

Abstract
Complexation of (tricarbonyl)iron to an acyclic diene serves to protect the ligand against oxidation, reduction and cycloaddition reactions while the steric bulk of this adjunct serves to direct the approach reagents to unsaturated groups attached to the diene onto the face opposite to iron. Furthermore, the Fe(CO)3 moiety can serve to stabilize carbocation centers adjacent to the diene (i.e. pentadienyl-iron cations). Recent applications of these reactivities to the synthesis of polyene, cyclopropane, cycloheptadiene and cyclohexenone containing natural products or analogs will be presented.
Keywords: Diene ligands, Iron, Synthetic methods, Regioselective nucleophilic addition
Introduction
While Reihlen and co-workers were the first to prepare an acyclic (butadiene)(tricarbonyl)iron (1, Figure 1) in 1930,[1a] the structure of this compound was not proposed until 1958 by Hallam and Pauson who were also the first to note that complexation of butadiene to iron protected the ligand towards catalytic reduction and cycloaddition reactions.[1b] Their structural assignment was eventually corroborated by X-ray crystallography in 1963.[1c] At about this same time, acyclic (pentadienyl)iron(1+) cations (2) were first reported by Pettit and co-workers.[2] Complexes of these types as well as the corresponding cyclic counterparts (3, 4) have found great utility in the synthesis of natural products. Numerous reviews have appeared concerning the use of complexes of type 3 and 4.[3] Similarly, reviews on the chemistry of complexes of type 1 and 2, covering up to 1999, have appeared.[4] For this reason this review will focus on chemistry related to complexes 1 and 2 from 2000 forward.
Figure 1.

Structures of diene- and dienyl-iron complexes.
Use of Fe(CO)3 as a Protecting and Stereodirecting Group
Synthesis of amphidinolide E
Amphidinolide E (5, Scheme 1) is a member of a family of macrolides isolated from the Amphidinium species of dinoflagellates.[5] Va and Roush have recently reported a synthesis of 5 which utilized Fe(CO)3 to protect a 3,5-hexadienoic acid against conjugation.[6] The synthesis begins with conversion of protected 4-penten-1,2,3-triol 6 into the tetrahydrofuranyl alcohol 7 in eight steps. Key steps in this sequence included a Johnson orthoester Claisen rearrangement to form the C9–C10 olefin and a [3+2] annulation[7] using an allylsilane to form the cis-tetrahydrofuranyl ring. Attempts at esterification of 7 with 2-methyl-3,5-hexadienoic acid were unsuccessful and generally led to recovery of 7 and the conjugated diene, 2-methyl-2,4-hexadienoic acid. Alternatively, esterification of (2S,3R) (2-methyl-3,5-hexadienoic acid)Fe(CO)3[8] (8) with 7 cleanly gave 9. In this case, iron serves as a protecting group such that the diene does not undergo isomerization. Oxidative decomplexation of 9, followed by ring closing metathesis[9] with Grubbs’ 1st generation catalyst afforded the macrolide ring 10 exclusively as the 3E,5E,9E-stereoisomer. Completion of the synthesis involved hydrostanallation of the alkyne, conversion to the 2-alkenyl iodide, cleavage of the protecting groups and Pd-catalyzed coupling.
Scheme 1.

Synthesis of amphidinolide E.
During the course of this work, Va and Roush discovered that the esterification of the diastereomeric (2S,3S) (2-methyl-3,5-hexadienoic acid)Fe(CO)3 (11) with 7 proceeded with complete inversion of the C2 methyl bond to afford 12 (Scheme 2).[10] These authors propose that the esterification of 11 proceeds via dehydration to generate the ketene intermediate 13; addition of the alcohol generates the ketene hemiacetal 14. Protonation of 14 occurs via the s-trans conformer and on the face opposite to the sterically bulky Fe(CO)3 group. Notably, the relative configurations at C2,C3 of 9 and 12 are the same (i.e. 2S,3R compared to 2R,3S), and thus it is likely that the transformation of 8 to 9 proceeds via the enantiomeric ketene (ent-13).
Scheme 2.

Esterification of (2S,3S)-(2-methyl-3,5-hexadienoic acid)Fe(CO)3.
Stereoselective synthesis of 11Z-retinal
Ito and co-workers have reported a highly stereoselective synthesis of 11Z-retinal (15a, Scheme 3), the chromophore of the visual pigment rhodopsin, which utilizes Fe(CO)3 complexation to facilitate generation of the 11Z-olefin.[11a–b] The synthesis begins with a nitrile aldol reaction of (β-ionone)Fe(CO)3 (16) with acetonitrile. This reaction proceeds with migration of the iron fragment to give 17. 1,3-Migration of the tricarbonyliron group have previously been observed.[4g,12] The presence of a terminal electron-withdrawing substituent (e.g. –CN) and the use of excess nucleophile generally lead to the more thermodynamically stable (diene)iron complex. Reduction of 17 gives the trienal 18, which upon Peterson olefination with ethyl trimethylsilylacetate affords a separable mixture of Z- and E-19 (77:15). Notably, Wittig or Horner-Emmons olefination of 18 gave only the E-stereoisomer. Conversion of Z-19 into nitrile 20, followed by decomplexation and nitrile reduction gave 15a. Nakanishi’s group has recently used this route to prepare the isotopically labelled 11Z-retinals 15b d; examination of the labelled retinals by solid state 2H NMR spectroscopy provided information on the orientation of these molecules in the rhodopsin binding pocket.[11c]
Scheme 3.

