic pH (8). Under these conditions, the IF1 forms dimers in which the N-terminal inhibitory domains are brought into direct opposition. The implication is that IF1 dimers could bind two ATP synthase complexes, thus physically locking them into a deactivated conformation. However, it has been shown that the required conditions for dimerization of the IF1 are not always present in mitochondria (9). We suggest that 14-3-3 protein has a comparable role to the IF1, but rather than being pH-dependent, the binding is phosphorylation-dependent. Otherwise, the similarities between 14-3-3 and IF1 are quite striking. 14-3-3s form dimers under physiological conditions and are able to bind two targets simultaneously (40). 14-3-3s are known to physically block enzymatic activity, as in the case of NR (41). It will be most interesting to find out exactly how 14-3-3 proteins regulate the ATP synthases. One approach is to investigate which residues of the β-subunit are involved in 14-3-3 binding.
The question of how 14-3-3 isoforms traverse the mitochondrial and chloroplasts' membranes remains contentious. It has been known for several years that 14-3-3 plays an important role in the targeting of proteins to animal mitochondria (14). Recently, it has been shown that 14-3-3 forms part of a guidance complex targeting nuclear-encoded proteins to the chloroplast (15). It is known that many 14-3-3 isoforms possess N- and C-terminal amphipathic α-helices (42, 43), but these α-helices carry a negative charge, in contrast to the mitochondrial targeting presequences that form positively charged amphipathic α-helices (44). The elucidation of the exact mechanism of 14-3-3 import into organelles is a challenge for the future. We have conclusively shown that 14-3-3 proteins are present in the mitochondrial matrix of plant cells, through a number of experiments. First, mitochondria and mitoplasts free of other membrane and cytoplasmic contamination (as judged by marker enzyme assay) contain 14-3-3 (Fig. 1A). Second, through the use of classical protease protection assays we have shown that the 14-3-3 proteins are localized to the mitochondrial matrix or attached to the inside of the inner membrane of this organelle (Fig. 1B). Third, mitochondrial ATP synthase complexes copurify with 14-3-3 protein in a phosphorylation-dependent manner, and in the complex, the β-subunit is the interaction partner (Fig. 4).
In mammalian and yeast cells, many targets for 14-3-3 proteins can be classified as signaling proteins (45), cell cycle regulators (46), and proteins involved in apoptosis (47), whereas plant 14-3-3 targets are notably involved in primary metabolism (19, 33). Examples of the latter are the plasma membrane H+ ATPase, NR, SPS, trehalose-6-phosphate synthase, and also, as shown here, the primary ATP producers of the cell. Whether these targets fundamentally differ between plants and mammalian/yeast cells remains to be seen, because the focus of research in the respective fields is not alike. Also, mammalian metabolic enzymes and plant protein kinases have been shown to be 14-3-3 targets (48). The question of whether there is a wider regulatory theme linking 14-3-3 targets, or whether each interaction is an independently regulated event, is of interest in this respect (19). This question has never been more relevant, because changes in environmental conditions require the adaptation of enzymes in metabolic pathways that have now been identified as 14-3-3 target proteins. How the different 14-3-3 isoforms communicate to achieve the coordinate response of so many enzymes is the challenge for the near future.
Acknowledgments
We thank M. Wagner for excellent technical assistance, and J. Mol, A. Wagner and H. Lill for helpful discussions. This research was supported by the European Union by contract BIO4-CT97–2275 (Central Role in Adaptation of Fourteen Three Three Proteins) and by the Netherlands Organization for Scientific Research (Netherlands Organization for Scientific Research) by Contracts 805.22.765 and 790.43.850 (to A.H.d.B.)
