

Abstract
General or brain-region-specific decreases in spine number or morphology accompany major neuropsychiatric disorders. It is unclear, however, whether changes in spine density are specific for an individual mental process or disorder and, if so, which molecules confer such specificity. Here we identify the scaffolding protein IQGAP1 as a key regulator of dendritic spine number with a specific role in cognitive but not emotional or motivational processes. We show that IQGAP1 is an important component of NMDAR multiprotein complexes and functionally interacts with the NR2A subunits and the extracellular signal-regulated kinase 1 (ERK1) and ERK2 signaling pathway. Mice lacking the IQGAP1 gene exhibited significantly lower levels of surface NR2A and impaired ERK activity compared to their wild-type littermates. Accordingly, primary hippocampal cultures of IQGAP1−/− neurons exhibited reduced surface expression of NR2A and disrupted ERK signaling in response to NR2A-dependent NMDAR stimulation. These molecular changes were accompanied by region-specific reductions of dendritic spine density in key brain areas involved in cognition, emotion, and motivation. IQGAP1 knock-outs exhibited marked long-term memory deficits accompanied by impaired hippocampal long-term potentiation (LTP) in a weak cellular learning model; in contrast, LTP was unaffected when induced with stronger stimulation paradigms. Anxiety- and depression-like behavior remained intact. On the basis of these findings, we propose that a dysfunctional IQGAP1 gene contributes to the cognitive deficits in brain disorders characterized by fewer dendritic spines.
Introduction
In the CNS, specific patterns of molecular signals arise from localized scaffolding of signaling modules to distinct membrane receptors and determine neuronal morphology and function. IQGAP1 serves as a scaffold for several signaling pathways, such as Lis1, Cdc42, B-Raf, and extracellular signal-regulated kinase (ERK), which regulate spine density and show marked abnormalities in psychiatric illnesses (Kholmanskikh et al., 2006; Reiner et al., 2006; Ryu et al., 2008; Ide and Lewis, 2010; Yuan et al., 2010). We therefore hypothesized that IQGAP1 plays an important role in dendritic spine regulation and behavior. IQGAP1 belongs to the IQGAP family of molecular scaffolds named for their homology to GTPase-activating proteins (GAPs) and isoleucine/glutamine (IQ) (calmodulin-binding) motifs (Briggs and Sacks, 2003). Three isoforms have been described in mammals (Brown and Sacks, 2006; Brandt and Grosse, 2007; Wang et al., 2007), with IQGAP1 being identified first in 1994 (Weissbach et al., 1994). Mammalian IQGAP1 is a 190 kDa protein containing several domains that enable association with many binding partners (Brown and Sacks, 2006). These motifs include a calponin homology domain, responsible for actin binding; a WW motif, which is necessary for the association with ERK2; a calmodulin-binding IQ domain; and a RasGAP-related domain that binds the small GTPases Cdc42 and Rac1 (Fukata et al., 1997; Owen et al., 2008). In addition, IQGAP1 binds to and regulates the functions of E-cadherin, β-catenin, and cytoplasmic linker protein 170 (Ren et al., 2005). Based on findings from multiple cell lines, IQGAP1 is considered to play pivotal roles in cell adhesion, migration, and polarization (Noritake et al., 2005). IQGAP1 also regulates mitogen activated protein kinase signaling (Roy et al., 2005) by directly binding to B-Raf (Ren et al., 2007), mitogen-activated protein kinase kinase (MEK) (Roy et al., 2005), and ERK (Roy et al., 2004). ERK is activated when Ca2+ influx promotes direct binding of IQGAP1 to the ERK activator B-Raf (Ren et al., 2008).
Despite its broadly recognized roles in various cell types, the function of IQGAP1 and associated signaling in the adult CNS is unknown. Yet, it has become increasingly recognized that the specific cellular actions of signal transduction pathways critically depend on their compartmentalized regulation by scaffolding proteins. We showed recently that relocation of IQGAP1 from dendritic spines to the shafts parallels impairments of N-cadherin-mediated ERK signaling in the dendritic cytoskeleton (Schrick et al., 2007). Given that signal transduction mediated by the ERK pathway has been implicated in cognitive (Chandler et al., 2001; Sweatt, 2004), emotional (Ailing et al., 2008), and motivational (Gourley et al., 2008) behavior, IQGAP1 may significantly contribute to these processes by regulating ERK signaling.
Here we identify IQGAP1 as a novel interacting partner of NR2 subunits of NMDARs that contributes to NR1/NR2A trafficking and NR2A-mediated ERK signaling. Using inducible or constitutive deletion of the IQGAP1 gene, we show that IQGAP1 is a key regulator of dendritic spine number in the hippocampus and lateral amygdala. Mice lacking IQGAP1 exhibited significant impairments in memory formation and long-term potentiation (LTP) but intact anxiety- and depression-like behavior. These findings unravel important roles of IQGAP1 in spine regulation, signal transduction, and NMDAR trafficking and suggest a preferential involvement of the NR2A/IQGAP1/ERK signaling module in cognitive versus emotional and motivational behavior.
Materials and Methods
Mice.
Knock-out mice lacking the IQGAP1 gene were obtained from Dr. Andre Bernards (Harvard Medical School, Boston) and backcrossed to the C57BL/6N strain (Harlan) for nine generations. Male 4- to 6- and 9-week-old IQGAP1−/− and IQGAP1+/+ littermates were used for electrophysiological recordings and behavioral experiments, respectively. Immunoblots were performed with brain tissue of mice of different ages, as described below. Primary hippocampal neurons were prepared from postnatal day 1 (P1) mice. All studies were approved by the Animal Care and Use Committee of Northwestern University in compliance with National Institutes of Health standards.
Hippocampal cultures and treatments.
The hippocampi from IQGAP1−/− and IQGAP1+/+ mice were isolated, dissociated, and stimulated as described previously (Gao et al., 2010). Briefly, cells were plated on coverslips coated with poly-d-lysine in 24-well culture plates at a density of 60,000–80,000 cells per well or six-well culture plates coated with poly-d-lysine at a density of 300,000–350,000 cells per well and grown in Neurobasal medium containing 2 mm GlutaMax, 0.5% gentamicin, and 2% B27. One-half of the medium was replaced with identical medium every 4 d. Under these conditions, >85% of cells were viable, >90% of cells were neurons, and cultures could be maintained for 3 weeks. Neurons were cultured for 12–14 d in vitro (DIV) before stimulation and fixation. One hour before stimulation, the extracellular (perfusion or bathing) solution [containing the following (in mm): 140 NaCl, 1.3 CaCl2, 5 KCl, 25 HEPES, pH 7.4, 33 glucose, 0.001 TTX, and 0.001 strychnine] with 40 μm CNQX and 5 μm nimodipine was changed. During the stimulation, the TTX, CNQX, and nimodipine were removed from the bath solution. To activate the NMDAR/ERK pathway, neurons were treated with 20 μm bicuculline and 100 μm glycine for 3 min, whereas to activate the NMDAR/CamKII pathway, neurons were treated with 20 μm NMDA and 20 μm glycine for 3 min.
shRNA and transfection.
SiRNA directed against IQGAP1 was cloned into the pGSuper plasmid (Kojima et al., 2004) to coexpress GFP and small hairpin RNA (shRNA) simultaneously. The sequences of four siRNA (21 nucleotide) were 1227 5′-GGAGCTAATGAATCCTGAAGC-3′ (shIQ1), 2078 5′-GGTACCATTACTACCACAACC-3′ (shIQ2), 2398 5′-GCTTACCTGCACTCCCATAAA-3′ (shIQ3), and 4437 5′-GGAACTCATCAACGACATTGC-3′ (shIQ4), respectively. Neurons were transfected at 11 DIV using Lipofectamine 2000 (Invitrogen). Hippocampal neurons were transfected with GFP-NR2A (a gift from Dr. Stefano Vicini, Georgetown University, Washington, DC) at 9–10 DIV for 4 d using Lipofectamine 2000.
Immunocytochemistry and quantitative immunofluorescence.