Stereoselective synthesis of 11Z-retinal via organoiron chemistry.
The rationale for Z-selective Peterson olefination rests on approach of the anion of trimethylsilylacetate to 18 in its s-trans-conformer (about the C10–C11 bond) on the face opposite to the sterically bulky (tricarbonyl)iron moiety. Of the two synclinal transition states of lowest presumed energy, TS–1 and TS–2 (Figure 2), only TS–1 avoids the steric repulsions between the bulky TMS group and the (diene)Fe(CO)3 group. Syn-elimination from the resultant β-silylalcohol,[13] as is known for anionic conditions, results preferentially in the 11Z-stereoisomer.
Figure 2.

Rationale for Z-selective Peterson olefination of 18.
Reactivity of In situ Generated Transoid (Pentadienyl)iron(1+) Cations
Acyclic (pentadienyl)iron(1+) cations 2 are most commonly prepared by ionization of (pentadienol)- or (pentadienyl ether)iron complexes under protic or Lewis acid conditions (Scheme 4).[2, 4] Ionization of the hydroxyl group occurs with anchimeric assistance from iron to generate the transoid pentadienyl cation 21; subsequent isomerization of 21 occurs with retention of configuration about the C1–C2 bond.[14] In certain cases, the in situ generated transoid pentadienyl iron cation can undergo attack by weak nucleophiles present in the reaction mixture. These reactions generally proceed via attack at C1 on the face opposite to iron.
Scheme 4.

Preparation of acyclic (pentadienyl)iron cations.
In situ generated transoid (pentadienyl)iron cations as initiators for polyene cyclization
Both the groups of Pearson[15] and of Franck-Neumann[16] reported polyene cyclizations initiated by in situ generated transoid (pentadienyl)iron cations (Scheme 5). These cyclizations may be terminated by attack of fluoride ion, formate ion or pendant electron-rich aromatic groups. For example, reaction of dienol complexes 22a or b,[16b] or the conjugated triene 23[15c] under either protic or Lewis acidic conditions resulted in the diastereoselective formation of the octahydrophenanthrene skeletons 24a–c. The relative configurations of 24a/b were determined by X-ray crystallography, while the relative configuration of 24c was assigned on the basis of extensive NMR spectral analysis of the free ligand (prepared by oxidation of 24c with excess Me3NO). The cyclizations were found to occur in a diastereoselective fashion; initial C-C bond formation occurred on the transoid (pentadienyl)iron cation (25) on the face opposite to the sterically bulky Fe(CO)3 group.
Scheme 5.

Polyene cyclizations initiated by a trans (pentadienyl)iron cation.
Diastereoselective Preparation of Dienyl Pyrrolidines and Piperidines
Cox and co-workers have reported on the diastereoselective preparation of dienyl pyrrolidine and dienyl piperidine complexes (26 and 27 respectively, Scheme 6) by the reductive amination of ketoaldehydes 28a and 28b.[17] These reactions are proposed to proceed via reductive amination at the aldehyde, followed by generation of the iminium complex 29 (an alternative resonance contributor is the transoid pentadienyl iron cation 30). The iminium ion/pentadienyl cation complex is preferentially oriented in the s-trans conformer about the diene-to-iminium carbon so as to minimize repulsion between the diene and the substituent R on nitrogen. Approach of hydride to the face opposite to iron (followed by rotation about the diene to pyrrolidine/piperidine bond) generated the products with excellent diastereoselective control (ψ-exo diastereomer).[18] The relative configurations of 26 and 27 were confirmed by X-ray crystal structures of one example each.
Scheme 6.

Reductive amination of diene ketoaldehyde complexes.
Preparation of Organoiron Nucleoside Analogs
Schmalz and co-workers reported on the preparation of organoiron containing nucleoside analogs by reaction of the dienyl ether complexes 31a or b (prepared in 5 steps from α-methyl glucopyranoside) with silylated nucleobases in the presence of trimethylsilyl triflate (Scheme 7).[19] This reaction presumably proceeds via intermediacy of the transoid pentadienyl cation 32. Nucleophilic attack on 32 occurs predominantly on the face opposite to the sterically bulky Fe(CO)3 group to afford exo-33a or b as the major product, along with a lesser amount of the diastereomeric endo complex. Complexes exo-33a and exo-33b were found to be cytotoxic against cultivated BJAB tumor cells (IC90 = 30 and 20 μM respectively). The cytotoxicity of exo-33b was attributed to its ability to induce apoptosis by DNA fragmentation. Notably, the free ligand of complex exo-33b exhibited considerably diminished cytotoxicity (IC90 > 100 μM), indicating a critical, but as yet undetermined, role for the metal.
Scheme 7.

Preparation of organoiron nucleoside analogs (TDS = thexyldimethylsilyl).
Reactivity of Isolable Cisoid (Pentadienyl)iron Cations
The acyclic (pentadienyl)iron(1+) cations 2 can act as excellent organometallic electrophiles toward a wide variety of nucleophiles. Nucleophilic attack can take place on the cisoid form of the pentadienyl cation at either termini to afford the E,Z-diene complexes 34 or 35, or on the internal atoms of the ligand (C2/C4) to afford complexes 36, or 37 (Scheme 8). Alternatively, since the transoid form exists in equilibrium with the cisoid form, nucleophilic attack on the transoid pentadienyl cation generates E,E-diene complexes 38 or 39 as a single diastereomer. The regioselectivity for nucleophilic attack depends on the nature of the substituents present on the pentadienyl ligand as well as the “spectator” ligand L, the nature of the nucleophile, and even the nucleophile counter ion. While not all of these factors are well understood, a few generalities can be made.[20] In general for tricarbonyl-ligated cations 2 (L = CO), weak neutral nucleophiles (e.g. H2O, alcohols, aryl amines, electron-rich aromatics, allyl silanes) reaction proceeds via the higher energy (and thus more reactive) transoid pentadienyl forms to afford products 38/39. Reaction of more reactive organocadmium reagents, organocuprates, phosphines and alkyl amines proceeds via attack at the terminal carbons of the cisoid conformer to give products 34/35. These reactions are believed to be under frontier orbital control. If the pentadienyl ligand bears a terminal electron-withdrawing group (e.g. R1 = CO2Me), reaction with methyl lithium, alkenyl Grignards, potassium phthalimide and stabilized carbon nucleophiles proceeds by attack at C2/C4, and this regioselectivity is believed to be due to charge control (i.e. nucleophilic attack at the pentadienyl carbon bearing the greatest partial positive charge). For cations in which the substituents are neither strongly electron-withdrawing or electon-donating, nucleophilic attack frequently does not occur in a regioselective fashion. There are considerably fewer cases of acyclic (pentadienyl)iron cations bearing a phosphine ligand (i.e. 2, L = PR3), however in these cases the regioselectivity is generally improved over that of their corresponding Fe(CO)3 cations due to the greater stability/decreased reactivity of the Fe(CO)2PR3 cations.
Scheme 8.

