

Abstract
Extensive research has unraveled the molecular basis of learning processes underlying contextual fear conditioning, but the mechanisms of fear extinction remain less known. Contextual fear extinction occurs when an aversive stimulus that initially caused fear is no longer present and depends on the activation of the extracellular signal-regulated kinase (ERK), among other molecules. Here we investigated how ERK signaling triggered by extinction affects its downstream targets belonging to the activator protein-1 (AP-1) transcription factor family. We found that extinction, when compared to conditioning of fear, markedly enhanced the interactions of active, phospho-ERK (pERK) with c-Jun causing alterations of its phosphorylation state. The AP-1 binding of c-Jun was decreased whereas AP-1 binding of JunD, Jun dimerization protein 2 (JDP2) and ERK were significantly enhanced. The increased AP-1 binding of the inhibitory JunD and JDP2 transcription factors was paralleled by decreased levels of the AP-1 regulated proteins c-Fos and GluR2. These changes were specific for extinction and were MEK-dependent. Overall, fear extinction involves ERK/Jun interactions and a decrease of a subset of AP-1-regulated proteins that are typically required for fear conditioning. Facilitating the formation of inhibitory AP-1 complexes may thus facilitate the reduction of fear.
Keywords: c-Fos; c-Jun; JunD; JDP2, context; hippocampus; fear conditioning; extinction
Introduction
Regulation of fear requires learning about the presence or absence of aversive reinforcement in a given environmental context. The learning processes causing conditioning or extinction of fear have been researched at multiple levels in humans (Grillon, 2009; Schiller and Delgado, 2010; Sehlmeyer et al., 2009) and rodents (Bouton et al., 2006; Maren and Quirk, 2004; Myers and Davis, 2002). Yet, the main molecular mechanisms have not been fully identified and and characterized (Radulovic and Tronson, 2010).
Contextual fear conditioning markedly depends on glutamatergic signaling in the hippocampus that triggers the major signalling pathways and expression of early and delayed genes contributing to memory storage. Some of these pathways converge to co-activate the extracellular signal-regulated kinase (ERK) (Ahi et al., 2004; Roberson et al., 1999). In turn, ERK regulates the activity of multiple downstream substrates, such as ion channels in the membrane (Schrader et al., 2006), cytoskeletal proteins (Sacks, 2006) in the neuropil, and downstream 90 kDa ribosomal S6 kinase (RSK) and mitogen- and stress-activated protein kinase (MSK) in the nucleus (Frodin and Gammeltoft, 1999). The histone H3 kinases RSK and MSK are thought to be the main activators of the expression of the immediate early gene c-Fos (Ha and Redmond, 2008) and are required for contextual fear conditioning (Chwang et al., 2006; Sananbenesi et al., 2002; Sindreu et al., 2007). Dimers formed between c-Fos and other members of the AP-1 family of transcription factors, most notably c-Jun, potently transactivate many genes containing the AP-1 consensus sequence in their promoters (Raivich and Behrens, 2006). Thus, by indirectly activating gene expression via MSK and RSK, the hippocampal ERK pathway mediates long-term storage of contextual memory (Sweatt, 2001; Thomas and Huganir, 2004).
The hippocampus also plays an important role in fear extinction (Fischer et al., 2007) (Ji and Maren, 2007), yet the molecular mechanisms of extinction have only recently begun to emerge (Peters et al., 2010; Radulovic and Tronson, 2010). Activation of nuclear ERK is robust, sustained and required for contextual fear extinction (Chen et al., 2005; Fischer et al., 2007; Ryu et al., 2008). Surprisingly, the immediate early gene c-Fos seems to be down-regulated after extinction of fear (Tronson et al., 2009) and other context-dependent behaviors (Marinelli et al., 2007; Neisewander et al., 2000). These findings suggest that the downstream effects and interactions of ERK with AP-1 transcription factors during extinction may differ from those observed for conditioning and indicate that the learning processes underlying acquisition and reduction of fear are substantially different at a molecular level.
We tested this hypothesis by analyzing the interactions of ERK with Jun members of the AP-1 transcription factor family. In addition to the AP-1 proteins known to transactivate gene expression, we also monitored the AP-1 repressor JDP2 (Katz et al., 2001). We showed that fear extinction, when compared to conditioning, markedly enhanced the interactions of pERK with c-Jun causing alterations of its phosphorylation state and decreased AP-1 binding. In contrast, the AP-1 binding of JunD, JDP2 and ERK was significantly enhanced in a MEK-dependent manner. The observed changes were paralleled by decreased levels of c-Fos and GluR2. These findings demonstrate that, contrary to conditioning, the effects of the MEK-ERK signaling pathway activated by fear extinction involve ERK/Jun interactions and decrease of a subset of AP-1-regulated proteins.
Results
Enhanced pERK/c-Jun interactions during contextual fear extinction
Based on the proposed interactions between ERK and members of the AP-1 protein family [Fig. 1A, Strings 8.3 database, (Szklarczyk et al., 2010)], and lack of pERK/c-Fos interactions during fear extinction (Tronson et al., 2009), we hypothesized that pERK may interact with the Jun members of AP-1 transcription factors. Training (T) in the contextual fear conditioning paradigm in which mice were placed in a box (context) and received a footshock, induced freezing behavior during re-exposure to the box without shock (tests 1-3). Freezing significantly declined after several (4-5) nonreinforced, indicating extinction (E) (Fig. 1B). One hour after the last test (E5), brains of randomly selected mice (n = 6) were removed, and pERK and c-Jun immunostaining was performed. Control groups consisted of naïve mice (N) and mice whose brains were collected 1 hour after training in the fear conditioning paradigm (T) (n = 6/group). As shown earlier (Huh et al., 2009), only mice of the E group exhibited prominent nuclear pERK signals (data not shown). We therefore quantified the co-localization pattern of pERK and c-Jun only in the E group. On average, 48% ± 8 of pERK-positive neurons co-localized with c-Jun within the nuclei of hippocampal CA1 neurons (Fig. 1C). In order to examine whether this co-localization is also accompanied by functional interaction between pERK and c-Jun, we performed co-immunoprecipitation experiments with nuclear dorsohippocampal lysates obtained from separate mice of the N, T and E groups (n = 6/group). In the E group, pERK, more specifically pERK2, exhibited an enhanced interaction with c-Jun when compared to the other groups. This interaction was specific for c-Jun but not other members of the Jun family (Fig. 1D).
Fig. 1.