The neurons were fixed with 4% formaldehyde in PBS containing 4% sucrose for 15 min at room temperature (RT). Cells were permeabilized and blocked simultaneously in PBS containing 0.1% Triton X-100 and 5% serum for 1 h at RT. Primary antibodies in blocking solution were added at 4°C overnight: rabbit anti-phospho-p44/42 ERK (pERK1/2) antibody (Cell Signaling Technology; 1:200), mouse anti-IQGAP1(BD Biosciences; 1:200), rabbit anti-NR2A (Millipore; 1:500), mouse anti-synaptophysin (Millipore; 1:1000), or rabbit anti-GFP (Millipore; 1:1000). After rinsing with PBS, rhodamine-conjugated (1:500) or fluorescein-isothiocyanate (FITC)-conjugated (1:300; Vector Laboratories) secondary antibodies were added in blocking solution for 1 h at RT. After four rinses, coverslips were mounted using an antifading reagent.
Live hippocampal neurons expressing GFP-NR2A were first labeled with anti-GFP (1:500) antibody for 15 min at RT. After three washes with PBS, neurons were fixed and incubated with a rhodamine-labeled secondary antibody. This procedure was used to label surface NR2A. To label intracellular NR2A, cells were subsequently permeabilized, blocked, and incubated with anti-GFP (1:1000) antibody overnight at 4°C, followed by an FITC secondary antibody for 1 h at RT.
Images were acquired and analyzed as described previously (Gao and Wolf, 2007) using a Nikon inverted microscope, CoolSNAPEZ digital camera, and MetaMorph software.
Golgi staining.
Animals were deeply anesthetized before being killed. The brain was fixed and stained with an FD Rapid Golgistain kit (FD NeuroTechnologies) according to the instructions.
Protein extraction and immunoblot.
Cultures were harvested by scraping in ice-cold buffer containing protease and phosphatase inhibitors as described previously (Gao et al., 2010). The hippocampus and other brain regions were lysed with the same method. Protein concentration was determined by Bio-Rad assay. The lysates (5–20 μg/well) were subjected to SDS-PAGE and subsequently blotted to PVDF membranes and developed as described previously (Sananbenesi et al., 2002). Four hundred microgram hippocampal lysates of IQGAP1−/− and IQGAP1+/+ mice were coimmunoprecipitated by using the Catch and Release v2.0 reversible immunoprecipitaion system (Millipore). Anti-pERK1/2, (Sigma-Aldrich; 1:5000), anti-ERK (Santa Cruz Biotechnology; 1:5000), anti-c-Fos (Santa Cruz Biotechnology; 1:500), anti-NR2A (Millipore, 1:3000), anti-NR2B (Millipore; 1:500), ant-NR3A (Millipore, 1:1000), anti-NR3B (Millipore; 1:1000), anti-IQGAP1 (BD Biosciences, 1:1000), and anti-PSD-95 (Sigma-Aldrich; 1:3000) antibodies were used for immunodetection. For coimmunoprecipitation studies, 4 μg of antibody was used per reaction. ImageJ software (NIH) was used for quantification.
Cell surface protein labeling and detection.
Hippocampal neurons were isolated from P1 IQGAP−/− mice and their IQGAP+/+ littermates as described above and cultured for 14 d. Cell surface proteins were labeled and isolated using Cell Surface Protein Isolation kit (Thermo Scientific) according to the manufacturer's instruction. Briefly, cells were washed with PBS and incubated at 4°C in the presence of sulfosuccinimdyl-2-(biotinamido)-ethyl-1, 3'-dithiopropionate (sulfo-NHS-SS-biotin) and disrupted in ice-cold lysis buffer to which protease inhibitors were added (Complete; Roche) with occasional sonication. A fraction of the cell lysate was saved as total protein extract. Biotinylated proteins were separated by incubation with NeutrAvidin agarose for 1 h at room temperature, washed four times with wash buffer (with Complete protease inhibitors), and eluted with sample buffer at 95°C. Protein extracts were separated on SDS-PAGE gels and transferred to PVDF membranes. Proteins were detected by Western blot analysis using the Snap ID system (Millipore). Anti-NR-2A (Millipore) was diluted at 1:200, and anti-actin antibody (Santa Cruz Biotechnology) at 1:333.
Hippocampal slice preparation and recordings.
Horizontal hippocampal slices (350 μm) were made from 4- to 6-week-old (P24–P36) IQGAP1+/+ or IQGAP1−/− mice using a Vibratome 3000 Plus. Mice were anesthetized with a ketamine/xylazine mixture before being perfused with ice cold sucrose-rich slicing solution (SRSS) equilibrated with 95% O2/5% CO2 containing 85 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm glucose, 75 mm sucrose, 25 mm NaHCO3, 10 μm dl-APV, 100 μm kynurenate, 0.5 mm CaCl2, and 4 mm MgCl2. The mice were then rapidly decapitated and the brains removed under ice-cold SRSS. Slices were slowly warmed to 30°C and allowed to return to room temperature while exchanging the SRSS for oxygenated slicing artificial CSF (ACSF) solution containing 125 mm NaCl, 2.4 mm KCl, 1.2 mm NaH2PO4, 25 mm NaHCO3, 25 mm glucose, 1 mm CaCl2, 2 mm MgCl2, 10 μm dl-APV, and 100 μm kynurenate. After a 1 h incubation period, slices were transferred to a recording chamber and continuously perfused with oxygenated ACSF solution containing (in mm) 125 NaCl, 2.4 KCl, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2 and 1 MgCl2 at 25°C. Glass electrodes were pulled from borosilicate glass to resistances of 3–6 MΩ and then filled with external ACSF recording solution. Field EPSPs (fEPSPs) were evoked by stimulating the Schaffer collaterals with a monopolar glass electrode filled with ACSF at a basal stimulation frequency of 0.05 Hz. fEPSPs were recorded in the CA1 apical dendrite layer using a glass pipette (3–6 MΩ) filed with ACSF recording solution. The stimulus intensity of the first pulse was adjusted to induce an fEPSP amplitude of 40% of the maximal fEPSP. Three stimulation protocols were used to induce LTP. In each case, we initially recorded fEPSPs at the basal frequency for 20 min before inducing LTP. LTP was induced with (1) two theta burst stimulations (TBSs), (2) a 100 Hz high-frequency stimulation (HFS), or (3) a single TBS preceded by a priming stimulus. TBSs consisted of two theta burst patterns separated by 10 s, where each theta burst consisted of a series of 10 short 100 Hz pulse trains of five 1 ms pulses each delivered at a frequency of 5 Hz. HFS consisted of a single 100 Hz train (100 stimuli, 10 ms each) for a 1 s duration. Priming-induced LTP consisted of two priming stimuli at 10 Hz for 10 s separated by 20 s, followed by an interval of 10 min of stimulation at basal frequency, and then a single theta burst stimulation. fEPSPs were recorded for 120 min after LTP induction in the TBS and HFS experiments, whereas the priming-induced plasticity was measured at 60 min after induction. The amount of LTP was determined by normalizing the averaged fEPSP slope during the last 20 min of each experiment to the 20 min of basal control. fEPSP waveforms were acquired with pClamp 10.2 software (Molecular Devices) at a sampling rate of 20 kHz and filtered at 1 kHz. The initial slope and peak amplitude were measured and analyzed with Origin 7.0 (OriginLab). Values are represented as the mean ± SEM, and n values represent the number of recordings from individual slice preparations. Only a single recording was taken from each slice preparation. All electrophysiological recordings and analyses were performed in a blind fashion. For presentation purposes, data were normalized and averaged in one-minute bins. However, all statistical analyses were performed on the raw data set.
Fear conditioning.
Contextual and tone-dependent fear conditioning was performed in an automated system (TSE Systems) and consisted of a single exposure to a context (3 min) followed by a 30 s tone (10 kHz; 75 dB SPL) and a foot shock (2 s; 0.7 mA; constant current) as described previously (Radulovic et al., 1998). Context-dependent freezing was measured 24 h later every 10th second over 180 s by two observers unaware of the experimental conditions and expressed as percentage of total number of observations. Freezing to the tone was scored every fifth second in a novel context during a 30 s exposure.
Object discrimination.