Abbreviations
- NR
nitrate reductase
- SPS
sucrose phosphate synthase
- ECL
enhanced chemiluminescence
- MFoF1 and CFoF1
mitochondrial and chloroplast FoF1
- HRP
horseradish peroxidase
- ΔμH+
proton motive force
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Abrahams J P, Leslie A G W, Lutter R, Walker J E. Nature (London) 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 2.Junge W, Lill H, Engelbrecht S. Trends Biochem Sci. 1997;22:420–423. doi: 10.1016/s0968-0004(97)01129-8. [DOI] [PubMed] [Google Scholar]
- 3.Sambongi Y, Iko Y, Tanabe M, Omote H, Iwamoto-Kihara A, Ueda I, Yanagida T, Wada Y, Futai M. Science. 1999;286:1722–1724. doi: 10.1126/science.286.5445.1722. [DOI] [PubMed] [Google Scholar]
- 4.Stock D, Leslie A G, Walker J E. Science. 1999;286:1700–1705. doi: 10.1126/science.286.5445.1700. [DOI] [PubMed] [Google Scholar]
- 5.Groth G, Strotmann H. Physiol Plant. 1999;106:142–148. [Google Scholar]
- 6.Schmidt R A, Qu J, Williams J R, Brusilow W S. J Bacteriol. 1998;180:3205–3208. doi: 10.1128/jb.180.12.3205-3208.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Oster G. Nature (London) 1998;396:279. doi: 10.1038/24409. [DOI] [PubMed] [Google Scholar]
- 8.Cabezon E, Arechaga I, Jonathan P, Butler G, Walker J E. J Biol Chem. 2000;275:28353–28355. doi: 10.1074/jbc.C000427200. [DOI] [PubMed] [Google Scholar]
- 9.St-Pierre J, Brand M D, Boutilier R G. Proc Natl Acad Sci USA. 2000;97:8670–8674. doi: 10.1073/pnas.140093597. . (First Published July 11, 2000; 10.1073/pnas.140093597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mills J, Mitchell P. In: Advances in Photosynthesis Research. Sybesma C, editor. II. Dordrecht, The Netherlands: Kluwer; 1984. pp. 523–526. [Google Scholar]
- 11.Bihn E A, Paul A L, Wang S W, Erdos G W, Ferl R J. Plant J. 1997;12:1439–1445. doi: 10.1046/j.1365-313x.1997.12061439.x. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Goodman H M, Zhang H. FEBS Lett. 1999;443:282–284. doi: 10.1016/s0014-5793(98)01739-6. [DOI] [PubMed] [Google Scholar]
- 13.Sehnke P C, Henry R, Cline K, Ferl R J. Plant Physiol. 2000;122:235–242. doi: 10.1104/pp.122.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alam R, Hachiya N, Sakaguchi M, Kawabata S, Iwanaga S, Kitajima M, Mihara K, Omura T. J Biochem (Tokyo) 1994;116:416–425. doi: 10.1093/oxfordjournals.jbchem.a124541. [DOI] [PubMed] [Google Scholar]
- 15.May T, Soll J. Plant Cell. 2000;12:53–64. doi: 10.1105/tpc.12.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierrat B, Ito M, Hinz W, Simonen M, Erdmann D, Chiesi M, Heim J. Eur J Biochem. 2000;267:2680–2687. doi: 10.1046/j.1432-1327.2000.01285.x. [DOI] [PubMed] [Google Scholar]
- 17.Baunsgaard L, Fuglsang A T, Jahn T, Korthout H A, de Boer A H, Palmgren M G. Plant J. 1998;13:661–671. doi: 10.1046/j.1365-313x.1998.00083.x. [DOI] [PubMed] [Google Scholar]
- 18.Booij P P, Roberts M R, Vogelzang S A, Kraayenhof R, de Boer A H. Plant J. 1999;20:673–683. doi: 10.1046/j.1365-313x.1999.00643.x. [DOI] [PubMed] [Google Scholar]
- 19.Mackintosh C. Curr Opin Plant Biol. 1998;1:224–229. doi: 10.1016/s1369-5266(98)80108-8. [DOI] [PubMed] [Google Scholar]
- 20.McDonnell E, Farrar J F. J Exp Bot. 1993;44:1485–1490. [Google Scholar]
- 21.Li Y M, Casida J E. Proc Natl Acad Sci USA. 1992;89:11867–11870. doi: 10.1073/pnas.89.24.11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakels R H A, van Walraven H S, Scholts M J C, Krab K, Kraayenhof R. Biochim Biophys Acta. 1991;1058:225–234. [Google Scholar]
- 23.Ames B N. Methods Enzymol. 1966;8:115–118. [Google Scholar]
