

Abstract
Background & objectives
Fibrous dysplasia (FD) is a rare metabolic bone disease and information available from India is limited to only anecdotal case reports. We describe the clinical profile and therapeutic outcome of 25 patients with FD observed over a period of 14 yr in a tertiary care centre from north India.
Methods
In this retrospective study patients (n = 25) with diagnosis of fibrous dysplasia based on either classical radiological features and/or histological evidence on bone biopsy, were analyzed. Associated endocrinopathies if any, were evaluated. The diagnosis of McCune Albright syndrome (MAS) was considered when fibrous dysplasia was accompanied by either café-au-lait macules and/or endocrinopathies. The clinical presentation, biochemical parameters and imaging were analysed. Seven patients received bisphosphonate therapy. The final outcome and side effects were noted.
Results
Age of the patients ranged from 7 to 48 yr (mean ± SD, 24.2 ± 11.4 yr) with a lag time between onset of symptoms and presentation ranging from 1 to 20 yr (mean ± SD, 6.6 ± 6.2 yr). The mean duration of follow up was 3.5 ± 2.1 yr. Eighteen (72%) patients had polyostotic disease while the remaining had monostotic FD. Eight patients had endocrinopathies: five had acromegaly, one each had gonadotropin independent precocious puberty (GIPP), hyperthyroidism and hypophosphatemic rickets. One child with GIPP later developed hyperthyroidism. McCune Albright syndrome was observed in 10 (40%) patients. A majority of the patients underwent various minor or major surgical procedures and seven patients received bisphosphonates for recurrent pathological fractures. Bone pain was reduced in all bisphosphonate treated patients with a decrease in subsequent fractures.
Interpretation & conclusions
This series of FD patients from north India shows the varying presentations of this rare disease. Medical treatment with bisphosphonates appears to be potentially rewarding.
Keywords: Bisphosphonates, endocrinopathies, fibrous dysplasia, McCune-Albright syndrome
Fibrous dysplasia (FD) is a congenital but non-inheritable benign disorder in which medullary bone is replaced by fibro-osseous tissue which causes distortion and overgrowth of the affected bone1. FD usually presents with bone pains, deformities, recurrent pathological fractures of the affected site and sometimes associated with endocrine hyperfunction2. There is no gender predilection for FD but it is more common in children and adolescents as compared to adults and older patients3.
Clinically FD is divided into three groups3 (1) monostotic: single bone involvement, which is most common (70%) (2) polyostotic: multiple bone involvement, a less common form (30%), and (3) McCune-Albright syndrome (MAS), a rare variant of mono/polyostotic disease in which FD is associated with cafι-au-lait macules and/or endocrinopathies4,5. Till recently, treatment of FD has been confined to orthopaedic surgeon for external fixation, bone grafting and curettage6–8. In the last two decades, bisphosphonates have been used extensively for management of various metabolic bone disorders including FD9,10.
Several studies are available on FD from western countries, but from developing countries, information is only available as short report or anecdotal case reports. We report clinical profile and treatment outcome of 25 patients with fibrous dysplasia from a tertiary care centre of north India.
Material & Methods
In this retrospective study medical records of 25 patients of documented fibrous dysplasia (January 1995 to October 2009) were retrieved from the Central Medical Records of Nehru Hospital at the Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh. The study protocol was approved by the Institute's Ethics Committee and informed consent was obtained from all the patients who received bisphosphonates. These records were reviewed in a structured proforma for age, sex, symptoms, signs, relevant biochemistry, hormonal profile, treatment modality and outcome. The diagnosis of fibrous dysplasia was based on one or more of the following criteria: classical radiological features of fibrous dysplasia and/or histological evidence of fibrous dysplasia on bone biopsy. McCune Albright syndrome (MAS) was diagnosed if patients with mono/ polystotic FD had either cafι-au-lait macules and/or endocrinopathy.
Fasting blood samples were collected on three consecutive days for estimation of serum calcium, inorganic phosphate, total serum alkaline phosphatase and albumin. Calcium was adjusted for albumin. Relevant skeletal survey was done. In addition,99mTc methylene diaphosphonate (MDP) bone scan was performed to assess the extent of disease. Patients who had symptoms and signs of acromegaly were subjected to growth hormone (GH) suppression test with 75 glucose and MRI head was done to localize the source of GH excess. Other endocrinopathies were investigated including thyroid function test for those who had hyperthyroidism, leutenizing hormone (LH), follicle stimulating hormone (FSH), and estradiol for those who had precocious puberty and appropriate work-up for hypophosphatemia in patients who presented with hypophosphatemic rickets.