Modes of reactivity for isolable (pentadienyl)iron cations.
Synthetic Studies on Diterpenes with a 3-Methyl-1,3Z-pentadienyl Side-chain
Heteroscyphic acids A and B are novel clerodane-type diterpenes isolated from cultured cells of the liverwort Heteroscyphus planus, whose structural assignments (40a/b) were based on their MS and NMR spectral data (Figure 3).[21a–c] In particular the 12Z-stereochemistry for 40b was assigned on the basis of NOEs between Me-16 and H-12 and between H-14 and H11. While no biological activity was reported for 40a or b, these compounds are nonetheless structurally related to the clerodane caseargrewiin D[21d] (41) which exhibits both antimalarial and antitumor activity.
Figure 3.

Proposed (40a) and revised (41) structures for heteroscyphic acid A, and structure for caeswaregiin D (42).
Donaldson’s group envisioned introduction of the 3-methyl-1,3Z-dienyl side-chain by nucleophilic addition to a (3-methylpentadienyl)iron cation.[22] To this end, 5-hexen-1-ol (43) was transformed into the decahydronaphthalene ester 44 (Scheme 9); the fused bicyclic skeleton was formed by a Mn-mediated oxidative radical cyclization.[23] Generation of the ester enolate anion from 44 and addition to the Fe(CO)2PPh3 ligated pentadienyl cation 45 gave complex 46. This was produced as a mixture of diastereomers due to nucleophilic attack at one or the other pentadienyl terminal carbons of the symmetrical cation. Decomplexation of 46, followed by purification by AgNO3 impregnated silica gel gave 47 as a single diastereomer. It was surprising to note that the NMR spectral data for the dienyl sidechain of 47 (confirmed as Z by comparison of its NMR spectral data to that of other known diterpenes possessing a 3-methyl-1,3Z-dienyl group) did not match well with that reported for the heteroscyphic acids A and B. In fact, the chemical shifts reported for heteroscyphic acids A and B are more consistent with those observed for a number of diterpenes possessing a 3-methyl-1,3E-dienyl group, and thus it was suggested that the heteroscyphic acids have this geometry for the sidechain (c.f. 41, Figure 3). This methodology might prove useful for the introduction of the 3-methyl-1,3Z-dienyl sidechain in 42.
Scheme 9.

Synthesis of a 3-methyl-1,3Z-pentadienyl diterpene skeleton.
Synthetic Studies on Macrolactin A
Macrolactin A (48, Figure 4) is a polyene macrolide aglycon originally isolated from a taxonomically unidentified marine bacterium.[24] More recently, other members of this family of 24-membered macrolides have been isolated from Bacillus sp. Sc026, Bacillus sp. PP19-H3, and Actinomadura sp.[25] Initial screening revealed that 48 displayed antibacterial, antiviral and antitumor activity. The complex structure of macrolactin A presents several synthetic challenges, including four sp3 asymmetric centers and three conjugated dienes. Several groups have reported synthetic studies,[26] including total syntheses by the groups of Smith,[27b] Carreira,[27c] and Marino.[27d]
Figure 4.
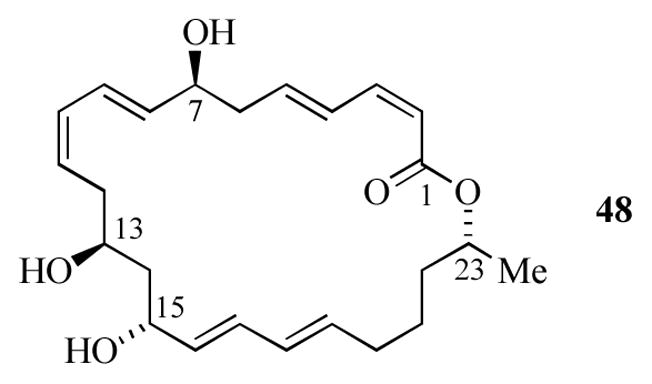
Structure of macrolactin A (48).
Takemoto’s group has prepared the C1–C15 segment of macrolactin A, in racemic form, utilizing the Fe(CO)3 group as a mobile chiral auxiliary.[28] The synthesis begins with the achiral (2,4-hexadiendial)Fe(CO)3 complex 49 (Scheme 10). Condensation of 49 with the enolate anion derived from ethyl acetate proceeded in a diastereoselective fashion to afford a separable mixture of predominantly the ψ-exo β-hydroxyester rac-50 along with the ψ-endo alcohol rac-51. Reaction of the derived TBS ether 52 with diethyl phosphorocyanidate gave the crude cyanophosphate 53 as a mixture of diastereomers, which were used in the next step without further purification. Protonation of 53 with HBF4 in the presence of 4-fluorobenzenethiol afforded the E,Z-dienylnitrile complex 55, along with a minor amount of the corresponding E,E-diene complex. This 1,2-migration of iron presumably proceeds via the intermediacy of the cisoid (pentadienyl)iron cation 54. The success of this reaction was highly dependent on the solvent and acid used; use of BF3-etherate gave greatly diminished yields of 55 at the expense of a variety of other nucleophilic addition products. Similarly, attempts to use hydride nucleophiles (Et3SiH or NaBH3CN) in the in situ formation of 54 were unsuccessful. Treatment of 54 with 6 equivalents of DIBAL, followed by quenching with aqueous NH4Cl, resulted in reduction of the nitrile and ester to an aldehyde and 1° alcohol respectively. After protection of the 1° alcohol as an acetate, addition of the organozinc reagent prepared from propargyl bromide and zinc in the presence of NH4Cl gave an equimolar mixture of the diastereomeric dienol complexes ψ-exo 56 and ψ-endo 57. Separation of the diastereomers was possible after protection as their TBS ethers 58/59. Rh-catalyzed hydroboration of ψ-exo 58 with pinacolborane gave the crude E-vinylboronate 60 in modest yield. Pd-catalyzed coupling of 60 with ethyl (Z)-3-iodopropenoate afforded a mixture of acetate 61 and alcohol 62. Hydrolysis of 61 afforded the (2Z,4E,8E,10Z-pentadecatetraenyl complex 62 in 60% overall yield from 60. Unfortunately, while the 1° alcohol of 62 could be oxidized with IBX, attempts at coupling the resultant aldehyde with an alkenylzirconium reagent, to generate the C15–C16 bond, were unsuccessful.
Scheme 10.