Animals were habituated for 5 min in an 80 × 80 cm arena on day 1. During the following 2 consecutive days, mice were exposed for 5 min to two different objects, object 1 (O1) and object 2 (O2), placed in two neighboring quadrants. On test day 4, O1 remained at the same location, whereas O2 was either moved to a new location or replaced by a new object [object 3 (O3)]. The percentage of time that mice spent visiting O1, O2, or O3 was measured during the 5 min exposure.
Anxiety.
Anxiety-like behavior was evaluated using the dark/light (D/L) emergence task. A 10 × 10 × 10 cm shelter was placed in the middle of the 80 × 80 cm arena. Latency to come out from the shelter and time spent in the dark area during a 5 min test were recorded automatically by VideoMot (TSE Systems).
Depression.
For the novelty-suppressed feeding test, mice were deprived of food overnight and water for 3 h before testing, sufficient to produce a state of mild hunger but little discomfort. For the testing exposure, mice were placed in a brightly lit open field (80 × 80 cm) enclosed by 20 cm walls. A single food pellet was placed in the center of the field and a video tracking system monitored the patterns of feeding and exploration during the 5 min trial. Latency to approach the food was recorded. For the forced swim test, mice were placed in an upright cylinder (20 cm diameter) filled with lukewarm water (26°C) up to 5 cm below its opening. Mice were observed for 6 min, and the amount of time spent in an immobile posture during the last 5 min was scored.
The experimenters performing the behavioral assays were always blind to genotype, and most behavioral measures were captured automatically.
Statistical analyses.
Statistical comparisons between groups were performed by two-tailed multiple t test with Bonferroni's correction.
Results
IQGAP1 is abundant in the CNS, but IQGAP1 deletion does not affect gross neuronal function
In primary hippocampal cultures, IQGAP1 was mainly detected in the soma on DIV 3, but by DIV 21 it also became abundant in neuronal processes (Fig. 1A). In hippocampal lysates, a strong IQGAP1-specific band at the expected size of 190 kDa was observed at different postnatal ages (Fig. 1B). Quantification revealed a decrease of IQGAP1 level after day 3 and steady levels afterward (Fig. 1C). Cultures from IQGAP1 knock-out mice did not show specific signals for this protein by immunoblot (Fig. 1D, left) or immunofluorescence (right). We did not notice any obvious deficits of neuronal polarity in the cultures of IQGAP1 knock-outs. In lysates of different brain regions of adult wild-type but not knock-out mice, IQGAP1 was expressed in the hippocampus, cortex, cerebellum, and striatum (Fig. 1E). Despite the disrupted interaction between IQGAP1 and CaMKII in IQGAP1 knock-outs, CaMKII phosphorylation remained intact (Fig. 1F). Similarly, Raf1-dependent Ras activity (Fig. 1G) and N-cadherin/β-catenin levels (Fig. 1H) were similar in IQGAP1 knock-outs and their wild-type littermates.
Figure 1.

IQGAP1 is abundant in the brain but its deficiency does not affect CaMKII signaling, Ras-GTPase activity, or levels of N-cadherin and β-catenin. A, IQGAP1 in cultured hippocampal neurons derived from wild-type mice, as detected by immunocytochemistry from DIV 3–21. B, Immunoblot showing the IQGAP1 levels in the hippocampus of wild-type mice of different postnatal age. C, Quantification of the immunoblots reveals a drop of IQGAP1 level after postnatal day 3 but steady levels thereafter. D, IQGAP1 signals were absent in hippocampal extracts (left) and neurons (right) of IQGAP1−/− mice. E, IQGAP1 signals were strong in the hippocampus, cortex, cerebellum, and striatum of 9-week-old IQGAP1+/+ mice but lacking in their IQGAP1−/− littermates. F, Immunoblots showing similar levels of pCaMKII or CaMKII in the hippocampus of IQGAP1+/+ and IQGAP1−/− mice. Coimmunoprecipitations (IP) of pCaMKII and IQGAP1 confirmed the interaction of these proteins in wild-type mice and its absence in IQGAP1 knock-outs (right). Stimulation (Stim) of NMDAR significantly increased the level of pCaMKII versus control (Cont) in both genotypes (middle). Quantification of the fold pCaMKII/CaMKII change of control (right) is shown (n = 3–5; *p < 0.05 when compared to unstimulated Cont). Data are represented as mean ± SEM. G, Ras pull-down test showed similar Ras-GTPase activity in hippocampal extracts of IQGAP1−/− when compared to IQGAP1+/+ mice. H, Similar levels of N-cadherin and β-catenin in wild-type and IQGAP1 knock-out mice as revealed by immunoblot. Scale bars: A, 20 μm; D, 20 μm. OD, Optical density.
Impaired neuronal signal transduction in IQGAP1−/− mice
Contrary to the other signaling pathways, ERK activation was markedly impaired under in vitro and in vivo conditions that typically trigger the ERK/c-fos pathway. In cultured hippocampal IQGAP1−/− neurons, NMDAR stimulation with bicuculline and glycine failed to induce ERK phosphorylation (p < 0.05) (Fig. 2A,B) and c-Fos production when compared to their wild-type littermates (p < 0.05) (Fig. 2C). Similarly, in hippocampi of IQGAP1 knock-outs, fear conditioning did not induce upregulation of phosphorylated ERK (pERK; p < 0.001) (Fig. 2D) or c-fos (p < 0.05) (Fig. 2E). These results suggest that IQGAP1 plays a critical role in ERK signaling.
Figure 2.

Downregulation of ERK signaling in IQGAP1−/− mice. A, Hippocampal neurons derived from IQGAP1+/+ or IQGAP1−/− mice were treated with bicuculline (20 μm) and glycine (100 μm) for 3 min to activate Syn NMDARs. IQGAP1 and pERK colocalize in stimulated IQGAP1+/+ neurons. Double-label immunostaining for IQGAP1 (red, top), pERK1/2 (green, middle), and merge (yellow, bottom) revealed significantly reduced level of pERK1/2 in IQGAP1−/− neurons. Scale bar, 20 μm. B, Representative immunoblots showing decreased pERK1/2 (normalized to total ERK1/2) levels after stimulation (Stim) of NMDARs in IQGAP1−/− neurons with bicuculline and glycine. Quantification of fold change of immunoreactivity (pERK/ERK) from unstimulated control (Cont) is presented below (n = 3; *p < 0.05 compared to corresponding IQGAP1+/+). C, Representative immunoblots showing decreased c-fos versus actin levels in IQGAP1−/− versus IQGAP1+/+ neurons. Quantification of the fold change of immunoreactivity (c-fos/actin) from control is presented below (n = 3; *p < 0.05 compared to IQGAP1+/+). D, Representative immunoblots showing decreased pERK1/2 in the adult hippocampus after fear conditioning (FC) of IQGAP1−/− versus IQGAP1+/+ mice. Quantification of the fold change of immunoreactivity (pERK2/ERK2) is presented below (n = 4; ***p < 0.001 compared to IQGAP1+/+). E, The levels of c-fos were also reduced as revealed by decreased c-fos versus actin (representative immunoblots; top) and quantification of the fold change of immunoreactivity (c-fos/actin; bottom; n = 3–4; *p < 0.05 compared to IQGAP1+/+). Data are represented as mean ± SEM.
Impaired NR2A-mediated ERK signaling and NR2A surface expression in IQGAP1−/− neurons
ERK/c-fos signaling in response to NMDAR stimulation is predominantly mediated by the NR2A subunit (Gao et al., 2010). We therefore hypothesized that the IQGAP1 knock-out affects the function of NR2A. Both NR2A and NR2B, but not NR3, coimmunoprecipitated with IQGAP1 (Fig. 3A). However, NR2A/IQGAP1 signals were consistently stronger than NR2B/IQGAP1 signals across three replicates. IQGAP1 also associated with PSD-95 (Fig. 3B), a key scaffolding protein of NMDARs. We next examined whether the trafficking of NMDAR subunits was affected in IQGAP1 knock-outs. Membrane levels of NR1 (Fig. 3C) and NR2A (Fig. 3D) were significantly decreased in IQGAP1−/− when compared to IQGAP1+/+ mice (p < 0.05), whereas NR2B levels were not changed (Fig. 3E). The surface expression of NR2A was also examined using hippocampal neurons transfected with GFP-tagged NR2A at the N terminus (Luo et al., 2002). The levels of surface GFP-NR2A and the ratio between surface and total GFP-NR2A were significantly decreased (p < 0.001) in IQGAP1−/− compared to IQGAP1+/+ cultures (Fig. 3F,G). Similarly, a reduction of endogenous surface NR2A, revealed as a significantly lower (p < 0.001) ratio of surface versus total NR2A (Fig. 3H), was demonstrated using a biotinylation assay. These experiments identify NR2A as a novel interacting partner of IQGAP1. Moreover, this interaction seems to be highly relevant for the regulation of surface expression of NR2A-containing NMDARs in hippocampal neurons.