Patients with history of fracture in last one year from the time of presentation were offered bisphosphonate therapy. Overall, seven patients received bisphosphonates. Four patients received pamidronate according to protocol (<2 yr of age, 0.5 mg/kg/day for three days; 2 to 3 yr, 0.75 mg/kg/day for three days and greater than 3 yr, 1 mg/kg/day for three days every three monthly for a year). Because of subsequent unavailability of pamidronate, three patients received other bisphosphonates (1 patient received intravenous zoledronate, once yearly, 1 patient received oral alendronate daily and another patient received combined intravenous and intralesional zoledronate which is still experimental)11. Side effects if any, were noted. Bisphosphonates were continued till there was a fracture free period of 1 year. Patients with endocrinopathies were treated accordingly.
Statistical analysis: The statistical program for the social sciences (10.0 PC windows; SPSS Inc. Chicago, IL) was used for data analysis.
Results
Age of the patients ranged from 7 to 48 yr (mean ± SD, 24.2 ± 11.4 yr) with a median age of 22 yr. Majority (68%) of the patients had childhood onset of disease. There were 13 males and 12 females with no gender predilection. The lag time from first reported symptom to diagnosis of FD ranged from 1 to 20 yr (mean ± SD, 6.6 ± 6.2 yr) and the mean duration of follow up was 3.5±2.1 yr.
Bone pains (64%), bony deformities (56%), fracture(s) (52%) and facial asymmetry (16%) (Fig. 1) were the common presenting manifestations. There was no significant difference in the frequency of clinical manifestations between males and females. The clinical characteristics of these 25 patients are summarized in the Table.
Fig. 1.
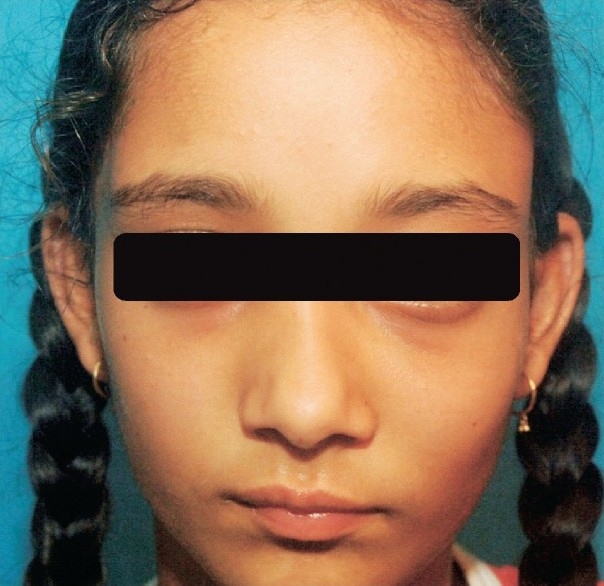
Craniofacial fibrous dysplasia showing asymmetry of face.
Table.
Summary of patients with fibrous dysplasia (n=25)
The diagnosis of FD was based on classical radiological features and/or histological evidence on bone biopsy. X-ray of the involved bone in all the patients showed typical expansile lytic lesions and some lesions with trabeculated area of radiolucency with or without ground glass appearance (Fig. 2). However, dysplastic lesions in the facial bone were radiodense. Based on these radiological findings, 18 patients (72%) had polyostotic and the remaining seven (28%) had monostotic lesions. Among polyostotic FD, the most commonly involved bones were craniofacial followed by long bones, ribs and orbit. Paranasal sinuses were involved in 11 (44%) patients and maxillary sinus was the most commonly involved. Among monostotic FD, the most commonly involved bone was femur followed by tibia and humerus. Classical Shepherd's Crook deformity was seen in three patients. Thirteen (52%) patients had one or more fractures.
Fig. 2.