Takemoto synthesis of the C1–C15 segment of macrolactin A (Ar = p-FC6H4).
Li and Donaldson have also applied diene-iron complexes to the synthesis of the C7–C24 segment of macrolactin A in enantiomerically enriched form (≥ 90% ee) (Scheme 11).[29a] Generation of the 8E,10Z-diene segment of macrolactin utilized nucleophilic addition to the enantiomerically enriched Fe(CO)2PPh3 ligated cation 62. This cation was prepared by standard procedures from enantiomerically pure methyl 6-oxo-2,4-hexadienoate complex.[30] Addition of nitroacetate anion 63 proceeds at an internal pentadienyl carbon under kinetic control, however a brief work-up of the initially formed (pentenediyl)iron complex with aqueous NH4Cl gave the E,Z-dienoate complex 64 as a mixture of diastereomers. This isomerization is proposed to proceed via protonation at the ester carbonyl, dissociation to the (pentadienyl)Fe(CO)2PPh3+ cation, followed by attack at the terminal position to generate the more thermodynamically E,Z-dienoate complex. Cleavage of the trimethylsilylethoxy ester from 64 and subsequent decarboxylation generated the C7–C13 segment (+)-65. Generation of the nitrile oxide from (+)-65 using Mukiayama conditions[31] in the presence of 1.5 equivalents of the enantiomerically enriched triene complex (+)-66[32] (≥ 90% ee) gave the bimetallic tetraene isoxazoline (+)-67 as a single diastereomer. The selective formation of the ψ-exo diastereomer in this intermolecular cycloaddition is due to approach of the nitrile oxide on the less hindered face of the s-trans triene rotomer.[33] Reductive hydrolysis of isoxazoline 67, using commercially purchased Raney-nickel, gave the bimetallic β-hydroxyketone (+)-68. Using this less reactive form of the catalyst, the two iron adjuncts serve to protect the diene segments against hydrogenation.[34] Diastereoselective reduction[35] of 68 gave the diol (+)-69. Generation of the acetonide followed by oxidative decomplexation with CAN gave the tetraene (−)-70. Oxidative removal of the two iron moieties was accompanied by cleavage of the acetonide group due to the acid generated under these reaction conditions. The diminished yield for this last step may be due to the lability of this tetraenyldiol as others have reported that removal of hydroxyl protecting groups from intact macrolactin A has proven to be difficult.[27b] In this synthesis of the C7–C24 segment, the iron-carbonyl adjuncts are responsible for i) stereoselective preparation for the C8–C11 E,Z-diene, ii) diastereoselective generation of the C23 alcohol by remote asymmetric induction, iii) introduction of the C15 stereocenter by a highly diastereoselective intermolecular nitrile oxide-olefin cycloaddition, and iv) protection of the C8–C11 and C16–C19 dienes during reductive hydrolysis of the isoxazoline group.
Scheme 11.

Synthesis of the enantiomerically enriched C7–C24 segment of macrolactin A (R = CH2CH2CH2CH(OTBS)Me, R′ = CH2CH2TMS).
Synthesis of Vinylcyclopropanes
(3-Pentene-1,5-diyl)iron complexes 71a, bearing an electron withdrawing group at C1, have been prepared by addition of carbon nucleophiles to (pentadienyl)iron(1+) cations (Scheme 12).[36] Alternatively, the thermal reaction of (vinylketene)iron complex 72 with dimethylfumarate generated the (pentenediyl)iron complex 71b.[37] Oxidation of either 71a or 71b with ceric ammonium nitrate gave the vinylcyclopropanecarboxylates 73a or b.[36a, 37] Since this is formally an oxidatively induced-reductive elimination, the reaction generally proceeds with retention of configuration at the two centers undergoing C–C bond formation.
Scheme 12.

Synthesis of vinylcyclopropanes via oxidative decomplexation of (3-pentene-1,5-diyl)iron complexes (a, R = CH(CO2Me)2, R′ = R″ = H; b, R = CO2Me, R′ = tBu, R″ = Ph).
Synthesis of 2-(2-Carboxycyclopropyl)glycines and Dysibetaine CPa
The selective activation of different glutamate receptors may depend on recognition of a particular conformer of this flexible molecule. For this reason, the synthesis and evaluation of a number of 2-(2′-carboxycyclopropyl)glycines (e.g. 74a–f, Figure 5), as conformationally restricted analogs of glutamate, has led to the discovery of ligands with mGluR specificity.[38] In particular the extended conformation, as exemplified by compounds 74a–d, is believed to be a requirement for binding to the mGluR1 and mGluR2 receptors. Recently, Sakai and co-workers isolated a novel water-soluble cyclopropane containing betaine from D. herbacea which they termed dysibetaine CPa (75, Figure 5).[39] Compound 75 displaced kinate from the NMDA-type glutamate receptor with IC50 = 13 μM.
Figure 5.