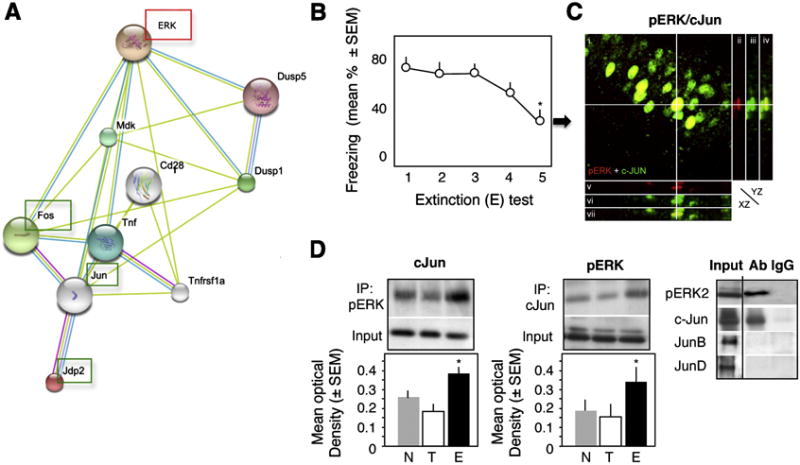
Interaction of pERK and c-Jun during fear extinction. (A) The proposed pathway linking ERK and AP-1 proteins. (B) Extinction of conditioned fear, as revealed by a significant reduction of freezing behavior (F1,4 = 7.23, p < 0.01 vs test 1), after repeated exposure to a context without shock. (C) Co-localization of ERK and c-Jun within hippocampal CA1 pyramidal neurons 1 hr after the 4th extinction (E) test. i) Confocal image (overlay) of pERK (red) and c-Jun (green) immunofluorescence in CA1. ii-vii) Orthogonal projections along the pERK-positive cell marked with crosshair. ii-iv) Orthogonal projection (YZ plane) of pERK (red), c-Jun (green), and pERK + c-Jun (overlay). v-vii) Orthogonal projection (XZ plane) of pERK (red), c-Jun (green), and pERK + c-Jun (overlay). (D) pERK2 interacts with c-Jun but not JunB or JunD in nuclear extracts of the dorsal hippocampus (left panel). This interaction is significantly stronger in the E (middle and right panels) when compared to the other groups (F2,15 = 11.47, p < 0.001 vs T and N). Statistically significant differences: *p < 0.01 vs test 1.
Enhanced c-Jun phosphorylation
It is thought that pERK primarily phosphorylates the Ser243 site of c-Jun mediating a decrease of c-Jun activity (Karin, 1994). However, sustained increase of pERK, as observed during fear extinction (Fischer et al., 2007), may also phosphorylate the Ser63/73 sites thought to contribute to c-Jun-mediated transactivation (Behrens et al., 1999). We therefore examined the levels of p-c-Jun Ser63/73 and Ser243 after fear extinction and compared them to the T and N groups. Immunoblot using nuclear dorsohippocampal lysates revealed that the level of p-c-Jun Ser63/73 was markedly and specifically elevated in mice of the E when compared to the N and T groups. The level of p-c-Jun Ser243 was decreased in the T group, and this decrease was reversed in the E group (Fig. 2A,B). Thus, when compared to T, phosphorylation on both sites was significantly increased in the E group (Fig. 2B, p < 0.01).
Fig. 2.
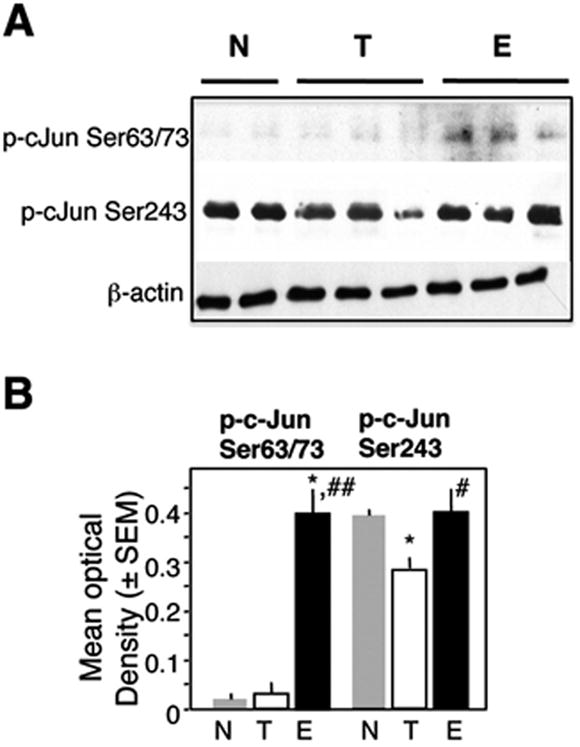
Phosphorylation of c-Jun during extinction. (A) The levels of p-c-Jun Ser63/73 were undetectable in the N and T groups and significantly increased in the E group (F2,15 = 9.65, p < 0.001). p-c-Jun Ser243 was decreased in the T group and returned to control, levels in the E group (F2,15 = 4.12, p < 0.01). (B) Quantification of the immunoblot data. Statistically significant differences: *p < 0.01 vs N, #p < 0.01 vs E, ##p < 0.001 vs E.
MEK-dependent decrease of c-Jun binding to AP-1
In order to determine whether phoshorylation of c-Jun affected its DNA-binding activity, we first performed AP-1 binding assays (TransAM, Active Motif). Dorsohippocampal nuclear lysates were incubated with the AP-1 consensus sequence immobilized on microplates. The interactions between c-Jun and AP-1 were visualized using colorimetric detection of the rabbit anti-c-Jun antibody/c-Jun/AP1 complex. The mean optical density is directly proportional to the amount of c-Jun bound to AP-1. The increase of c-Jun phosporylation and ERK/c-Jun interaction were paralleled by a significant decrease of c-Jun binding to the AP-1 consensus sequence (Fig. 3, left panel) in the E when compared to the T group. Injection of the MEK inhibitor U0126 immediately after individual extinction tests, which is known to block fear extinction (Tronson et al., 2008), restored the AP-1 binding of c-Fos and c-Jun to AP-1 (Fig. 3, right panel).
Fig. 3.
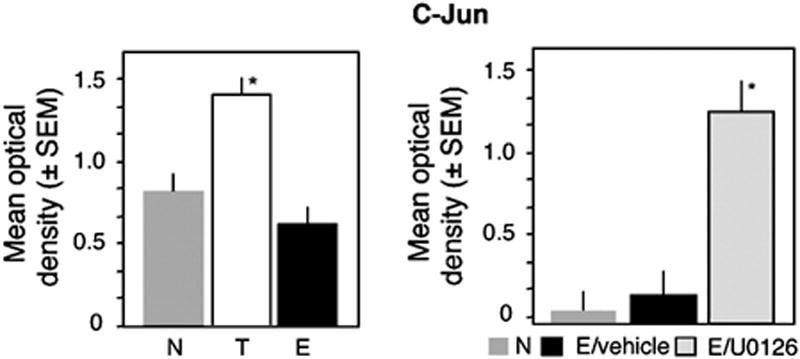
AP-1 binding of c-Jun during extinction. (A) The AP-1 binding activity of c-Jun was triggered in the T but not E group (F2,15 = 11.23, p < 0.001 T vs E and N). (B) After inhibition of the MEK/ERK pathway, extinction enhances the AP-1 binding of c-Jun, as it is observed after training. Statistically significant differences: *p < 0.001 vs N and E/vehicle.