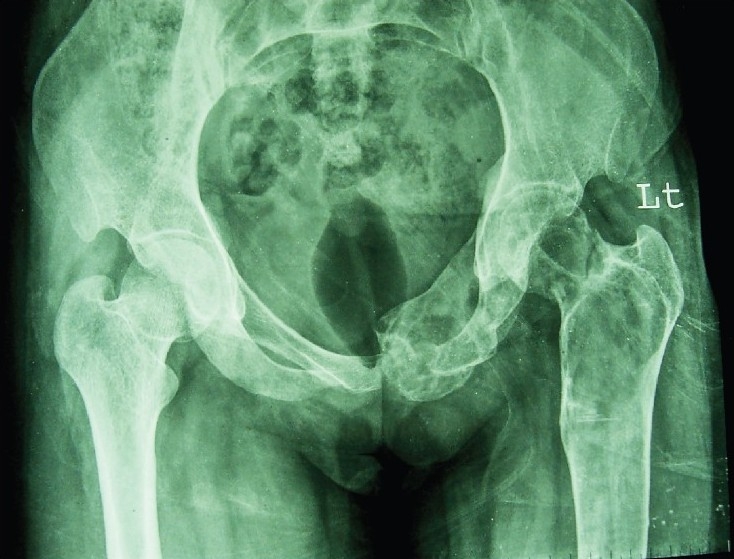
X-ray pelvis AP view showing multiple expansile lytic lesion with fish net appearance involving left ischium, pubis, left neck of femur and greater trochanter.
99mTcMDP bone scan was available in 20 patients and 13 of them had polyostotic disease which corresponded with the radiology. Bone biopsy was available in 18 patients and showed irregular trabeculae of woven bones (chinese letter pattern) without osteoblastic rimming and surrounded by fibrous connective tissue, features characteristic of FD (Fig. 3) and in the remaining patients diagnosis was based on characteristic radiology.
Fig. 3.
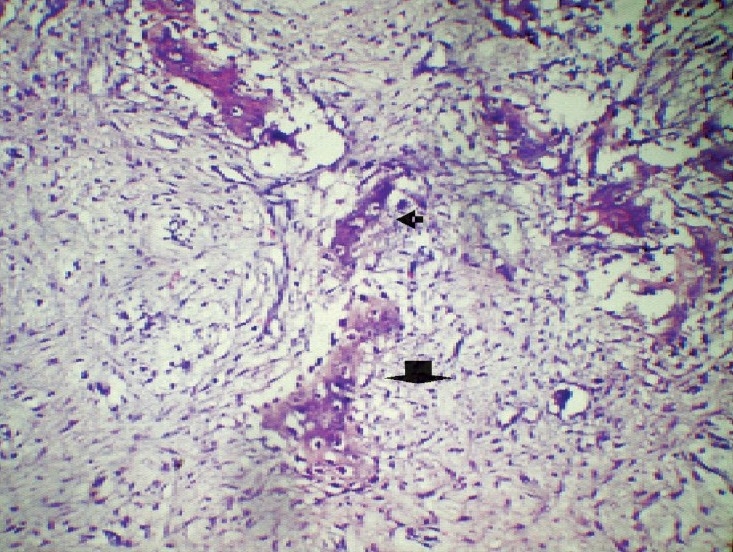
Histopathology of bone biopsy specimen showing irregular trabeculae of woven bones (chinese letter pattern; small arrow) without osteoblastic rimming (big arrow), surrounded by fibrous connective tissue, features characteristic of FD (H&E, 20X).
Of the 25 patients, eight (33%) had endocrinopathies. Five patients had acromegaly, one each had gonadotropin independent precocious puberty (GIPP), hyperthyroidism and hypophosphatemic rickets. The child with GIPP later also developed hyperthyroidism. Of the five patients with growth hormone excess, one patient had gigantism while the others had acromegaly. All of them had facial asymmetry and hyperprolactinaemia and four of them had documented pituitary tumour on imaging. One patient with hyperthyroidism had nodular goiter and another had diffuse goiter. Five patients with FD had cafι-au-lait macules (Fig. 4). MAS was observed in 10 (40%) patients.
Fig. 4.