Structure of conformationally restricted glutamate analogs (74) and dysibetaine CPa (75).
Reaction of the enantiomerically enriched tricarbonyl-ligated cation (1R)-76 (≥ 80% ee) with the anion generated from methyl nitroacetate gave (pentenediyl)iron complex 77 as a mixture of diastereomers at the nitroacetate carbon (Scheme 13).[36a] Decomplexation of the mixture of diastereomers afforded vinylcyclopropanecarboxylate (2′S)-78 as an inseparable mixture of diastereomers at the nitroacetate carbon. Transformation of the diastereomeric mixture (2′S)-78 into the individual 3-ethyl CCGs (−)-79 and (+)-80 required reduction of the vinyl and nitro groups, conversion of the amines into a separable mixture of diphenylmethylene imines,[40] hydrolysis of the separate diphenylmethylene imines and the methyl esters and finally generation of the free bases.
Scheme 13.

Synthesis of 2-(2′-carboxycyclopropyl)glycines and dysibetaine CPa (E = CO2Me).
For the preparation of dysibetaine CPa, reaction of the dicarbonyl(triphenylphosphine)-ligated cation rac-62 with the anion generated from nitromethane gave (pentenediyl)iron complex 81 in excellent yield (Scheme 13).[41] Oxidative decomplexation of 81 gave the vinylcyclopropanecarboxylate 82. Transformation of 82 into rac-75 required conversion of the vinyl functionality to an ester, subsequent reduction of the 1° nitro group, hydrolysis and exhaustive methylation.
Synthesis of the C9–C16 Segment of Ambruticin
Ambruticin (83, Figure 6) is a structurally unique carboxylic acid isolated from Polyangium cellulosum var fulvum, which exhibits potent oral antifungal activity against Coccidioides immitis, Histoplasma capsulatum, and Blastomyces dermititidis.[42] The complex structure of ambruticin presents several synthetic challenges, including a tetrahydropyranyl ring, a dihydropyranyl ring and a divinylcyclopropane ring. Several groups have reported synthetic studies,[43] including total syntheses by the groups of Kende,[44a] Jacobsen,[44b] Martin.[44c] and Lee.[44d]
Figure 6.

Structure of the antifungal agent ambruticin (83).
Reaction of (1S)-76 in CH2Cl2 with a ethereal solution of methyl lithium gave the (pentenediyl)iron complex (−)-84 along with a minor amount of tricarbonyl(methyl-3,5-hexadienoate)iron (Scheme 14).[45] It was found that use of CH2Cl2 as solvent was crucial to the success of this reaction. Use of either ether or THF gave reduced yields of the (pentenediyl)iron complex. Oxidative decomplexation of (−)-84 cleanly gave the stereodefined vinylcyclopropanecarboxylate (+)-85. Cross metathesis of 85 with a nine fold excess of (R)-86 in presence of 5 mol % of Grubbs’ 2nd generation catalyst gave 87 as a mixture of E- and Z-isomers (6:1 ratio).[46]
Scheme 14.

Synthesis of the C9–C16 segment of ambruticin (E = CO2Me).
Synthesis of Divinylcyclopropanes and Cope rearrangement
Donaldson and co-workers have demonstrated that the reaction of (2-methoxycarbonylpentadienyl)iron(1+) cations 62 or 76 with alkenyl Grignard reagents gave primarily the corresponding (2-alkenyl-3-pentene-1,5-diyl)iron complexes 88 or 89 respectively (Scheme 15).[47] The yield of these products was dependent on the reaction media; use of methylene chloride gave the best results while THF or toluene led to diminished yields of 88/89. Nucleophilic attack on the face opposite to the metal was corroborated by X-ray crystal structure of the parent complex 89 (R1 = RE = RZ = H).[47b] Oxidative decomplexation of 88/89 gave the divinylcyclopropane 90. In most cases CAN gave good yields of the 2,3-divinylcyclopropanecarboxylate, however for complexes with an electron rich 2-alkenyl group secondary oxidation of the resultant divinylcyclopropane product led to diminished yields. In these cases, oxidation with alkaline hydrogen peroxide provided superior yields, but led to mixtures of both cis- and trans-divinylcyclopropanes. Reduction of 90, followed by [3,3]-sigmatropic rearrangement[48] afforded the 2,6-cycloheptadienylmethanol 91. While the temperature required for the Cope rearrangement varied depending on the alkenyl substituents and olefin geometry, good overall yields were obtained from complexes 88/89.
Scheme 15.

Synthesis of divinylcyclopropanes and Cope rearrangement.
Synthesis of a Guianolide Skeleton
The guianolides are a family of sesquiterpenes characterized by a 5,7,5-fused tricyclic skeleton. The majority of these compounds possess a trans-γ-butyrolactone ring, but differ with respect to the oxygenation and oxidation state(s) of carbons 2–5, 8, 10, and 11.[49] Representative members of this family include chinesiolide B (92, Figure 7),[49d] cynaropikrin (93),[49e] and cladantholide (94).[49f]
Figure 7.