Figure 3.
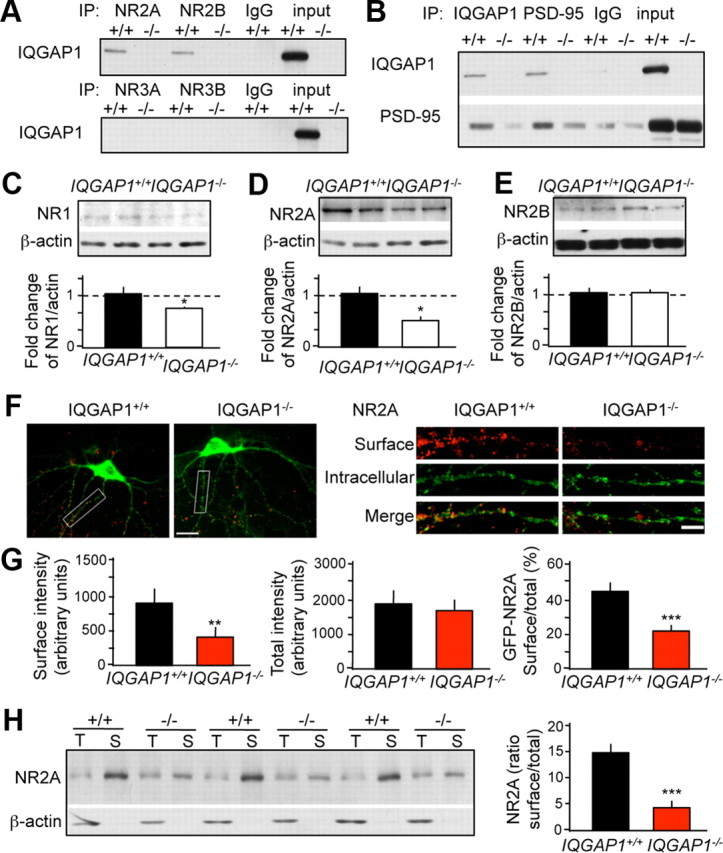
IQGAP1 interacts with NR2 subunits and regulates their surface expression. A, IQGAP1 interacted with NR2A, and to a lesser extent NR2B (top) but not NR3A or NR3B (bottom). B, IQGAP1 also interacted with PSD-95. C–E, Representative immunoblots showing the levels of NR1 (C), NR2A (D), and NR2B (E) in membrane fractions derived from IQGAP1+/+ and IQGAP1−/− mice. The ratios of NR/actin were quantified and presented below the corresponding immunoblots. Membrane levels of NR1 and NR2A were significantly reduced in IQGAP1−/− versus IQGAP1+/+ membranes (n = 3–4; *p < 0.05 compared to IQGAP1+/+). F, Double staining of surface NR2A in hippocampal neurons (left). Analyses of dendritic surface (red), intracellular (green), and total (red and green) NR2A reveal reduced surface NR2A expression in IQGAP1 knock-outs (right). G, Quantification of GFP-NR2A surface intensity (left), GFP-NR2A total intensity (middle), and the ratio of surface/total GFP-NR2A (right; n = 8 for IQGAP1+/+, 12 for IQGAP1−/− neurons. H, Biotinylation assays revealed a significant reduction of the ratio between surface (S) and total (T) endogenous NR2A in cultured hippocampal neurons (left, immunoblot; right, quantification). Total NR2A levels were determined in input lysates whereas surface NR2A were determined after elution from the avidin column (the total band thus appears weaker because the sample is more dilute). **p < 0.01; ***p < 0.001 compared to IQGAP1+/+. Scale bar: F, left, 20 μm; right, 5 μm. Data are represented as mean ± SEM.
Region-specific decrease of spine numbers in IQGAP1−/− mice
By regulating the cytoskeleton of neuronal cells, IQGAP1 contributes to neuronal outgrowth (Li et al., 2005) and promotes spine formation. Accordingly, inducible downregulation of IQGAP1 with shIQGAP14 (shIQ4, an shRNA that reduced the level of IQGAP1 by ∼48%) (Fig. 4A) significantly decreased spine density when compared to the pGSuper vector control (p < 0.001) (Fig. 4B). Hippocampal neurons in cultures prepared from IQGAP1−/− mice also exhibited lower spine density than IQGAP+/+ neurons (p < 0.001) (Fig. 4C).
Figure 4.

Region-specific decrease of spine density after deletion of IQGAP1. A, Human embryonic kidney 293 cells were transfected for 3 d with pGSuper (express GFP) and shIQGAP1 (shIQ) that coexpress both GFP and siRNA against IQGAP1. Downregulation of IQGAP1 was achieved by shIQ4. B, Spine density in IQGAP1+/+ neurons transfected with pGSuper or shIQ4 and in IQGAP1 knock-outs. Representative immunostainings for GFP (top) and quantification of spine number (bottom) revealed a significant decrease of spine number by shIQ4 and IQGAP1 knock-out (n = 23 pGSuper neurons, 29 shIQ4 neurons; ***p < 0.001, shIQ4 compared to pGSuper control). Quantification of spine number also showed a significant reduction in IQGAP1−/− (***p < 0.001) when compared to IQGAP1+/+ neurons neurons (n = 22 IQGAP1+/+ and 15 IQGAP1−/− neurons). C, Golgi stain of the hippocampus and outline of the areas used for spine analyses. D, Reduced spine density in the hippocampus and lateral amygdala (LA) of 9-week-old IQGAP1+/+ and IQGAP1−/− mice (***p < 0.001 compared to corresponding IQGAP1+/+ littermates). E, Intact spine density in the medial prefrontal cortex (mPFC) and basal amygdala (BA) of the same mice. The numbers of analyzed dendrites were 24 IQGAP1+/+ and 27 IQGAP1−/− for hippocampus, 22 IQGAP1+/+ and 23 IQGAP1−/− for mPFC, 25 IQGAP1+/+ and 29 IQGAP1−/− for the LA, and 16 IQGAP1+/+ and 20 IQGAP1−/− for the BA. Scale bars: B, 5 μm; C, left panels, 100 μm; right panels, 50 μm; D, E, 5 μm. Data are represented as mean ± SEM..
In the adult brain, IQGAP1 knock-out also resulted in reduced spine numbers, as revealed by Golgi staining; however, the effect was region specific. Significantly lower spine densities were found in the hippocampus (Fig. 4D) (p < 0.001) and lateral amygdala (p < 0.01), but not medial prefrontal cortex or basal amygdala of IQGAP1 knock-outs when compared to their wild-type littermates (Fig. 4E). Together, these findings show that inducible and constitutive IQGAP1 deficiency results in enduring reduction of dendritic spine density.