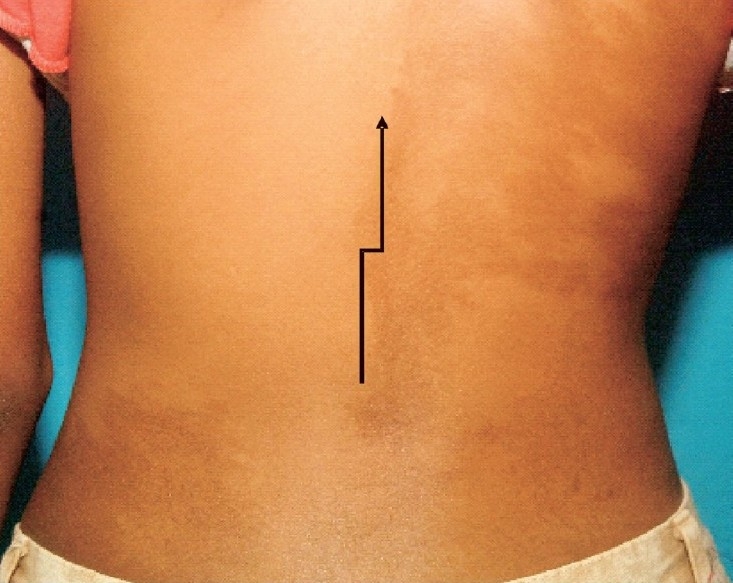
A large café-au-lait macule (arrow) measuring 10×7 cm over the back with irregular margin and not extending beyond the midline.
Twenty three of the 25 patients underwent minor/major reconstructive surgery for fibrous dysplasia. For endocrinopathies, 4 out of 5 patients with acromegaly underwent pituitary surgery (2 transfrontal and 2 transsphenoidal) and one with toxic adenoma underwent total thyroidectomy and the other was planned for radioablation. Other endocrinopathies were treated accordingly.
Seven patients received bisphosphonates for pathological fractures. Bone pain was reduced in all bisphosphonate treated patients but none of the patients showed improvement in bone deformity. However, on X-ray the bone lesions showed signs of healing. Five out of seven (72%) patients did not develop any new fracture after bisphosphonate therapy. Three patients who received injectable bisphosphonates developed fever on day 2 of infusion and one patient developed hypocalcemia.
Discussion
This study describes the varying presentations of fibrous dysplasia with various endocrinopathies and successful use of bisphosphonates in a subset of these patients.
Patients with FD usually present in childhood or early adolescence3 as was seen in our study, in that more than two third of patients had onset of disease before 20 yr of age. The usual lag time between onset of symptoms and clinical presentation varied from months to years in various studies similar to our observation3. Majority of our patients remained undiagnosed for a long period of time and presented later with fractures and endocrinopathies. The most common presenting manifestations are bone pains, deformity and fractures12 and sometimes isolated endocrinopathies like GIPP3, thyrotoxicosis and acrogigantism. Usually patients with monostotic FD have delayed presentation unless accompanied with endocrinopathies as compared to polyostotic variants. This was not observed in our study as many patients even with endocrinopathies presented at a later age. The male to female ratio was almost same in our study, these findings are similar to others12.
One of the common presenting manifestation of FD is skeletal fracture(s) and 52 per cent of our patients had one or more fractures, compared to 65 per cent in another study12. Bone deformity and facial asymmetry were more common in our patients compared to others12. The high incidence of bone deformities was probably due to delayed presentation. Most commonly involved bones in our study were craniofacial followed by femur, tibia, humerus and ribs as reported by others3,13. Commonly involved paranasal sinus in our study was maxillary sinus followed by sphenoid sinus, however, in one study sphenoid sinus (83%) was more commonly involved compared to maxillary sinus (11%)14.