Representative guianolide natural products.
Donaldson and co-workers have applied organoiron methodology to the synthesis of the 5,7,5 ring system of the guianolides (Scheme 16).[50] Reaction of the Grignard reagent derived from known[49] cyclopentenyl bromide 95 with the (dienyl)Fe(CO)3+ cation 96[50] gave the (pentenediyl)iron complex 97 as a mixture of diastereomers at the silyl ether carbon (*). Oxidative decomplexation, ester reduction and Cope rearrangement at elevated temperatures gave 98. The hexahydroazulene 98 was transformed into the epoxydiol 99 via i) selective hydrogenation of the less substituted olefin, ii) extension of the C3 sidechain by tosylation and cyanide displacement, iii) cleavage of the silyl ether, iv) epoxidation and finally, v) twofold reduction of the nitrile sidechain. Oxidation of 99 with catalytic TPAP and NMO (3.2 equiv) gave a single lactone 100. This transformation presumably proceeds via oxidation of both the 1° and 2° alcohols, followed by β-elimination of the epoxide, generation of a lactol and further oxidation to the lactone. Reduction of 100 afforded 101, which possesses the relative stereochemistry about the seven-membered ring of cladantholide.
Scheme 16.

Preparation of the 5,7,5 ring system of the guianolides.
Synthesis of Cyclohexenones
The (pentenediyl)iron complexes discussed in Schemes 12–16 are stable, isolable species. This is believed to be due to the fact that the presence of an electron withdrawing group attached to a carbon-metal σ-bond slows the rate of carbonyl insertion.[53] In contrast, (pentenediyl)iron complexes lacking an electron withdrawing group at C1 (e.g. 36 or 37, Scheme 17) may be generated by nucleophilic attack on acyclic (pentadienyl)iron cations at the internal C2 position.[54] These complexes are generally unstable and undergo CO insertion to generate the (acyl)iron complexes 102/103. Reductive elimination of 102/103, followed by conjugation of the olefin, gives cyclohexenones 104/105 respectively.
Scheme 17.

Generation of cyclohexenones from (pentadienyl)iron cations.
An alternative route to cyclohexenones is the photochemically initiated ring rearrangement carbonylation of alkenylcyclopropanes (Scheme 18).[55] While this reaction does not formally involve a (diene)iron complex or (dienyl)iron cation, it is nonetheless related by the presumed intermediates. This reaction is believed to proceeds via oxidative insertion of iron into one of the proximal vinylcyclopropane bonds (b or a) to generate (pentenediyl)iron intermediates 106 or 107 (respectively). Carbonyl insertion, followed reductive elimination and conjugation gives 108 or 109. Isolation of 108 as the major cyclohexenone product indicates that insertion into the cyclopropane bond “b” is favored. Since the major product arises by cleavage of the less substituted vinylcyclopropanes bond “b”, beginning with enantiomerically enriched (> 99% ee) vinylcyclopropane 110 (R = CH2OBn, R4 = R5 = H) led to 108 in enantiomerically enriched form (> 95% ee). The enantiomeric excess and absolute configuration of the minor product 109 was not identified.
Scheme 18.

Generation of cyclohexenones via iron-mediated carbonylation of alkenylcyclopropanes.
Taber and co-workers have applied this methodology to the enantioselective synthesis of (−)-delobanone (111) beginning with geraniol (Scheme 19).[55d]
Scheme 19.

Taber and co-workers synthesis of (−)-delobanone.
Miscellaneous
Christie and co-workers have reported the 1,3-dipolar cycloaddition of aldehydes with the racemic dienylcyclopropane complex 112 in the presence of ZnBr2 (Scheme 20).[56] This reaction affords the dienylfurans 113 as a mixture of two (of the four possible) diastereomers. In all cases, the relative configuration at the iron-diene and the tetrahydrofuranyl carbon adjacent to the diene were found to be as indicated (i.e. ψ-exo), and thus the formed are due to the cis- and trans-2,4-disubstituted furan ring. at the dienyl at the indicated carbon (*) in moderate yield. The authors propose that this reaction proceeds via formation of the zwitterionic intermediate 114 which reacts with the aldehyde on the face opposite to the sterically bulky (tricarbonyl)iron adjunct.
Scheme 20.