MEK-dependent increase of JunD, JDP2 and ERK binding to AP-1
Contrary to the decreased binding of c-Jun, the AP-1 binding of Jun D and JDP2 was significantly increased in the E when compared to the T group (Fig. 4A). This increase was completely reversed by the MEK inhibitor U0126 (Fig. 4B). In addition to JunD and JDP2, ERK itself exhibited enhanced, MEK-dependent (Figure 5A) and specific (Fig. 5B) binding to AP-1 in vitro. The specificity of ERK binding to AP-1 was confirmed by the finding that only a consensus but not mutated AP-1 sequence effectively and dose-dependently displaced ERK from the immobilized AP-1 complex (as revealed by decreased optical density in Fig. 5B). In order to determine whether such interactions also occur during extinction in vivo on AP-1-regulated promoters, we next performed chromatin immunoprecipitation (ChIP) assays. ChIP assays confirmed that ERK binds to the c-Fos promoter containing, among others, an AP-1-like site (Fig. 5C). Only the use of antibodies against non-phosphorylated ERK showed clear signals (Fig. 5D, upper panel) whereas anti-pERK antibodies were ineffective (data not shown). ERK binding was observed only in hippocampal extracts of the E, but not T or N groups (Fig. 5D, lower panel). In order to determine whether extinction enhanced the formation of ERK-containing c-Fos repressors, mSin3A or HDAC1, which are known to bind to AP-1 we performed additional co-immunoprecipitation assays. We found no evidence of ERK/mSin3A interaction and only a slight increase of ERK/HDAC1 complexes (Fig. 5E, upper panel) in both in the T and E groups (Fig. 5E, lower panel).
Fig. 4.
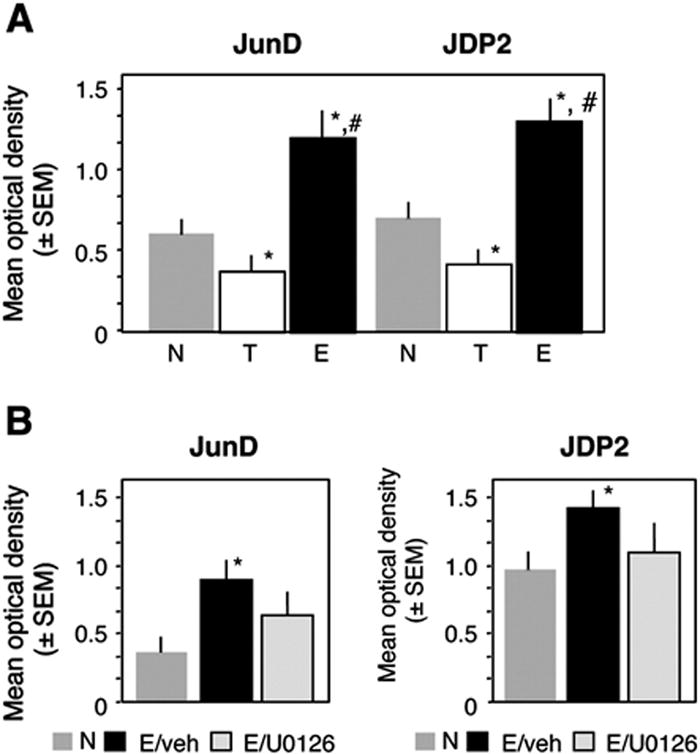
AP-1 binding of JunD and JDP2 during extinction. (A) Contrary to c-Jun, the AP-1 binding of JunD (F2,15 = 9.7, p < 0.001) and JDP2 (F2,15 = 8.91, p < 0.001) was increased during extinction. (B) This enhancement was completely reversed by intrahippocampal inhibition of the MEK/ERK pathway. Statistically significant differences: *p < 0.001 vs N and E/vehicle.
Fig. 5.
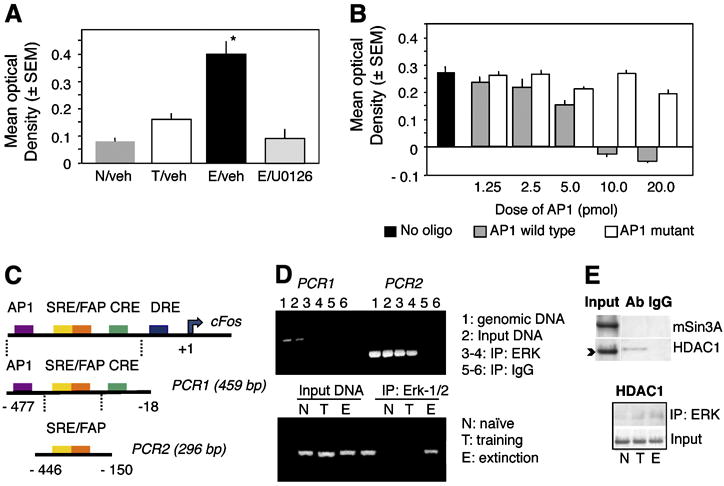
Biding of ERK to AP-1 and the c-Fos promoter. (A) ERK exhibits enhanced binding to the AP- consensus sequence after extinction. (B) This effect is specific because only addition of the consensus but not mutated AP-1 sequence displaces ERK from the immobilized AP-1 oligonucleotide. (C) Schematic representation of the c-Fos promoter. (D) ChIP assays showing amplification of the c-Fos promoter after the nested PCR (PCR2) from genomic DNA, input DNA and DNA co-immunoprecipitated by ERK but not control IgG (right half of the upper panel). PCR1 alone did not yield significant amplification. ERK loaded on the c-Fos promoter only in the E but not N and T groups (lower panel, representative of 6 independent replicates). (E) ERK did not show strong interaction with the c-Fos repressors mSin3A or HDAC1. A slight increase of ERK/HDAC1 (upper panel) was seen both in the T and E groups (lower panel). Statistically significant differences: *p < 0.001 vs all other groups.
MEK-dependent decrease of the AP-1 regulated proteins
In order to determine how the observed changes of AP-1 binding affect the expression and translation of exemplary AP-1-regulated gene products involved in memory processes, we determined the hippocampal AP-1 activity and level of c-Fos and levels of GluR2. Consistent with the levels of c-Fos (Tronson et al., 2009), c-Fos AP-1 activity was significantly reduced by E when compared to T (Fig. 6A, left panel). The MEK inhibitor U0126 completely reversed this effect and increased the number of c-Fos-positive nuclei (Fig. 6A, right panel). Similarly, the level of GluR2 significantly increased (Fig. 6A) in the T group, but returned to baseline in the E group. Injection of U0126 prevented the decrease of GluR2 levels caused by extinction (Fig. 6B). These findings suggested that activation of MEK during fear extinction results in reduced levels of a subset of AP-1-regulated proteins.
Fig. 6.