Of the 25 patients, eight had endocrinopathy. Most common endocrine abnormality documented in our series was hypersomatotropism (20%)5 and hyperprolactinemia (20%) followed by hyperthyroidism15 (8%), hypophosphatemia16 (4%) and precocious puberty (4%). However, in a study done in paediatric age group, asymptomatic hypophosphatemia was the most common endocrine abnormality (38.5%) followed by sexual precocity (16.5%)17. The acromegaly associated with fibrous dysplasia differs from classical acromegaly by its presentation at a younger age, facial asymmetry, hyperprolactinaemia and lack of demonstratable adenoma on imaging in majority of patients5. However, in our study 4 out of 5 patients with acromegaly had pituitary adenoma and all had hyperprolactinaemia. Hyperthyroidism associated with FD is due to autonomous thyroid nodule with constitutive activation of Gsα subunit of TSH receptor18. Ablative therapy is the treatment of choice as was done in our patients. Patient who had GIPP later developed hyperthyroidism emphasizing the fact that these patients need continuous surveillance for evolving endocrinopathies. Patients with FD sometimes may present with manifestations of rickets and osteomalacia during childhood and adolescence. Hypophosphatemia is the characteristic abnormality in these patients and is attributed to increased secretion of fibroblast growth factor 23 (FGF23), a phosphatonin secreted from dysplastic bone lesions, hence leading to phosphaturia16. Only one of our patients had low serum phosphate and florid features of hypophosphatemic rickets16 in contrast to the findings in another study which reported asymptomatic hypophosphatemia (38.5%) as a more common entity in FD12.
The diagnosis of FD is based on classical radiological findings substantiated by bone scans and characteristic pathological findings on histopathology. However, sometimes bone lesions particularly in craniofacial regions are not accessible to biopsy. Therefore, the diagnosis rests on classical radiological findings. In our study, histopathology data were available in more than two third of patients.
The pathogenesis of FD involves somatic activating mutation of the gene encoding the alpha subunit of the stimulatory G protein in the bone marrow cells, resulting in locally increased stimulatory activity of adenyl cyclase and cAMP. This mutation leads to increased production of C-fos protein and interleukin-6 (IL-6) that result in classic dysplastic bone of FD3. The associated endocrinopathies are the result of constitutive activation of G protein coupled receptor by hormones acting through it including LH, FSH, thyroid stimulating hormone (TSH) and growth hormone relating hormone (GHRH) thereby manifesting as GIPP, hyperthyroidism and acromegaly respectively4,5,15.
Till recently, the treatment of FD was only restricted to symptomatic orthopaedic management like correction of fractures, internal fixation, curettage and grafting. The use of bisphosphonate ( intravenous pamidronate ) in FD showed promising results with remarkable improvement in bone pain and healing of bone lesions12. Similar effects were later shown by oral alendronate and intravenous zoledronate. The possible mechanism of action of bisphosphonates in FD is related to suppressed osteoclastic activation which occurs in FD due to constitutive activation of Gsα subunit in the bone tissue. In our study, those who received bisphosphonates showed symptomatic improvement in bone pain and reduction of fracture incidence, although there was no improvement in bony deformities, a finding similar to other studies9,12. Long term (> 5 yr) bisphosphonates therapy is associated with severe suppression of bone turn over (SSBT) and results in atypical fracture of shaft of long bones19. The proposed mechanisms for fracture in SSBT include impaired healing of microfractures and altered bone quality though bone mineral density progressively rises19. SSBT is unlikely to be encountered in patients with FD as prolonged use of bisphosphonates is not recommended.
The major limitations of our study were: it being a retrospective analysis, follow up data were not robust, and limited number of patients received bisphosphonates.
In conclusion, fibrous dysplasia has varied presentation including different endocrine manifestations. Therapy with bisphosphonates may reduce the co-morbidities.
Acknowledgments
Authors thank Drs Ravikiran Muthuswamy, Padala Ravikumar and Shanmugasundar G for editorial assistance.