- 24.Dignam J D, Strobel H W. Biochemistry. 1977;16:1116–1123. doi: 10.1021/bi00625a014. [DOI] [PubMed] [Google Scholar]
- 25.Pick U, Racker E. J Biol Chem. 1979;254:2793–2799. [PubMed] [Google Scholar]
- 26.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 27.Towbin H, Staehlin T, Gordon J. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Testerink C, van der Meulen R M, Oppedijk B J, de Boer A H, Heimovaara-Dijkstra S, Kijne J W, Wang M. Plant Physiol. 1999;121:81–87. doi: 10.1104/pp.121.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradford M M. Anal Biochem. 1970;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 30.Bruinsma J. Biochim Biophys Acta. 1961;52:576–578. doi: 10.1016/0006-3002(61)90418-8. [DOI] [PubMed] [Google Scholar]
- 31.Krab K, Bakels R H A, Scholts M J C, van Walraven H S. Biochim Biophys Acta. 1993;1141:197–205. [Google Scholar]
- 32.Lemasters J J, Hackenbrock C R. Eur J Biochem. 1976;67:1–10. doi: 10.1111/j.1432-1033.1976.tb10625.x. [DOI] [PubMed] [Google Scholar]
- 33.Moorhead G, Douglas P, Cotelle V, Harthill J, Morrice N, Meek S, Deiting U, Stitt M, Scarabel M, Aitken A, MacKintosh C. Plant J. 1999;18:1–12. doi: 10.1046/j.1365-313x.1999.00417.x. [DOI] [PubMed] [Google Scholar]
- 34.Yaffe M B, Rittinger K, Volinia S, Caron P R, Aitken A, Leffers H, Gamblin S J, Smerdon S J, Cantley L C. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 35.Burkey K O, Mathis J N. Plant Physiol. 1998;116:703–708. doi: 10.1104/pp.116.2.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanekatsu M, Saito H, Motohashi K, Hisabori T. Biochem Mol Biol Int. 1998;46:99–105. doi: 10.1080/15216549800203602. [DOI] [PubMed] [Google Scholar]
- 37.Kanekatsu M, Ezumi A, Nakamura T, Ohtsuki K. Plant Cell Physiol. 1995;36:1649–1656. [PubMed] [Google Scholar]
- 38.Kagawa Y, Hamamoto T. J Bioenerg Biomembr. 1996;28:421–431. doi: 10.1007/BF02113984. [DOI] [PubMed] [Google Scholar]
- 39.Green D W, Grover G J. Biochim Biophys Acta. 2000;1458:343–355. doi: 10.1016/s0005-2728(00)00085-2. [DOI] [PubMed] [Google Scholar]
- 40.Ikeda Y, Koizumi N, Kusano T, Sano H. J Biol Chem. 2000;275:31695–31700. doi: 10.1074/jbc.M004892200. [DOI] [PubMed] [Google Scholar]
- 41.Kanamaru K, Wang R, Su W, Crawford N M. J Biol Chem. 1999;274:4160–4165. doi: 10.1074/jbc.274.7.4160. [DOI] [PubMed] [Google Scholar]
- 42.Rittinger K, Budman J, Xu J, Volinia S, Cantley L C, Smerdon S J, Gamblin S J, Yaffe M B. Mol Cell. 1999;4:153–166. doi: 10.1016/s1097-2765(00)80363-9. [DOI] [PubMed] [Google Scholar]
- 43.de Vetten N C, Lu G, Feri R J. Plant Cell. 1992;4:1295–1307. doi: 10.1105/tpc.4.10.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee C M, Sedman J, Neupert W, Stuart R A. J Biol Chem. 1999;274:20937–20942. doi: 10.1074/jbc.274.30.20937. [DOI] [PubMed] [Google Scholar]
- 45.Bonnefoy-Berard N, Liu Y C, von Willebrand M, Sung A, Elly C, Mustelin T, Yoshida H, Ishizaka K, Altman A. Proc Natl Acad Sci USA. 1995;92:10142–10146. doi: 10.1073/pnas.92.22.10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalal S N, Schweitzer C M, Gan J, DeCaprio J A. Mol Cell Biol. 1999;19:4465–4479. doi: 10.1128/mcb.19.6.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Chen J, Fu H. Proc Natl Acad Sci USA. 1999;96:8511–8515. doi: 10.1073/pnas.96.15.8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sehnke P C, Ferl R J. Science's STKE. 2000. http://www.stke.org/cgi/content/full/OC_sigtrans , http://www.stke.org/cgi/content/full/OC_sigtrans; 2000/56/pe1. ; 2000/56/pe1. [DOI] [PubMed] [Google Scholar]