Formation of polysubstituted dienyltetrahydrofurans.
Conclusions
Complexation of diene and dienyl ligands to iron facilitates the stereoselective preparation of conjugated E,E- and E,Z-1,3-dienes, trisubstituted cyclopropanes, 1,4-cycloheptadienes and cyclohexenones. These features of the (tricarbonyl)iron adjunct have been exploited by a number of research groups in the synthesis of polyene macrolides, optical pigment chromophores, heterocycles, terpenes, conformationally restricted ligands for glutamate receptors, and antifungal agents.
Acknowledgments
WAD is grateful to his co-workers whose research accomplishments are described in this review. Their experimental skill, dedication, and keen observation turned ideas into reality. Financial support over the years from the National Institutes of Health (GM-42641) and the National Science Foundation (CHE-0415771) is gratefully acknowledged.
Biographies
William Donaldson was born near Philadelphia, Pennsylvania. He received his B.A. degree in Chemistry from Wesleyan University (1977), and his Ph.D. in Organometallic Chemistry from Dartmouth College (1981) working with Prof. Russell Hughes, before conducting postdoctoral research with Prof. Myron Rosenblum at Brandeis University (1981–1982). Following a one-year position at Wesleyan University, he joined the faculty at Marquette University in 1983. His research has focused on the application of organoiron complexes to organic synthesis, as well as the synthesis of hydropyran natural products.
Subhabrata Chaudhury is a Bengali from the southern part of Bengal. After receiving his B.Sc. from the University of Calcutta (1997) and M.Sc. degree from the Indian Institute of Technology, Kharagpur (1999), he joined the group of Professor Donaldson at Marquette University where he worked on the development of organoiron methodologies for the total synthesis of natural products. He obtained his Ph.D. degree in 2006 and was a postdoctoral fellow in the Department of Medicinal Chemistry at University of Kansas. Before he started his second period of postdoctoral research at the Department of Biophysics, Medical College of Wisconsin, he returned to Marquette and spent an interim period at Prof. Donaldson’s laboratory. In September 2008 he moved to Scotland to join the group of Professor J. S. Clark at the University of Glasgow as a research associate. His research interests involve developing methodologies for the preparation of organic building blocks using transition metal complexes and their application in organic synthesis.
References
- 1.(a) Gruhl A, Hessling G, Pfrengle O, Reihlen H. Justus Liebigs Ann Chem. 1930;482:161–182. [Google Scholar]; (b) Hallam B, Pauson P. J Chem Soc. 1958;168:642–645. [Google Scholar]; (c) Mills OS, Robinson G. Acta Cryst. 1963;16:758–762. [Google Scholar]
- 2.(a) Mahler JE, Pettit R. J Am Chem Soc. 1963;85:3955–3959. [Google Scholar]; (b) Mahler JE, Gibson DH, Pettit R. J Am Chem Soc. 1963;85:3959–3963. [Google Scholar]
- 3.Choi TA, Czerwonka R, Forke R, Jaeger A, Knoell J, Krahl MP, Krause T, Reddy KR, Franzblau SG, Knoelker HJ. Med Chem Res. 2008;17:374–385. [Google Scholar]; Knoelker HJ. Curr Org Syn. 2004;1:309–331. [Google Scholar]; Knoelker HJ, Braier A, Brocher DJ, Cammerer S, Frohner W, Gonser P, Hermann H, Herzberg D, Reddy KR, Rohde G. Pure Appl Chem. 2001;73:1075–1086. [Google Scholar]; Knoelker HJ. Chem Soc Rev. 1999;28:151–157. [Google Scholar]; Stephenson GR. In: Handbook of Functionalized Organometallics. Knochel P, editor. Vol. 2. Wiley-VCH; Weinheim: 2005. pp. 569–627. [Google Scholar]; Ong CW, Lia MC. Trends in Organomet Chem. 2002;4:47–58. [Google Scholar]; Pearson AJ. In: Advances in Metal-Organic Chemistry. Liebeskind LS, editor. Vol. 1. JAI Press; Greenwich: 1989. pp. 1–49. [Google Scholar]
- 4.(a) Gree R. Synthesis. 1989:341–355. [Google Scholar]; (b) Gree R, Lellouche JP. In: Advances in Metal-Organic Chemistry. Liebeskind LS, editor. Vol. 4. JAI Press Inc; Greenwich, CT: 1995. pp. 129–273. [Google Scholar]; (c) Iwata C, Takemoto Y. Chem Commun. 1996:2497–2504. [Google Scholar]; (d) Donaldson WA. Aldrichimica Acta. 1997;30:17–24. [Google Scholar]; (e) Donaldson WA. Curr Org Chem. 2000;4:837–868. [Google Scholar]; (f) Cox LR, Ley SV. Chem Soc Rev. 1998;27:301–314. [Google Scholar]; (g) Salzer A. In: Organometallics in Organic Synthesis. Werner H, Erker G, editors. Vol. 2. Springer-Verlag; Berlin, Germany: 1989. pp. 291–309. [Google Scholar]
- 5.(a) Kobayashi J, Ishibashi M, Murayama T, Takamatsu M, Iwamura M, Ohizumi Y, Sasaki T. J Org Chem. 1990;55:3421–3423. [Google Scholar]; (b) Kubota T, Tsuda M, Kobayashi J. J Org Chem. 2002;67:1651–1656. doi: 10.1021/jo016326n. [DOI] [PubMed] [Google Scholar]
- 6.(a) Va P, Roush WR. Tetrahedron. 2007;63:5768–5796. doi: 10.1016/j.tet.2007.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Va P, Roush WR. J Am Chem Soc. 2006;128:15960–15961. doi: 10.1021/ja066663j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Micalizio GC, Roush WR. Org Lett. 2000;2:461–464. doi: 10.1021/ol9913082. [DOI] [PubMed] [Google Scholar]