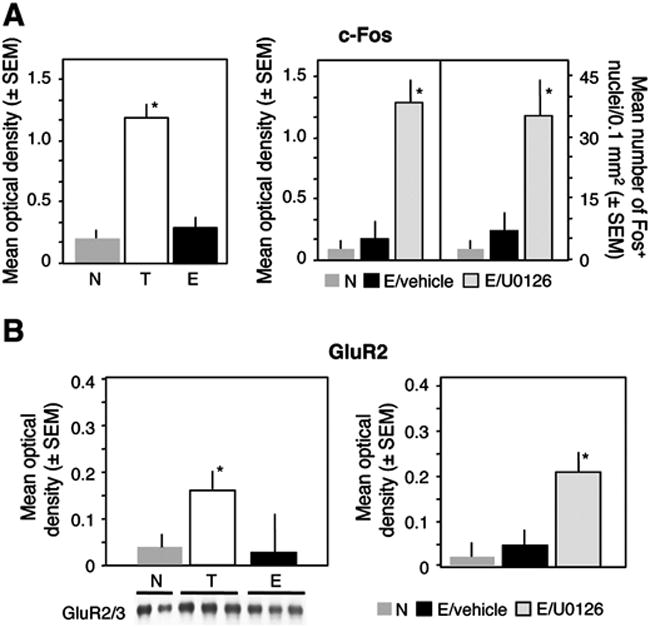
MEK-dependent decrease of c-Fos and GluR2 levels after fear extinction. (A) The activity of c-Fos in dorsohippocampal nuclear extracts was significantly lower in the E when compared to the T group (F2,15 = 11.19, p < 0.001). The AP-1 binding and c-Fos levels were increased after inhibition of the MEK/ERK pathway. (B) The level of GluR2 increased in the T group (F2,15 = 6.67, p < 0.01) but this increase was reversed in the E group. After inhibition of the hippocampal MEK/ERK pathway by U0126, the level of GluR2 remained elevated (F2,15 = 15.67, p < 0.001. Statistically significant differences: *p < 0.001 all other groups.
Discussion
Here we show that learning processes contributing to conditioning and extinction of contextual fear involve differential regulation of hippocampal ERK/AP-1 complexes with opposite outcomes on c-Fos and GluR2 levels. These effects may involve, at least in part, extinction-specific interactions of ERK with members of the Jun transcription factor family.
Our previous data show that nuclear levels of pERK, but not c-Fos, increase in CA1 pyramidal neurons of the hippocampus during contextual fear extinction. This finding was surprising given that the ERK/MSK/RSK pathway mediating fear conditioning (Chwang et al., 2007; Sananbenesi et al., 2003; Sindreu et al., 2007) triggers c-Fos production (Gao et al., 2010). Increasing evidence demonstrates, however, that ERK activity can also down-regulate the expression of the c-Fos gene, in particular when ERK phosphorylation is sustained (Cook et al., 1999; Grooms et al., 2006; Nakakuki et al., 2010), as it is observed during fear extinction (Fischer et al., 2004). These bidirectional actions may depend on the effects of ERK on protein phosphorylation, regulation of multiprotein transcriptional complexes, and binding to gene promoters (Denhardt, 1996; Johnson and Lapadat, 2002). Our findings confirmed two alternatives to ERK-mediated signaling that occur during fear extinction versus conditioning: (i) Instead of c-Fos, ERK interacts with Jun AP-1 members; and (ii) ERK may be responsible for the down-regulation of c-Fos.
Despite the-well established role of ERK in phosphorylation and regulation of c-Jun activity in various cell types (Davis, 1995; Murphy et al., 2004; Tian et al., 2007), the role of ERK-Jun interactions underlying memory processes in the brain has remained unexplored. It was previously found in vivo that active ERK2 forms stable complexes with c-Jun and JunD but not JunB but disengage after Jun transcription factors bind to DNA (Bernstein et al., 1994). These findings may explain why we observed ERK2 only in complexes with c-Jun and not JunD, which exhibits significantly increased AP-1 binding during extinction. In addition, it was proposed that ERK2/AP-1 complexes repress DNA binding (Kumar and Bernstein, 2001), suggesting that the formation of ERK2/c-Jun interactions could contribute to the marked decrease of c-Jun AP-1 binding after fear extinction.
In vitro, ERK phosphorylates the Ser243 site of c-Jun, which is known to attenuate c-Jun-mediated transactivation by decreasing its DNA binding (Boyle et al., 1991; Huang et al., 2008; Lin et al., 1992). We found that the phosphorylation of this site was reduced during fear conditioning but returned to baseline during extinction. The direct interaction of ERK and c-Jun may contribute to this effect. The phosphorylation of the Ser 63/73 site was also markedly increased, and not only in comparison with fear conditioning but also versus the naive group. This modification enhances some (Behrens et al., 1999), but not all types of c-Jun activity (Besirli et al., 2005; Cruzalegui et al., 1999) and is thought to be specifically mediated by the c-Jun N-terminal kinase (JNK) (Derijard et al., 1994; Kallunki et al., 1994). It is possible that ERK counteracts the JNK-induced activation of c-Jun by preserving phosphorylation at the Ser243 site. Alternatively, increased phosphorylation on both sites may not be related to c-Jun transactivation properties (Baker et al., 1992) or may induce a new pattern of gene regulation.
To date, the effects of MEK/ERK on gene expression have mainly been viewed as indirect (Chwang et al., 2007) and mediated by RSK, MSK or other downstream targets. This pathway has been successfully confirmed in models of fear conditioning. Learning to extinguish fear, on the other hand, seems to involve a different MEK/ERK pathway. Several studies reported that ERK binds to the AP-1 (Benkoussa et al., 2002) or serum response element (SRE) (Zhang et al., 2008) sequences of gene promoters, suggesting that the actions of ERK on gene expression may also be direct. Our findings support the latter observations by identifying significant loading of ERK on the c-Fos promoter associated with MEK-dependent down-regulation of c-Fos production. The question why this phenomenon is only observed after extinction needs further investigation. Our hypothesis that ERK may form complexes with HDAC1 or mSin3A, which are known to repress c-Fos (Usenko et al., 2003; Yang et al., 2001) was not confirmed. The possibility remains that pERK targets JunD and JDP2 to AP-1 and subsequently dephosphorylates and dissociates from them. Such mechanism is consistent with the findings that active ERK does not interact with Jun transcription factors when they are bound to DNA (Kumar and Bernstein, 2001). This may also explain the lack of detectable pERK/JunD complexes or direct pERK binding to the c-Fos promoter.
In quiescent cells, juxtaposition of SRE and AP-1/ATF-like sequences (homologous to AP-1) is required for repression of the c-Fos gene. The proximity of these sites is influenced by transcriptional regulators, most notably Jun proteins, which co-operate to repress c-Fos (Konig et al., 1989; Morgan and Birnie, 1992). In addition, interactions of JDP2 with JunD and JunB generate inhibitory AP-1 complexes (Heinrich et al., 2004). Here we show that fear extinction similarly enhances, in an ERK-dependent fashion, JunD and JDP2 AP-binding along with decreased levels of two AP-1-regulated proteins, c-Fos and GluR2. The later observation is consistent with the finding that N-methyl-D-aspartate receptor (NMDAR) activation leads to ERK-mediated transcriptional arrest of GluR2 mRNA and long-lasting reduction in synaptic GluR2 number (Grooms et al., 2006). It remains to be determined whether NMDAR are causing the molecular alterations we observed and thus mediate fear extinction processes (Szapiro et al., 2003).