Impaired LTP of IQGAP1 knock-outs in response to weak stimuli
We next tested whether the marked effects of IQGAP1 deletion on NR2A distribution and ERK signaling were reflected in altered NMDAR-dependent LTP in acute hippocampal slice preparations. Input–output relationships for fEPSPs evoked by stimulation of Schaffer collateral–CA1 pyramidal cell synapses recorded from wild-type and IQGAP1 knock-outs were equivalent (Fig. 5A). Three LTP induction paradigms were used to compare fEPSP potentiation in response to both strong and weak stimulation. Slices from IQGAP1 mice exhibited similar levels of TBS LTP as their wild-type counterparts (fEPSP potentiation, 133 ± 10% for wild-type, 142 ± 16% for knock-outs; n = 11 and 13 slices, respectively) (Fig. 5B). Similarly, LTP induced by high-frequency tetanic stimulation was equivalent in wild-type and knock-out mice (121 ± 6% and 132 ± 11%, respectively; n = 9 slices each) (Fig. 5C). In contrast, a single theta burst train delivered 10 min after an initial priming stimulation elicited potentiation in recordings from wild-type slices (128 ± 9%; n = 6) but was not induced in IQGAP1 knock-out slices (92 ± 12%; p < 0.05) (Fig. 5D). These results suggest that cellular plasticity induced by a weak stimulation of excitatory synapses on CA1 pyramidal neuron is selectively abrogated in the IQGAP1−/− mice, whereas more robust forms of plasticity remain intact.
Figure 5.

IQGAP1−/− mice exhibit a deficit in cellular plasticity in response to weak induction stimuli. A, Input–output curves for CA1 fEPSPs evoked by Schaffer collateral stimulation are equivalent in wild-type and IQGAP1−/− mice. B, Theta burst LTP was equivalent in wild-type and IQGAP−/− mice. Plots show the averaged data over all the recordings, with the black arrow indicating when TBS (as defined in Materials and Methods) was induced in the experiments (n = 11 and 13 slices from each genotype, respectively). C, LTP induced by HFS was equivalent in wild-type and IQGAP−/− fEPSP recordings (n = 9 slices from each genotype). D, LTP induced by priming a single theta burst with two 10 Hz stimulations induces LTP of fEPSPs in wild-type slices but not in IQGAP−/− hippocampal slices (n = 6 slices from each genotype; p < 0.05). Data are represented as mean ± SEM.
Memory deficits in IQGAP1−/− mice
IQGAP1 knock-outs had a significant deficit in contextual fear conditioning (t = 3.346; p < 0.01) (Fig. 6A), tone-dependent fear conditioning (t = 2.375; p < 0.05) (Fig. 6B), and discrimination between the fear conditioning and novel contexts (wild type, t = 3.946, p < 0.01 vs context 1; knock-out, t = 0.144, p = 0.888 vs context 1) (Fig. 6C) when compared to their wild-type littermates. Lack of IQGAP1 did not affect activity in response to context (t = 1.089; p = 0.297) or electric shock (t = 0.785; p = 0.447) (Fig. 6D,E).
Figure 6.

IQGAP1 deletion causes memory deficits. Impaired fear conditioning and context discrimination in IQGAP1−/− mice when compared to IQGAP1+/+ littermates. A, Context-dependent fear conditioning (**p < 0.01; n = 8 for IQGAP1+/+; n = 10 for IQGAP1−/−). B, Tone-dependent fear conditioning (*p < 0.05; n = 8 for IQGAP1+/+; n = 10 for IQGAP1−/−). C, Freezing to context 1 where shock was delivered during training versus freezing to a novel context 2 (n = 9 for IQGAP1+/+; n = 7 for IQGAP1−/−; **p < 0.01 when compared to context 1). D, Similar locomotor activity of IQGAP1+/+ mice and IQGAP−/[minus] mice (n = 7 for IQGAP1+/+; n = 8 for IQGAP−/−). E, Similar shock responses were recorded for IQGAP1+/+ and IQGAP1−/− mice (n = 7 for IQGAP1+/+; n = 8 for IQGAP1−/−). F, No preference for object 1 (black) or object 2 (gray) during training. G, Impaired object discrimination in IQGAP1−/− mice when compared to IQGAP1+/+ littermates as revealed by a similar percentage of visiting time for objects 1 and 2 when object 2 is presented in a new spatial location. H, Impaired object discrimination in IQGAP1−/− mice when compared to IQGAP1+/+ littermates as revealed by similar percentage of visiting time for object 1 and a novel object 3 (n = 9 for IQGAP1+/+; n = 7 for IQGAP1−/−). For G and H, *p < 0.05, ***p < 0.001 compared to object 1. I, Similar exploration of objects 1, 2, and 3 by IQGAP−/− mice (n = 7 per group). Data are represented as mean ± SEM.
In the object recognition/discrimination paradigm, both IQGAP1+/+ and IQGAP1−/− mice showed similar exploration of novel objects 1 and 2 during training (wild type, t = 0.121, p = 0.905, O2 vs O1; knock-out, t = 0.099, p = 0.922, O2 vs O1) (Fig. 6F). During testing, IQGAP1 wild types spent significantly more time with the object whose location was changed (t = 2.593; p < 0.05 vs O1) (Fig. 6G), whereas IQGAP1 knock-outs did not (t = 0.298; p = 0.772). Similarly, IQGAP1 wild types spent significantly more time with a novel object 3 (t = 4.400; p < 0.001 vs O1) (Fig. 6H), whereas IQGAP1 knock-outs equally explored the familiar (O1) and novel (O3) objects (t = 1.459; p = 0.175). This was not because of differences in activity in response to O3 (t = 0.099; p = 0.922) (Fig. 6I). There were also no changes in gross neuronal morphology or body weight (data not shown), indicating that developmental or sensory–motor deficits did not contribute to the observed memory impairments. Most of these impairments were reproduced in female mice and could be overcome by massed training (data not shown). Together, IQGAP1 knock-outs show impairments in contextual, spatial, cued, and object learning tasks.
Normal anxiety- and depression-like behavior in IQGAP1−/− mice
We next examined whether deletion of IQGAP1 affects anxiety or depression-like behavior. These behaviors were evaluated using the dark/light emergence, novelty-suppressed feeding, and forced swim tests. In the D/L test, there were no significant differences between IQGAP1+/+ and IQGAP1−/− mice in the latency to exit (t = 0.602; p = 0.553) (Fig. 7A) and percentage of time spent in the dark area (t = 1.441; p = 0.163) (Fig. 7B). In the novelty-suppressed feeding test, the latency to eat the food pellet placed in the center of an open field was similar in IQGAP1−/− and IQGAP1+/+ mice (t = 0.766; p = 0.456) (Fig. 7C). Similarly, the time spent in an immobile posture in the forced swim test was not significantly different between IQGAP1+/+ and IQGAP1−/− mice (t = 0.541; p = 0.596) (Fig. 7D). These results demonstrate that deletion of IQGAP1 does not affect anxiety or depression-like behaviors.
Figure 7.

IQGAP1 deletion does not affect anxiety- or depression-like behavior. A, B, Dark/light test showing similar latency to exit the dark area (A) and the percentage of time spent in dark area (B) in IQGAP1−/− and IQGAP1+/+ mice; n = 12 (IQGAP1+/+) and 13 (IQGAP1−/−). C, Novelty-suppressed feeding test showing similar latency to start consumption of food pellets located in the center of an open field of IQGAP1−/− and IQGAP1+/+ mice; n = 6 (IQGAP1+/+) and 10 (IQGAP1−/−). D, Forced swim test showing similar time spent floating in IQGAP1−/− versus IQGAP1+/+ mice; n = 8 (IQGAP1+/+) and 10 (IQGAP1−/−). Data are represented as mean ± SEM.
Discussion
We showed that IQGAP1 is required for ERK signaling, region-specific dendritic spine density, synaptic plasticity, and memory. We also demonstrated that IQGAP1 interacts with PSD-95 and NR2A and regulates the surface expression of NR2A on hippocampal neurons. Some of the observed molecular, cellular, and behavioral effects of the IQGAP1 knock-out may thus be mediated by the newly described interactions between IQGAP1, PSD-95, and NR2 subunits. Surprisingly, motivational and emotional behaviors remained intact. On the basis of these findings, we propose that IQGAP1 plays a nonredundant role in a subset of mechanisms underlying memory formation.