Footnotes
Conflict of Interest: Nothing to declare
References
- 1.Schoenau E, Rauch F. Fibrous dysplasia. Horm Res. 2002;57(Suppl 2):79–82. doi: 10.1159/000058106. [DOI] [PubMed] [Google Scholar]
- 2.Bhadada S, Sharma DC, Gupta LK, Gupta A, Gupta R. Fibrous dysplasia. J Indian Med Assoc. 1998;96:100. [PubMed] [Google Scholar]
- 3.Chapurlat RD, Orcel P. Fibrous dysplasia of bone and McCune-Albright syndrome. Best Pract Res Clin Rheumatol. 2008;22:55–69. doi: 10.1016/j.berh.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Albright F, Butler AM, Hampton AO, Smith PH. Syndrome characterized by osteitis fibrosa disseminate, areas of pigmentation and endocrine dysfunction with precocious puberty in females. Report of five cases. N Engl J Med. 1937;216:727–46. [Google Scholar]
- 5.Bhansali A, Sharma BS, Sreenivasulu P, Singh P, Vashisth RK, Dash RJ. Acromegaly with fibrous dysplasia: McCune-Albright Syndrome. Clinical studies in cases and brief review of literature. Endocr J. 2003;50:793–9. doi: 10.1507/endocrj.50.793. [DOI] [PubMed] [Google Scholar]
- 6.Edgerton MT, Persing JA, Jane JA. The surgical treatment of fibrous dysplasia.With emphasis on recent contributions from cranio-maxillo-facial surgery. Ann Surg. 1985;202:459–79. doi: 10.1097/00000658-198510000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris WH, Dudley HR, Jr, Barry RJ. The natural history of fibrous dysplasia.An orthropaedic, pathological and roentgenographic study. J Bone Joint Surg Am. 1962;44-A:207–33. [PubMed] [Google Scholar]
- 8.Maher CO, Friedman JA, Meyer FB, Lynch JJ, Unni K, Raffel C. Surgical treatment of fibrous dysplasia of the skull in children. Pediatric Neurosurg. 2002;37:87–92. doi: 10.1159/000065110. [DOI] [PubMed] [Google Scholar]
- 9.Liens D, Delmas PD, Meunier PJ. Long-term effects of intravenous pamidronate in fibrous dysplasia of bone. Lancet. 1994;343:953–4. doi: 10.1016/s0140-6736(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 10.Chapurlat RD. Medical therapy in adults with fibrous dysplasia of bone. J Bone Miner Res. 2006;21(Suppl 2):114–9. doi: 10.1359/jbmr.06s222. [DOI] [PubMed] [Google Scholar]
- 11.Bhadada SK, Das S, Bhansali A, Sen R, Bhattachrya A. Visual vignette. Endocr Pract. 2009;15:655. doi: 10.4158/EP09168.VVR. [DOI] [PubMed] [Google Scholar]
- 12.Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab. 2003;88:4569–75. doi: 10.1210/jc.2003-030050. [DOI] [PubMed] [Google Scholar]
- 13.Ricalde P, Horswell BB. Craniofacial fibrous dysplasia of the fronto-orbital region: a case series and literature review. J Oral Maxillofac Surg. 2001;59:157–67. doi: 10.1053/joms.2001.20487. discussion 167-8. [DOI] [PubMed] [Google Scholar]
- 14.Tehranzadeh J, Fung Y, Donohue M, Anavim A, Pribram HW. Computed tomography of Paget disease of the skull versus fibrous dysplasia. Skeletal Radiol. 1998;27:664–72. doi: 10.1007/s002560050456. [DOI] [PubMed] [Google Scholar]
- 15.Bhat MH, Bhadada S, Dutta P, Bhansali A, Mittal BR. Hyperthyroidism with fibrous dysplasia: an unusual presentation of McCune-Albright syndrome. Exp Clin Endocrinol Diabetes. 2007;115:331–3. doi: 10.1055/s-2007-960497. [DOI] [PubMed] [Google Scholar]
- 16.Chattopadhyay A, Bhansali A, Mohanty SK, Khandelwal N, Mathur SK, Dash RJ. Hypophosphatemic rickets and osteomalacia in polyostotic fibrous dysplasia. J Pediatr Endocrinol Metab. 2003;16:893–6. doi: 10.1515/jpem.2003.16.6.893. [DOI] [PubMed] [Google Scholar]
- 17.Biermann JS. Common benign lesions of bone in children and adolescents. J Pediatr Orthop. 2002;22:268–73. [PubMed] [Google Scholar]
- 18.Styne DM, Grumbach M. Puberty: ontogeny, neuroendocrinology, physiology and disorders. In: Wilson JD, Foster DW, Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, editors. Williams textbook of endocrinology. 11th ed. Philadelphia Pa: Elsevier Saunders; 2008. pp. 1090–5. [Google Scholar]
- 19.Odvina CV, Levy S, Rao S, Zerwekh JE, Rao DS. Unusual mid-shaft fractures during long-term bisphosphonate therapy. Clin Endocrinol (Oxf) 2010;72:161–8. doi: 10.1111/j.1365-2265.2009.03581.x. [DOI] [PubMed] [Google Scholar]