In summary, our findings identify ERK-mediated alterations of the phosphorylation, AP-1 binding, and composition of Jun complexes, as one of the important and extinction-specific responses of hippocampal neurons. In concert with other signaling pathways, ERK may thus cause a decrease of the levels of the AP-1-regulated proteins c-Fos and GluR2. The significance of the inhibitory of AP-1 complexes and individual roles of c-Jun, JunD and JDP2 in extinction remains to be further evaluated in the framework of the alterations of other molecules involved in fear extinction. It will be particularly important to elucidate whether extinction of fear generally decreases transcription, or only down-regulates the subset of molecules typically required for fear conditioning, such as c-Fos (Fleischmann et al., 2003) and GluR2 (Wiltgen et al., 2010), while up-regulating other extinction-specific genes.
Experimental methods
Animals
Male 9-week-old C57BL/6J (Jackson Laboratories) were individually housed after 8 weeks of age and maintained in enclosed animal cubicles with their own ventilation system (15 air exchanges/hr), 12/12 dark light cycle (7 am-7 pm), 40-50% humidity, and 20 ± 2°C. All studies were approved by the Animal Care and Use Committee of Northwestern University in compliance with National Institutes of Health standards.
Chemicals
The MEK inhibitor U0126 was purchased from Promega. Specific antibodies employed for immunoblot included rabbit polyclonal c-Fos (1:20,000, Oncogene), ERK (1:5000, Santa Cruz), rabbit polyclonal mSin3A (1:500, Santa Cruz), HDAC1 (1:1000, Santa Cruz), c-Jun (1:1,000, Santa Cruz), JunB (1:500, TransAM), JunD (1:500, TransAM), and mouse monoclonal pERK (1:15,000, Sigma). Rabbit (1:500) and mouse (1:500) antibodies against JDP2 were obtained from Dr. Ami Aronheim (Rappaport Faculty of Medicine, Haifa, Israel) and Dr. Kazunari Yokoyama (RIKEN, Ibaraki, Japan), respectively.
Immunohistochemical analyses were performed with pERK and c-Jun antibodies.
Cannulation and treatment
Double cannulae were placed into the dorsal hippocampus (AP - 1.5 mm, lateral 1 mm, depth 2 mm) as described (Radulovic et al., 1998b). The gauge of the guide and injection cannulae was 26 and 28, respectively. U0126 (0.5 μg/site) was dissolved in 2% DMSO and diluted in artificial cerebrospinal fluid (aCSF). The inhibitor was delivered bilaterally (0.25 μl/side) over a 15 s period immediately after individual extinction trials testing unless indicated otherwise.
Fear conditioning and extinction
The behavioral experiments were performed as described earlier (Fischer et al., 2004). Briefly, one-trial contextual fear consisted of exposing the mice to a conditioning context (3 min) followed by a 2-sec 0.7mA electric footshock (constant current). Extinction trials consisted of re-exposure of the animals to the context (3 min) without shock once a day for 4 consecutive days. Freezing, defined as lack of movement besides heart beat and respiration (Blanchard and Blanchard, 1969), was recorded every 10th second by trained observers unaware of the experimental condition. Activity and shock responses were automatically recorded by an infrared beam system coupled to the analysis software (TSE).
Design
Behavioral studies involved naive mice (N) or mice exposed to training (T) or training followed by extinction (E). This group was further split by treatment (vehicle, U0126) in indicated experiments. For molecular studies, brain tissue or dissected dorsal hippocampi were obtained from N mice or 1 hr after training or 4th extinction test for the T and E groups, respectively. This time point was selected from earlier experiments showing maximal ERK activation (Fischer et al., 2007; Tronson et al., 2009).
Imunohistochemistry, image analyses and data quantification
Mice were anesthetized with an intraperitoneal injection of 240 mg/kg of Avertin, and transcardially perfused with ice-cold 4% paraformaldehyde in phosphate buffer (pH 7.4, 150 ml/mouse). Brains were post-fixed for 48 hours in the same fixative and then immersed for 24 hrs each in 10%, 20% and 30% sucrose in phosphate buffer. After the tissue was frozen by liquid nitrogen, 50μm thick coronal sections were used for performing free-floating immunocytochemistry with corresponding primary antibodies as described previously (Radulovic et al., 1998a). For co-immunolabeling studies the TSA Fluorescence System (NEN Life Science Products) was employed, using fluorescein and rhodamine as substrates. Multicolor immunofluorescence was captured and analyzed with a fluorescent microscope (Leica) provided with a CCD camera connected to the Spot software (Macintosh). Cell counts from the dorsohippocampal CA1 subfield was performed using three consecutive dorsohippocampal sections per mouse (Brown et al., 1998). For double labeling, the separate FITC and rhodamine captures were digitally combined to produce composite images. Equal cutoff thresholds were applied to all captures to remove background autofluorescence. For each capture, cell counts were performed within 100μm2 grids (~7 grids/section) for three sections of each CA1. Counts of individual signals were first performed separately, followed by identification and counting of double positive cells on the composite images. An overlapping signal of FITC and rhodamine fluorescence in nuclei that were sharply in focus in a single focal plane was used as a criterion for nuclear colocalization of c-Jun and pERK (Patterson et al., 2001; Tronson et al., 2009). The measures for each capture were averaged to give the number of pERK and c-Jun-positive nuclei and expressed per 0.1 mm2 area. Finally, the proportional number (percentage) of double positive neurons from total pERK immunopositive neurons was calculated. Representative images (4–5 per group) were captured using a Zeiss LSM5 Pascal confocal microscope.
Quantification of immunostaining signals was performed separately for the CA1 hippocampal layers (stratum pyramidale and stratum radiatum) as described previously (Lalonde et al., 2004). Digital images were captured with a cooled color CCD camera (RTKE Diagnostic Instruments) and SPOT software for Macintosh. Adobe Photoshop 5.0 for Macintosh was used for image processing. Cell counts as well as signal intensity measures were performed with ImageJ. The separate fluorescein and rhodamine captures were digitally combined to produce composite images. Equal cutoff thresholds were applied to all captures to remove background fluorescence digitally. For each capture of the pERK and c-Jun signals, nuclear cell counts were performed within a 100 μm2 grid three times for the CA1 area. An overlapping signal of FITC and rhodamine fluorescence was used as a criterion for co-localization of pERK and c-Jun. The number of c-Fos-positive nuclei was determined similarly except that we used diaminobenzidine for visualization and light microscopy for analyses. The measures for each capture were averaged to give the number of c-Fos-positive nuclei per 0.1 μm2 area.
Nuclear extract preparation
The hippocampi were dissected, frozen in liquid nitrogen and kept at -80°C. Cytoplasmic, membrane, cytoskeletal and nuclear fractions were prepared by using the ProteoExtract kit for subcellular proteome extraction (EMD Biosciences) according to the instructions. To control for the purity of the nuclear when compared to other fractions, control immunoblots determined the levels of cAMP-response element binding protein (CREB, using rabbit polyclonal anti-CREB antibody, 1:1,000, Cell Signaling) and histone 1 (H1, rabbit polyclonal anti-H1, 1:1,000, Santa Cruz) as described earlier (Gao et al., 2010). Aliquots of individual samples were used for AP-1 binding assays, immunoprecipitation or immunoblot.