In various cells and tissues, IQGAP1 has primarily been recognized as a scaffolding protein for the cadherin/catenin, calmodulin, Rac1/Cdc42, and MEK/ERK pathways (Brown and Sacks, 2006; Johnson et al., 2009). Nevertheless, the peripheral consequences of IQGAP1 deletion, in particular those associated with cadherin function and development, were minimal (Li et al., 2000). In the brain, the levels, distribution, and activation patterns of most IQGAP1-regulated pathways were similarly unaffected in IQGAP1 knock-outs, as were gross neuronal morphology, maturation, and sensory–motor behavior. This may be attributable to the constitutive lack of IQGAP1 triggering compensatory processes or, alternatively, to regulatory redundancy of cadherin/catenin, CaMKII, and Rac1/Cdc42 signaling by other proteins and scaffolds. A notable exception, however, was the ERK pathway, which exhibited a significantly impaired phosphorylation. Activation and nuclear propagation of ERK signaling is robustly induced by stimulation of NMDAR in vitro (Ivanov et al., 2006; Gao et al., 2010) or learning processes in vivo (Sananbenesi et al., 2002; Shalin et al., 2006; Schrick et al., 2007), and is predominantly mediated by the NR2A subunit (Gao et al., 2010). However, in the IQGAP1 knock-outs, both experimental conditions failed to trigger ERK phosphorylation despite normal ERK levels, possibly because of impaired NR2A trafficking, surface expression, or ineffective scaffolding to ERK.
IQGAP1 interacted with NR2A and PSD-95, to a lesser extent with NR2B, and not with NR3 subunits, which are known for their lack of binding to PSD-95 (Eriksson et al., 2007). This identifies IQGAP1 as a component of NR1/NR2/PSD-95 complexes, regulating NR2A trafficking and NR2A-induced ERK signaling. Given the key involvement of this NMDAR subunit in long-term memory formation, (Kishimoto et al., 1997; Sprengel et al., 1998), the role of IQGAP1 in cognition may be mediated by its interaction with NR2A. It should be noted, however, that NR2A have also been implicated in anxiety and depression (Boyce-Rustay and Holmes, 2006), two behaviors that remained unaffected in the IQGAP1 knock-out. Our findings thus suggest that anxiogenic and depressant effects of NR2A do not critically depend on IQGAP1/ERK interactions and are likely mediated by different scaffolding or signaling complexes.
Despite the ubiquitous distribution of IQGAP1 in the CNS, the decrease in spine numbers in IQGAP1 knock-out mice was region specific, with the hippocampus and lateral amygdala most affected. This neuroanatomical specificity resulting from a general genetic deficit is reminiscent of an earlier observation in mutant mice lacking kalirin, an important regulator of spine density and morphology (Cahill et al., 2009). These localized effects likely depend on the regional repertoire of functionally related molecular pathways that compensate for the deficiency of the knock-out. In the present study, it is very likely that regional actions of IQGAP2 or IQGAP3 overcame the IQGAP1 deficit, given the structural homology and functional redundancy of these family members. All IQGAP proteins share the ability to interact with actin filaments, a feature that is particularly important for their redundant effects on neuronal outgrowth (Fukata et al., 1997; Wang et al., 2007), and most likely spine formation, as observed in our study. Interestingly, whereas the involvement of IQGAP1 (but not IQGAP2 and IQGAP3) in the growth of hippocampal axons was dispensable (Wang et al., 2007), its role in hippocampal dendritic spine density observed here was nonredundant. Thus, in addition to regional specificity, IQGAP proteins may exert subcellular specificity in their actions. A detailed analysis of the neuroanatomical and cellular distribution of IQGAP proteins is critical for better understanding of their unique and redundant roles.
Comparative analysis of cellular plasticity in wild-type and IQGAP1 knock-out mice supports a role for the scaffolding protein in transducing weak but not strong stimuli into stable potentiation of CA1 excitatory transmission. Long-term plasticity was induced using three distinct models: a single TBS preceded by two 10 Hz priming trains, two trains of TBS, or a strong 100 Hz stimulus. LTP of CA1 fEPSPs occurred after the two stronger induction paradigms but was absent in hippocampi from IQGAPI mice after the single primed TBS. In principle, the lack of potentiation in this model could result from signaling deficits within the priming phase or simply as a result of the weaker (single TBS) induction stimulus compared to the other paradigms tested. TBS with a prior 10 Hz stimulation is permissive for NMDA-receptor-dependent LTP through complex homosynaptic and heterosynaptic pathways in which mGlu receptors play crucial roles (Chevaleyre and Castillo, 2004; Abraham, 2008). Postsynaptic priming follows activation of group I mGlu receptors, which enhances CA1 pyramidal cell excitability and facilitates induction of LTP (Cohen et al., 1999; Abraham, 2008). Heterosynaptic metaplasticity, in contrast, occurs through depression of inhibitory tone on pyramidal neurons subsequent to mGlu-receptor-driven synthesis of endocannabinoids, which activate cannabinoid receptors on interneuron presynaptic terminals and consequently reduce release of GABA (Chevaleyre and Castillo, 2004). Ablation of IQGAP1 might interfere with a variety of elements of these metaplastic signaling pathways, or alternatively, NMDA receptor signaling might be weakened in the hippocampus of IQGAP1 knock-outs such that threshold induction stimuli of any sort no longer evoke potentiation. In line with this view, the specific roles of individual scaffolding proteins in the regulation of LTP are thought to depend on their contribution to unique NMDAR signaling complexes (Carlisle et al., 2008). Stronger stimulation paradigms clearly elicited LTP in the IQGAP knock-out mice, as also seen in several studies using specific and limited functional disruption of the NMDAR scaffolds PSD-95 and SynGAP1 (Lim et al., 2003; Rama et al., 2008). Analogous to the observed effects of IQGAP1 knock-out on neuronal plasticity, we found marked learning deficits with one-trial training protocols in the associative-conditioning and object-recognition paradigms that were overridden by intensified training. These findings suggest that stronger stimuli recruit additional, redundant molecular mechanisms that are able to compensate for the observed LTP and learning deficits.
General or brain-region-specific decreases in spine density accompany major neuropsychiatric disorders, such as schizophrenia (Glantz and Lewis, 2000), major depression (Law et al., 2004), and mental retardation (Fiala et al., 2002). Commonly, deletion of a single gene results in reduction of spine density and multiple behavioral phenotypes (Cahill et al., 2009; Ayhan et al., 2010). It thus remains unclear why the IQGAP1 knock-outs selectively exhibited memory deficits. Reduced spine numbers in hippocampus and lateral amygdala, along with decreased ERK signaling in the hippocampus, could provide a neuroanatomical basis for selective cognitive deficits of contextual fear conditioning (Kim and Fanselow, 1992; Phillips and LeDoux, 1992), object recognition memory (Kelly et al., 2003; Fernandez et al., 2008), and tone-dependent fear conditioning (Schafe et al., 2000; Radley et al., 2006; Lamprecht et al., 2009). Alternatively, spine density may be specifically involved in the regulation of mnemonic rather than emotional and motivational processes, and changes seen in mood disorders (Li et al., 2010) could be related to the cognitive deficits accompanying these illnesses.
Contrary to NR2A, IQGAP1 did not seem to play a major role in NR2B function. Alternatively, preferential NR2B scaffolds, such as SynGAP1 (Kim et al., 2005), RACK1 (Yaka et al., 2003), SAP102 (Petralia et al., 2005), or DAPK1 (Tu et al., 2010), might have compensated for the loss of IQGAP1. However, this is unlikely given that the functional consequences of IQGAP1 deletion seemed opposite from those caused by NR2B/PSD-95 scaffolds. IQGAP1 knock-out resulted in a significant impairment of ERK signaling, in accordance with a positive MEK/ERK regulatory role of IQGAP1 (Brown and Sacks, 2006) and NR2A (Kim et al., 2005). On the other hand, lack of SynGAP1, a functionally related scaffold of the GAP protein family, increases the spine number and accelerates spine maturation in primary hippocampal cultures (Vazquez et al., 2004). Furthermore, reduced levels of SynGAP1 increase ERK phosphorylation (Komiyama et al., 2002), consistent with the negative regulation of the ERK pathway by SynGAP (Rumbaugh et al., 2006) and NR2B (Kim et al., 2005). It is possible that distinctive functions of GAP domains account for decreased ERK signaling by NR2B/SynGAP1 versus enhanced ERK signaling by NR2A/IQGAP1 complexes: SynGAP1 accelerates hydrolysis of bound GTP on Ras, whereas IQGAP1, using its GAP-related domain, binds to Cdc42 and Rac1 and stabilizes their GTP-bound states (Kurella et al., 2009). The different molecular and morphological actions of IQGAP1 and SynGAP1 probably mediate their discrete effects on behavior: unlike SynGAP1 deficiency, which affects working and reference memory, activity, anxiety, and object recognition memory (Muhia et al., 2010), IQGAP1 knock-out caused profound deficits of object recognition and other forms of memory without alterations of motivational or emotional behavior. These findings stress the importance of scaffolding proteins as generators of stimulus-specific neuronal signaling patterns (Sheng and Kim, 2002) underlying distinctive behavioral profiles.