AP-1 binding assays
The binding of AP-1 transcription factors to an immobilized AP-1 consensus sequence was determined by enzyme-linked immunosorbent assays (TransAM kits, Active Motif), according to the instructions. The AP-1 wild-type (5’ TGAGTCA 3’) and AP-1 mutated oligonucleotide (5’ GGAGTCG 3’) were used. AP-1 was bound to its consensus sequence by addition of binding buffer to each well of a 96-strip-well oligonucleotide-coated plate. Lysis buffer and 10 μg of nuclear extract diluted in lysis buffer were added; the plate was sealed and incubated at room temperature with mild agitation. Following each 1 hr incubation, wells were washed 1x wash buffer. Binding of the primary antibody occured when the antibody was diluted in 1x antibody binding buffer (phospho-c-Jun, 1:500 and c-Fos, JunB or JunD 1:10,000), added to each well and incubated for 1hr at room temperature without agitation. Binding of the secondary antibody occurred when the anti-IgG HRP-conjugate was added (1:10,000 in 1x antibody binding Buffer) and incubated for 1 hr at room temperature. Developing solution was added to all wells and incubated for 20 min at room temperature protected from direct light. Stop solution was added to terminate the reaction. Within 5 minutes, absorbance on a spectrophotometer was recorded at 450 nm with a reference wavelength of 655 nm. In addition to the antibodies provided within the kits, we employed mouse monoclonal anti-pERK (1:5,000, Sigma) and JDP2 (provided by Dr. Kaz Yokoyama, RIKEN, Japan, 1:200), and rabbit polyclonal anti-ERK (Santa Cruz, 1:1,000) and JDP2 (provided by Dr. Ami Aronheim, The Rappaport Institute, Israel, 1:500). For competitive binding experiments, the assay is performed in the presence of wild-type or mutated competitor oligonucleotides. As the wild-type oligonucleotide contains a viable binding site, its presence in the buffer reduces binding to the oligonucleotide adhered to the plate. The mutated competitor oligonucleotide does not affect binding to the plate because it contains three mutated bases. The mean optical density, reflecting displacement by AP-1 but not mutant AP-1, served as an indicator of specific AP-1 binding by AP-1 proteins, ERK or JDP2.
Immunoblot and iImmunoprecipitation
After determining the protein concentration (Bio-Rad), the lysates (5-20 μg/well) were subjected to 10% SDS polyacrylamide gel electrophoresis and subsequently blotted to PVDF membranes (Millipore) as described previously (Sananbenesi et al., 2002). The membranes were saturated with I-block (Tropix) and then incubated with the primary and corresponding secondary antibodies, enhancer (Nitro-block II, Tropix) and chemiluminescent substrate (CDP Star, Tropix). Rabbit polyclonal anti-c-Fos, HDAC1, mSin3A (Santa Cruz, 1:1,000) and GluR2 antibodies (obtained from Dr. Peter Penzes, 1:1,000) served as primary antibodies. Blots were exposed to X-ray films and developed in the range of maximal chemiluminescence emission (10 min). Molecular weight and densitometric calculations of were performed with the computer software ImageJ (NIH). The levels of individual proteins were normalized to beta-actin.
For co-immunoprecipitation, cell lysates were combined with 4mg of antibody and incubated 1 hour at 4°C on a rolling mixer. Protein A microbeads (Myltenyi Biotech) were added and incubated for 30 minutes on ice. The m column was placed on mMACS separator and rinsed with a high salt buffer (500mM NaCl, 1% Igepal CA630 NP-40, 50mM Tris HCl pH 8.0). The cell lysates were applied to the m column and the non-bound fraction was collected for flow-through analysis. The m column was treated with 6 high salt buffer and 3 low salt buffer (20mM Tris HCl pH 7.5) washes. 1x SDS reducing loading buffer was applied to column and the eluate was collected for SDS-PAGE analysis.
Chromatin immunoprecipitation (ChIP) assays
Hippocampi were obtained from the N,T, and E groups after perfusion of mice with 37% formaldehyde and kept frozen at −20°C. After tissue lysis, cross-linked chromatin was sheared by ultrasound with 10 pulses of 20 s on ice water and a 20 s rest on ice. This yielded DNA fragments of 200–600 bp. The samples were incubated with anti-pERK, anti-ERK or corresponding normal immunoglobulin (IgG). Following co-immunoprecipitation, input and immunoprecipitated samples were reverse cross-linked with proteinase K and DNA was subsequently purified using phenol/chloroform/isoamyl-alcohol extraction followed by ethanol precipitation. The fragment of the c-Fos promoter associated with these antibodies was identified by a two-step PCR. A 459 base-long fragment of the c-Fos promoter (- 477 to −18) was amplified first (PCR1) with the primers: forward, 5’- GAA CCG GGT CCA CAT TGA ATC - 3’, reverse, 5’- CGC TCT ATC CAG TCT TCT CAG - 3’. A second, nested PCR (PCR2) was performed to further amplify a 295 base-long fragment (-446 to −150) with the primers: forward, 5’- AAT GTT CGC TCG CCT TCT CTG CCT T - 3’, reverse 5’- ACC CCC GTC TTG GCA TAC ATC TTT C -3’. This part of the c-Fos promoter contains the AP1, SRE, cAMP-dependent response (CRE) and dyad symmetry (DSE) regulatory elements. The sequences of both fragments matched the originally published sequence in Genbank (accession number, K03231).
Statistical analyses
Experiments including behavior manipulations were analyzed by repeated measure or one-way ANOVA. Data analysis for pharmacological experiments was performed by two-way ANOVA with Group and Treatment as factors. Shefe’s test was used for post-hoc comparisons.
Fig. 7.
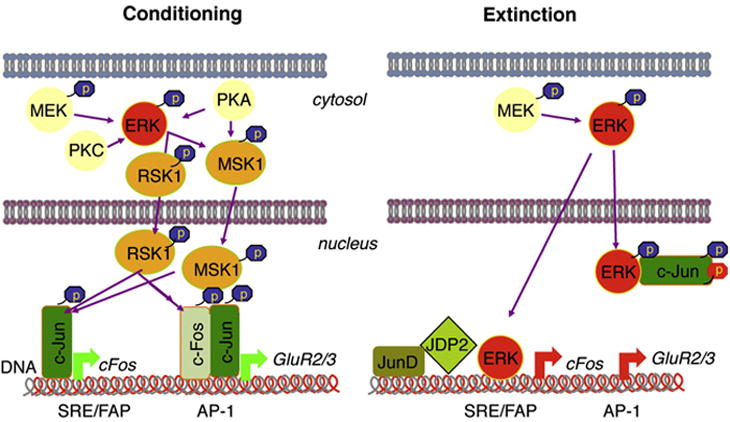
ERK-dependent signalling during conditioning and extinction of fear. Conditioning involves the ERK/MSK/RSK/c-Fos pathway. Extinction, on the other hand, induces nuclear pERK/c-Jun interactions and increases the inhibitory AP-1 complexes JunD and JDP2. The later changes may be involved, at least in part, in the down-regulation of c-Fos and GluR2.
Acknowledgments
We thank Dr. Ami Aronheim and Dr. Kaz Yokoyama for anti-JDP2 antibodies, Dr. Peter Penzes for anti-GluR2 antibodies, and, with Kelly Jones and Dr. Deepak Srivastava, for their help with confocal microscopy, and Dan Sylvester for helping with the preparation of the manuscript. This work was supported by the NIMH grant MH073669 and Dunbar Funds to J.R.
Abbreviations
- ERK
extracellular signal-regulated kinase
- AP-1
activator protein-1
- CRE
cAMP-dependent element
- DSE
dyad symmetry element
- HDAC1
histone deacetylase 1
- JDP2
Jun dimerization protein 2
- RSK
90 kDa ribosomal S6 kinase
- MEK
mitogen activated protein kinase kinase
- MSK
mitogen- and stress-activated protein kinase
- SRE
serum response element
- JNK
c-Jun N-terminal kinase
- NMDAR
N-methyl-D-aspartate receptor
References
- Ahi J, Radulovic J, Spiess J. The role of hippocampal signaling cascades in consolidation of fear memory. Behav Brain Res. 2004;149:17–31. doi: 10.1016/s0166-4328(03)00207-9. [DOI] [PubMed] [Google Scholar]
- Baker SJ, Kerppola TK, Luk D, Vandenberg MT, Marshak DR, Curran T, Abate C. Jun is phosphorylated by several protein kinases at the same sites that are modified in serum-stimulated fibroblasts. Mol Cell Biol. 1992;12:4694–4705. doi: 10.1128/mcb.12.10.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- Benkoussa M, Brand C, Delmotte MH, Formstecher P, Lefebvre P. Retinoic acid receptors inhibit AP1 activation by regulating extracellular signal-regulated kinase and CBP recruitment to an AP1-responsive promoter. Mol Cell Biol. 2002;22:4522–4534. doi: 10.1128/MCB.22.13.4522-4534.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein LR, Ferris DK, Colburn NH, Sobel ME. A family of mitogen-activated protein kinase-related proteins interacts in vivo with activator protein-1 transcription factor. J Biol Chem. 1994;269:9401–9404. [PubMed] [Google Scholar]
- Besirli CG, Wagner EF, Johnson EM., Jr The limited role of NH2-terminal c-Jun phosphorylation in neuronal apoptosis: identification of the nuclear pore complex as a potential target of the JNK pathway. J Cell Biol. 2005;170:401–411. doi: 10.1083/jcb.200501138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC. Crouching as an index of fear. J Comp Physiol Psychol. 1969;67:370–375. doi: 10.1037/h0026779. [DOI] [PubMed] [Google Scholar]
- Bouton ME, Westbrook RF, Corcoran KA, Maren S. Contextual and temporal modulation of extinction: behavioral and biological mechanisms. Biol Psychiatry. 2006;60:352–360. doi: 10.1016/j.biopsych.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, Hunter T. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991;64:573–584. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- Brown HE, Garcia MM, Harlan RE. A two focal plane method for digital quantification of nuclear immunoreactivity in large brain areas using NIH-image software. Brain Res Brain Res Protoc. 1998;2:264–272. doi: 10.1016/s1385-299x(98)00003-8. [DOI] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Chwang WB, Arthur JS, Schumacher A, Sweatt JD. The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci. 2007;27:12732–12742. doi: 10.1523/JNEUROSCI.2522-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwang WB, O’Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SJ, Aziz N, McMahon M. The repertoire of fos and jun proteins expressed during the G1 phase of the cell cycle is determined by the duration of mitogen-activated protein kinase activation. Mol Cell Biol. 1999;19:330–341. doi: 10.1128/mcb.19.1.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruzalegui FH, Hardingham GE, Bading H. c-Jun functions as a calcium-regulated transcriptional activator in the absence of JNK/SAPK1 activation. EMBO J. 1999;18:1335–1344. doi: 10.1093/emboj/18.5.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Transcriptional regulation by MAP kinases. Mol Reprod Dev. 1995;42:459–467. doi: 10.1002/mrd.1080420414. [DOI] [PubMed] [Google Scholar]
- Denhardt DT. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem J. 1996;318(Pt 3):729–747. doi: 10.1042/bj3180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Fischer A, Radulovic M, Schrick C, Sananbenesi F, Godovac-Zimmermann J, Radulovic J. Hippocampal Mek/Erk signaling mediates extinction of contextual freezing behavior. Neurobiol Learn Mem. 2007;87:149–158. doi: 10.1016/j.nlm.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann A, Hvalby O, Jensen V, Strekalova T, Zacher C, Layer LE, Kvello A, Reschke M, Spanagel R, Sprengel R, Wagner EF, Gass P. Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos in the CNS. J Neurosci. 2003;23:9116–9122. doi: 10.1523/JNEUROSCI.23-27-09116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Gao C, Gill MB, Tronson NC, Guedea AL, Guzman YF, Huh KH, Corcoran KA, Swanson GT, Radulovic J. Hippocampal NMDA receptor subunits differentially regulate fear memory formation and neuronal signal propagation. Hippocampus. 2010;20:1072–1082. doi: 10.1002/hipo.20705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C. D-cycloserine facilitation of fear extinction and exposure-based therapy might rely on lower-level, automatic mechanisms. Biol Psychiatry. 2009;66:636–641. doi: 10.1016/j.biopsych.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grooms SY, Noh KM, Regis R, Bassell GJ, Bryan MK, Carroll RC, Zukin RS. Activity bidirectionally regulates AMPA receptor mRNA abundance in dendrites of hippocampal neurons. J Neurosci. 2006;26:8339–8351. doi: 10.1523/JNEUROSCI.0472-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Redmond L. ERK mediates activity dependent neuronal complexity via sustained activity and CREB-mediated signaling. Dev Neurobiol. 2008;68:1565–1579. doi: 10.1002/dneu.20682. [DOI] [PubMed] [Google Scholar]
- Heinrich R, Livne E, Ben-Izhak O, Aronheim A. The c-Jun dimerization protein 2 inhibits cell transformation and acts as a tumor suppressor gene. J Biol Chem. 2004;279:5708–5715. doi: 10.1074/jbc.M307608200. [DOI] [PubMed] [Google Scholar]
- Huang CC, Wang JM, Kikkawa U, Mukai H, Shen MR, Morita I, Chen BK, Chang WC. Calcineurin-mediated dephosphorylation of c-Jun Ser-243 is required for c-Jun protein stability and cell transformation. Oncogene. 2008;27:2422–2429. doi: 10.1038/sj.onc.1210888. [DOI] [PubMed] [Google Scholar]
- Ji J, Maren S. Hippocampal involvement in contextual modulation of fear extinction. Hippocampus. 2007;17:749–758. doi: 10.1002/hipo.20331. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kallunki T, Su B, Tsigelny I, Sluss HK, Derijard B, Moore G, Davis R, Karin M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- Karin M. Signal transduction from the cell surface to the nucleus through the phosphorylation of transcription factors. Curr Opin Cell Biol. 1994;6:415–424. doi: 10.1016/0955-0674(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Katz S, Heinrich R, Aronheim A. The AP-1 repressor, JDP2, is a bona fide substrate for the c-Jun N-terminal kinase. FEBS Lett. 2001;506:196–200. doi: 10.1016/s0014-5793(01)02907-6. [DOI] [PubMed] [Google Scholar]