Together, we identified IQGAP1 as a component of NR2A/PSD-95 complexes playing a principal role in NR2A trafficking, ERK signaling, dendritic spine density, and memory. Given that region-specific alterations of dendritic spine morphology and memory deficits, as found during IQGAP1 deficiency, are commonly observed in Alzheimer's and other types of dementia (Aoki et al., 2007; Kasai et al., 2010; Schulz-Schaeffer, 2010), IQGAP1 malfunction may have significant implications in cognitive symptoms of human psychiatric disorders.
Footnotes
This work was supported by the National Alliance for Research on Schizophrenia and Depression (C.G.), a Diversifying Faculty in Illinois Award (S.F.F.), NINDS Grant NS071952 (G.T.S.), NIMH Grant MH073669 (J.R.), and Dunbar funds (J.R.). We thank Drs. Andre Bernards (Massachusetts General Hospital Cancer Center, Boston, MA) and Wadie Bahou [State University of New York (SUNY) at Stony Brook, Stony Brook, NY] for providing the IQGAP1 knock-outs, Dr. Stefano Vicini (Georgetown University, Washington, DC) and Gulcan Akgul (SUNY Stony Brook) for providing NR2A-GFP, Dr. Zhong Xie and Dr. Peter Penzes for providing the pGSuper plasmid, and Dan Sylvester for assistance with the preparation of this manuscript.
References
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Ailing F, Fan L, Li S, Manji S. Role of extracellular signal-regulated kinase signal transduction pathway in anxiety. J Psychiatr Res. 2008;43:55–63. doi: 10.1016/j.jpsychires.2008.01.018. [DOI] [PubMed] [Google Scholar]
- Aoki C, Mahadomrongkul V, Fujisawa S, Habersat R, Shirao T. Chemical and morphological alterations of spines within the hippocampus and entorhinal cortex precede the onset of Alzheimer's disease pathology in double knock-in mice. J Comp Neurol. 2007;505:352–362. doi: 10.1002/cne.21485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayhan Y, Abazyan B, Nomura J, Kim R, Ladenheim B, Krasnova IN, Sawa A, Margolis RL, Cadet JL, Mori S, Vogel MW, Ross CA, Pletnikov MV. Differential effects of prenatal and postnatal expressions of mutant human DISC1 on neurobehavioral phenotypes in transgenic mice: evidence for neurodevelopmental origin of major psychiatric disorders. Mol Psychiatry. 2011;16:293–306. doi: 10.1038/mp.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Holmes A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology. 2006;31:2405–2414. doi: 10.1038/sj.npp.1301039. [DOI] [PubMed] [Google Scholar]
- Brandt DT, Grosse R. Get to grips: steering local actin dynamics with IQGAPs. EMBO Rep. 2007;8:1019–1023. doi: 10.1038/sj.embor.7401089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs MW, Sacks DB. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003;4:571–574. doi: 10.1038/sj.embor.embor867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MD, Sacks DB. IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 2006;16:242–249. doi: 10.1016/j.tcb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Cahill ME, Xie Z, Day M, Photowala H, Barbolina MV, Miller CA, Weiss C, Radulovic J, Sweatt JD, Disterhoft JF, Surmeier DJ, Penzes P. Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc Natl Acad Sci U S A. 2009;106:13058–13063. doi: 10.1073/pnas.0904636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle HJ, Fink AE, Grant SG, O'Dell TJ. Opposing effects of PSD-93 and PSD-95 on long-term potentiation and spike timing-dependent plasticity. J Physiol. 2008;586:5885–5900. doi: 10.1113/jphysiol.2008.163469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Dorairaj NR, Norwood D. N-methyl-d-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J Biol Chem. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Coussens CM, Raymond CR, Abraham WC. Long-lasting increase in cellular excitability associated with the priming of LTP induction in rat hippocampus. J Neurophysiol. 1999;82:3139–3148. doi: 10.1152/jn.1999.82.6.3139. [DOI] [PubMed] [Google Scholar]
- Eriksson M, Samuelsson H, Samuelsson EB, Liu L, McKeehan WL, Benedikz E, Sundstrom E. The NMDAR subunit NR3A interacts with microtubule-associated protein 1S in the brain. Biochem Biophys Res Commun. 2007;361:127–132. doi: 10.1016/j.bbrc.2007.06.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez SM, Lewis MC, Pechenino AS, Harburger LL, Orr PT, Gresack JE, Schafe GE, Frick KM. Estradiol-induced enhancement of object memory consolidation involves hippocampal extracellular signal-regulated kinase activation and membrane-bound estrogen receptors. J Neurosci. 2008;28:8660–8667. doi: 10.1523/JNEUROSCI.1968-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fukata M, Kuroda S, Fujii K, Nakamura T, Shoji I, Matsuura Y, Okawa K, Iwamatsu A, Kikuchi A, Kaibuchi K. Regulation of cross-linking of actin filament by IQGAP1, a target for Cdc42. J Biol Chem. 1997;272:29579–29583. doi: 10.1074/jbc.272.47.29579. [DOI] [PubMed] [Google Scholar]
- Gao C, Wolf ME. Dopamine alters AMPA receptor synaptic expression and subunit composition in dopamine neurons of the ventral tegmental area cultured with prefrontal cortex neurons. J Neurosci. 2007;27:14275–14285. doi: 10.1523/JNEUROSCI.2925-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Gill MB, Tronson NC, Guedea AL, Guzman YF, Huh KH, Corcoran KA, Swanson GT, Radulovic J. Hippocampal NMDA receptor subunits differentially regulate fear memory formation and neuronal signal propagation. Hippocampus. 2010;20:1072–1082. doi: 10.1002/hipo.20705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Wu FJ, Kiraly DD, Ploski JE, Kedves AT, Duman RS, Taylor JR. Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol Psychiatry. 2008;63:353–359. doi: 10.1016/j.biopsych.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide M, Lewis DA. Altered cortical CDC42 signaling pathways in schizophrenia: implications for dendritic spine deficits. Biol Psychiatry. 2010;68:25–32. doi: 10.1016/j.biopsych.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M, Sharma M, Jamieson C, Henderson JM, Mok MT, Bendall L, Henderson BR. Regulation of beta-catenin trafficking to the membrane in living cells. Cell Signal. 2009;21:339–348. doi: 10.1016/j.cellsig.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Kelly A, Laroche S, Davis S. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J Neurosci. 2003;23:5354–5360. doi: 10.1523/JNEUROSCI.23-12-05354.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholmanskikh SS, Koeller HB, Wynshaw-Boris A, Gomez T, Letourneau PC, Ross ME. Calcium-dependent interaction of Lis1 with IQGAP1 and Cdc42 promotes neuronal motility. Nat Neurosci. 2006;9:50–57. doi: 10.1038/nn1619. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Kawahara S, Kirino Y, Kadotani H, Nakamura Y, Ikeda M, Yoshioka T. Conditioned eyeblink response is impaired in mutant mice lacking NMDA receptor subunit NR2A. Neuroreport. 1997;8:3717–3721. doi: 10.1097/00001756-199712010-00012. [DOI] [PubMed] [Google Scholar]