- Konig H, Ponta H, Rahmsdorf U, Buscher M, Schonthal A, Rahmsdorf HJ, Herrlich P. Autoregulation of fos: the dyad symmetry element as the major target of repression. EMBO J. 1989;8:2559–2566. doi: 10.1002/j.1460-2075.1989.tb08394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar NV, Bernstein LR. Ten ERK-related proteins in three distinct classes associate with AP-1 proteins and/or AP-1 DNA. J Biol Chem. 2001;276:32362–32372. doi: 10.1074/jbc.M103677200. [DOI] [PubMed] [Google Scholar]
- Lalonde J, Lachance PE, Chaudhuri A. Monocular enucleation induces nuclear localization of calcium/calmodulin-dependent protein kinase IV in cortical interneurons of adult monkey area V1. J Neurosci. 2004;24:554–564. doi: 10.1523/JNEUROSCI.1668-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Frost J, Deng T, Smeal T, al-Alawi N, Kikkawa U, Hunter T, Brenner D, Karin M. Casein kinase II is a negative regulator of c-Jun DNA binding and AP-1 activity. Cell. 1992;70:777–789. doi: 10.1016/0092-8674(92)90311-y. [DOI] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Funk D, Juzytsch W, Li Z, Le AD. Effects of opioid receptor blockade on the renewal of alcohol seeking induced by context: relationship to c-fos mRNA expression. Eur J Neurosci. 2007;26:2815–2823. doi: 10.1111/j.1460-9568.2007.05898.x. [DOI] [PubMed] [Google Scholar]
- Morgan IM, Birnie GD. The serum response element and an AP-1/ATF sequence immediately downstream co-operate in the regulation of c-fos transcription. Cell Prolif. 1992;25:205–215. doi: 10.1111/j.1365-2184.1992.tb01395.x. [DOI] [PubMed] [Google Scholar]
- Murphy LO, MacKeigan JP, Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol Cell Biol. 2004;24:144–153. doi: 10.1128/MCB.24.1.144-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Nakakuki T, Birtwistle MR, Saeki Y, Yumoto N, Ide K, Nagashima T, Brusch L, Ogunnaike BA, Okada-Hatakeyama M, Kholodenko BN. Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell. 2010;141:884–896. doi: 10.1016/j.cell.2010.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisewander JL, Baker DA, Fuchs RA, Tran-Nguyen LT, Palmer A, Marshall JF. Fos protein expression and cocaine-seeking behavior in rats after exposure to a cocaine self-administration environment. J Neurosci. 2000;20:798–805. doi: 10.1523/JNEUROSCI.20-02-00798.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic J, Kammermeier J, Spiess J. Relationship between fos production and classical fear conditioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci. 1998a;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic J, Sydow S, Spiess J. Characterization of native corticotropin-releasing factor receptor type 1 (CRFR1) in the rat and mouse central nervous system. J Neurosci Res. 1998b;54:507–521. doi: 10.1002/(SICI)1097-4547(19981115)54:4<507::AID-JNR8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Radulovic J, Tronson NC. Molecular specificity of multiple hippocampal processes governing fear extinction. Rev Neurosci. 2010;21:1–17. doi: 10.1515/revneuro.2010.21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Behrens A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog Neurobiol. 2006;78:347–363. doi: 10.1016/j.pneurobio.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J, Futai K, Feliu M, Weinberg R, Sheng M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal-regulated kinase signaling, enhanced long-term depression, and impaired spatial learning and fear extinction. J Neurosci. 2008;28:8178–8188. doi: 10.1523/JNEUROSCI.1944-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks DB. The role of scaffold proteins in MEK/ERK signalling. Biochem Soc Trans. 2006;34:833–836. doi: 10.1042/BST0340833. [DOI] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Mol Cell Neurosci. 2002;21:463–476. doi: 10.1006/mcne.2002.1188. [DOI] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory. J Neurosci. 2003;23:11436–11443. doi: 10.1523/JNEUROSCI.23-36-11436.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller D, Delgado MR. Overlapping neural systems mediating extinction, reversal and regulation of fear. Trends Cogn Sci. 2010;14:268–276. doi: 10.1016/j.tics.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anderson AE, Sweatt JD. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol. 2006;290:C852–861. doi: 10.1152/ajpcell.00358.2005. [DOI] [PubMed] [Google Scholar]
- Sehlmeyer C, Schoning S, Zwitserlood P, Pfleiderer B, Kircher T, Arolt V, Konrad C. Human fear conditioning and extinction in neuroimaging: a systematic review. PLoS ONE. 2009;4:e5865. doi: 10.1371/journal.pone.0005865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindreu CB, Scheiner ZS, Storm DR. Ca2+ -stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron. 2007;53:79–89. doi: 10.1016/j.neuron.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I. The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, Mering CV. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol. 2007;9:905–914. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Schrick C, Fischer A, Sananbenesi F, Pages G, Pouyssegur J, Radulovic J. Regulatory mechanisms of fear extinction and depression-like behavior. Neuropsychopharmacology. 2008;33:1570–1583. doi: 10.1038/sj.npp.1301550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Schrick C, Guzman YF, Huh KH, Srivastava DP, Penzes P, Guedea AL, Gao C, Radulovic J. Segregated populations of hippocampal principal CA1 neurons mediating conditioning and extinction of contextual fear. J Neurosci. 2009;29:3387–3394. doi: 10.1523/JNEUROSCI.5619-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenko T, Kukushkin A, Pospelova T, Pospelov V. Transient expression of E1A and Ras oncogenes causes downregulation of c-fos gene transcription in nontransformed REF52 cells. Oncogene. 2003;22:7661–7666. doi: 10.1038/sj.onc.1206975. [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ, Royle GA, Gray EE, Abdipranoto A, Thangthaeng N, Jacobs N, Saab F, Tonegawa S, Heinemann SF, O’Dell TJ, Fanselow MS, Vissel B. A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. PLoS ONE. 2010;5 doi: 10.1371/journal.pone.0012818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Vickers E, Brehm A, Kouzarides T, Sharrocks AD. Temporal recruitment of the mSin3A-histone deacetylase corepressor complex to the ETS domain transcription factor Elk-1. Mol Cell Biol. 2001;21:2802–2814. doi: 10.1128/MCB.21.8.2802-2814.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HM, Li L, Papadopoulou N, Hodgson G, Evans E, Galbraith M, Dear M, Vougier S, Saxton J, Shaw PE. Mitogen-induced recruitment of ERK and MSK to SRE promoter complexes by ternary complex factor Elk-1. Nucleic Acids Res. 2008;36:2594–2607. doi: 10.1093/nar/gkn099. [DOI] [PMC free article] [PubMed] [Google Scholar]