- Kojima S, Vignjevic D, Borisy GG. Improved silencing vector co-expressing GFP and small hairpin RNA. Biotechniques. 2004;36:74–79. doi: 10.2144/04361ST02. [DOI] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O'Carroll CM, Martin SJ, Morris RG, O'Dell TJ, Grant SG. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci. 2002;22:9721–9732. doi: 10.1523/JNEUROSCI.22-22-09721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurella VB, Richard JM, Parke CL, Lecour LF, Jr, Bellamy HD, Worthylake DK. Crystal structure of the GTPase-activating protein-related domain from IQGAP1. J Biol Chem. 2009;284:14857–14865. doi: 10.1074/jbc.M808974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, Dracheva S, Assoun S, LeDoux JE. Fear conditioning induces distinct patterns of gene expression in lateral amygdala. Genes Brain Behav. 2009;8:735–743. doi: 10.1111/j.1601-183X.2009.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law AJ, Weickert CS, Hyde TM, Kleinman JE, Harrison PJ. Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines. Am J Psychiatry. 2004;161:1848–1855. doi: 10.1176/ajp.161.10.1848. [DOI] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wang Q, Chakladar A, Bronson RT, Bernards A. Gastric hyperplasia in mice lacking the putative Cdc42 effector IQGAP1. Mol Cell Biol. 2000;20:697–701. doi: 10.1128/mcb.20.2.697-701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, McNulty DE, Marler KJ, Lim L, Hall C, Annan RS, Sacks DB. IQGAP1 promotes neurite outgrowth in a phosphorylation-dependent manner. J Biol Chem. 2005;280:13871–13878. doi: 10.1074/jbc.M413482200. [DOI] [PubMed] [Google Scholar]
- Lim IA, Merrill MA, Chen Y, Hell JW. Disruption of the NMDA receptor-PSD-95 interaction in hippocampal neurons with no obvious physiological short-term effect. Neuropharmacology. 2003;45:738–754. doi: 10.1016/s0028-3908(03)00276-4. [DOI] [PubMed] [Google Scholar]
- Luo JH, Fu ZY, Losi G, Kim BG, Prybylowski K, Vissel B, Vicini S. Functional expression of distinct NMDA channel subunits tagged with green fluorescent protein in hippocampal neurons in culture. Neuropharmacology. 2002;42:306–318. doi: 10.1016/s0028-3908(01)00188-5. [DOI] [PubMed] [Google Scholar]
- Muhia M, Yee BK, Feldon J, Markopoulos F, Knuesel I. Disruption of hippocampus-regulated behavioural and cognitive processes by heterozygous constitutive deletion of SynGAP. Eur J Neurosci. 2010;31:529–543. doi: 10.1111/j.1460-9568.2010.07079.x. [DOI] [PubMed] [Google Scholar]
- Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K. IQGAP1: a key regulator of adhesion and migration. J Cell Sci. 2005;118:2085–2092. doi: 10.1242/jcs.02379. [DOI] [PubMed] [Google Scholar]
- Owen D, Campbell LJ, Littlefield K, Evetts KA, Li Z, Sacks DB, Lowe PN, Mott HR. The IQGAP1-Rac1 and IQGAP1-Cdc42 interactions: interfaces differ between the complexes. J Biol Chem. 2008;283:1692–1704. doi: 10.1074/jbc.M707257200. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Sans N, Wang YX, Wenthold RJ. Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci. 2005;29:436–452. doi: 10.1016/j.mcn.2005.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Johnson LR, Janssen WG, Martino J, Lamprecht R, Hof PR, LeDoux JE, Morrison JH. Associative Pavlovian conditioning leads to an increase in spinophilin-immunoreactive dendritic spines in the lateral amygdala. Eur J Neurosci. 2006;24:876–884. doi: 10.1111/j.1460-9568.2006.04962.x. [DOI] [PubMed] [Google Scholar]
- Radulovic J, Kammermeier J, Spiess J. Relationship between fos production and classical fear conditioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci. 1998;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rama S, Krapivinsky G, Clapham DE, Medina I. The MUPP1-SynGAPalpha protein complex does not mediate activity-induced LTP. Mol Cell Neurosci. 2008;38:183–188. doi: 10.1016/j.mcn.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner O, Sapoznik S, Sapir T. Lissencephaly 1 linking to multiple diseases: mental retardation, neurodegeneration, schizophrenia, male sterility, and more. Neuromolecular Med. 2006;8:547–565. doi: 10.1385/NMM:8:4:547. [DOI] [PubMed] [Google Scholar]
- Ren JG, Li Z, Crimmins DL, Sacks DB. Self-association of IQGAP1: characterization and functional sequelae. J Biol Chem. 2005;280:34548–34557. doi: 10.1074/jbc.M507321200. [DOI] [PubMed] [Google Scholar]
- Ren JG, Li Z, Sacks DB. IQGAP1 modulates activation of B-Raf. Proc Natl Acad Sci U S A. 2007;104:10465–10469. doi: 10.1073/pnas.0611308104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren JG, Li Z, Sacks DB. IQGAP1 integrates Ca2+/calmodulin and B-Raf signaling. J Biol Chem. 2008;283:22972–22982. doi: 10.1074/jbc.M804626200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Li Z, Sacks DB. IQGAP1 binds ERK2 and modulates its activity. J Biol Chem. 2004;279:17329–17337. doi: 10.1074/jbc.M308405200. [DOI] [PubMed] [Google Scholar]
- Roy M, Li Z, Sacks DB. IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol Cell Biol. 2005;25:7940–7952. doi: 10.1128/MCB.25.18.7940-7952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Adams JP, Kim JH, Huganir RL. SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A. 2006;103:4344–4351. doi: 10.1073/pnas.0600084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J, Futai K, Feliu M, Weinberg R, Sheng M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal-regulated kinase signaling, enhanced long-term depression, and impaired spatial learning and fear extinction. J Neurosci. 2008;28:8178–8188. doi: 10.1523/JNEUROSCI.1944-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Mol Cell Neurosci. 2002;21:463–476. doi: 10.1006/mcne.2002.1188. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrick C, Fischer A, Srivastava DP, Tronson NC, Penzes P, Radulovic J. N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron. 2007;55:786–798. doi: 10.1016/j.neuron.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50:765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298:776–780. doi: 10.1126/science.1075333. [DOI] [PubMed] [Google Scholar]
- Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, Kim JJ, Thompson RF, Sun W, Webster LC, Grant SG, Eilers J, Konnerth A, Li J, McNamara JO, Seeburg PH. Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell. 1998;92:279–289. doi: 10.1016/s0092-8674(00)80921-6. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Hippocampal function in cognition. Psychopharmacology (Berl) 2004;174:99–110. doi: 10.1007/s00213-004-1795-9. [DOI] [PubMed] [Google Scholar]
- Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Lew F, Chan SL, Chen Y, Lu Y. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140:222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez LE, Chen HJ, Sokolova I, Knuesel I, Kennedy MB. SynGAP regulates spine formation. J Neurosci. 2004;24:8862–8872. doi: 10.1523/JNEUROSCI.3213-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Watanabe T, Noritake J, Fukata M, Yoshimura T, Itoh N, Harada T, Nakagawa M, Matsuura Y, Arimura N, Kaibuchi K. IQGAP3, a novel effector of Rac1 and Cdc42, regulates neurite outgrowth. J Cell Sci. 2007;120:567–577. doi: 10.1242/jcs.03356. [DOI] [PubMed] [Google Scholar]
- Weissbach L, Settleman J, Kalady MF, Snijders AJ, Murthy AE, Yan YX, Bernards A. Identification of a human rasGAP-related protein containing calmodulin-binding motifs. J Biol Chem. 1994;269:20517–20521. [PubMed] [Google Scholar]
- Yaka R, He DY, Phamluong K, Ron D. Pituitary adenylate cyclase-activating polypeptide (PACAP(1–38)) enhances N-methyl-D-aspartate receptor function and brain-derived neurotrophic factor expression via RACK1. J Biol Chem. 2003;278:9630–9638. doi: 10.1074/jbc.M209141200. [DOI] [PubMed] [Google Scholar]
- Yuan P, Zhou R, Wang Y, Li X, Li J, Chen G, Guitart X, Manji HK. Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J Affect Disord. 2010;124:164–169. doi: 10.1016/j.jad.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]