- 8.For a previous preparation of 8 see: Wasicak JT, Craig RA, Henry R, Dasgupta B, Li H, Donaldson WA. Tetrahedron. 1997;53:4185–4198.
- 9.(a) Grubbs RH. Tetrahedron. 1998;54:4413–4450. [Google Scholar]; (b) Schuster M, Blechert S. Angew Chem Int Ed Engl. 1997;36:2036–2056. [Google Scholar]
- 10.Va P, Roush WR. Org Lett. 2007;9:307–310. doi: 10.1021/ol0628098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Wada A, Fujioka N, Tanaka Y, Ito M. J Org Chem. 2000;65:2438–2443. doi: 10.1021/jo9916030. [DOI] [PubMed] [Google Scholar]; (b) Wada A, Hiraishi S, Takamura N, Date T, Aoe K, Ito M. J Org Chem. 1997;62:4343–4348. doi: 10.1021/jo961041+. [DOI] [PubMed] [Google Scholar]; (c) Tanaka K, Struts AV, Krane S, Fujioka N, Salgado GFJ, Martinez-Mayrga K, Brown MF, Nakanishi K. Bull Chem Soc Jpn. 2007;80:2177–2184. [Google Scholar]
- 12.(a) Hafner A, von Philipsborn W, Salzer A. Angew Chem, Int Ed Engl. 1985;24:126–127. [Google Scholar]; (b) Adams CM, Cerioni G, Hafner A, Kalchhauser K, von Philipsborn W, Prewo R, Schwenk A. Helv Chim Acta. 1988;71:1116–1142. [Google Scholar]; (c) Takemoto Y, Ishii K, Ibuka T, Miwa Y, Taga T, Nakao S, Tanaka T, Oshishi H, Kai Y, Kanehisa N. J Org Chem. 2001;66:6116–6123. doi: 10.1021/jo010434s. [DOI] [PubMed] [Google Scholar]
- 13.(a) Hudric PF, Peterson D. J Am Chem Soc. 1975;97:1464–1468. [Google Scholar]; (b) Ager DJ. Synthesis. 1984:384–398. [Google Scholar]
- 14.(a) Gresham DG, Kowalski DJ, Lillya CP. J Organomet Chem. 1978;144:71–79. [Google Scholar]; (b) Dobosh PA, Gresham DG, Kowalski DJ, Lillya CP, Magyar ES. Inorg Chem. 1978;17:1775–1781. [Google Scholar]
- 15.(a) Pearson AJ, Alimardanov A, Pinkerton AA, Rouchard DM, Kirschbaum K. Tetrahedron Lett. 1998;39:5919–5922. [Google Scholar]; (b) Pearson AJ, Alimardanov AR, Kerber WD. J Organomet Chem. 2001;630:23–32. [Google Scholar]; (c) Pearson AJ, Ghidu VP. J Org Chem. 2004;69:8975–8978. doi: 10.1021/jo048557a. [DOI] [PubMed] [Google Scholar]
- 16.(a) Franck-Neumann M, Geoffroy P, Hanss D. Tetrahedron Lett. 1999;40:8487–8490. [Google Scholar]; (b) Franck-Neumann M, Geoffroy P, Hanss D. Tetrahedron Lett. 2002;43:2277–2280. [Google Scholar]
- 17.(a) Williams I, Kariuki BM, Reeves K, Cox LR. Org Lett. 2006;8:4389–4392. doi: 10.1021/ol061132l. [DOI] [PubMed] [Google Scholar]; (b) Williams I, Reeves K, Kariuki BM, Cox LR. Org Biomol Chem. 2007;5:3325–3329. doi: 10.1039/b710898b. [DOI] [PubMed] [Google Scholar]
- 18.It should be noted that ψ-exo diastereomeric dienylpiperidenes have previously been prepared by intramolecular attack of tethered carbamate nucleophiles on in situ generated pentadienyl cations: Hachem A, Teniou A, Gree R. Bull Soc Chem Belg. 1991;100:625–626.Additionally, ψ-endo diastereomeric dienylpiperidone and 5-(dienyl)-2,3-dihydropyridinone complexes have been prepared by intramolecular attack of carbon nucleophiles or by cycloaddition of Danishefsky’s diene with (dienylimine)iron complexes: Ripoche I, Bennis K, Canet JL, Gelas J, Troin Y. Tetrahedron Lett. 1996;37:3391–3392.Ripoche I, Canet JL, Gelas J, Troin Y. Eur J Org Chem. 1999:1517–1521.Takemoto Y, Ueda S, Takeuchi J, Nakamoto T, Iwata C. Tetrahedron Lett. 1994;47:8821–8824. As some of these reactions have previously been reviewed [4b,d] they are only mentioned here in the references.
- 19.Schlawe D, Majdalani A, Velcicky J, Hessler E, Wieder T, Prokop A, Schmalz HG. Angew Chem Int Ed. 2004;43:1731. doi: 10.1002/anie.200353132. [DOI] [PubMed] [Google Scholar]
- 20.Donaldson WA, Shang L, Tao C, Yun YK, Ramaswamy M, Young VG., Jr J Organomet Chem. 1997;539:87–98. [Google Scholar]
- 21.(a) Nabeta K, Oohata T, Izumi N, Katoh K. Phytochem. 1994;37:1263–1268. [Google Scholar]; (b) Nabeta K, Ishikawa T, Kawae T, Okuyama H. J Chem Soc, Chem Commun. 1995:681–682. [Google Scholar]; (c) Nabeta K, Ishikawa T, Okuyama H. J Chem Soc Perkin Trans 1. 1995:3111–3115. [Google Scholar]; (d) Kanokmedhakul S, Kanokmedhakul K, Kanarsa T, Buayairaksa M. J Nat Prod. 2005;68:183–188. doi: 10.1021/np049757k. [DOI] [PubMed] [Google Scholar]
- 22.Chaudhury S, Li S, Donaldson WA. Chem Commun. 2006:2069–2070. doi: 10.1039/b602700h. [DOI] [PubMed] [Google Scholar]
- 23.(a) Zoretic PA, Ramchandani M, Caspar ML. Syn Comm. 1991;21:915–922. [Google Scholar]; (b) Snider B. Chem Rev. 1996;96:339–364. doi: 10.1021/cr950026m. [DOI] [PubMed] [Google Scholar]
- 24.(a) Gustofson K, Roman M, Fenical W. J Am Chem Soc. 1989;111:7519–7524. [Google Scholar]; (b) Rychnovsky SD, Skalitzky DJ, Pathirana C, Jensen PR, Fenical W. J Am Chem Soc. 1992;114:671–677. [Google Scholar]
- 25.(a) Jaruchoktaweechai C, Suwanborirus K, Tanasupawatt S, Kittakoop P, Menasveta P. J Nat Prod. 2000;63:984–986. doi: 10.1021/np990605c. [DOI] [PubMed] [Google Scholar]; (b) Nagao T, Adachi K, Sakai M, Nishijima M, Sano H. J Antibiot. 2001;54:333–339. doi: 10.7164/antibiotics.54.333. [DOI] [PubMed] [Google Scholar]; (c) Kim HH, Kim WG, Ryoo IJ, Kim CJ, Suk JE, Han KH, Hwang SY, Yoo ID. J Microbio Biotech. 1997;7:429–434. [Google Scholar]
- 26.For previous synthetic studies see: Benvegnu T, Schio L, Le Floc’h Y, Gree R. Synlett. 1994;7:505–506.Tanimori S, Morita Y, Tsubota M, Nakayama M. Synth Commun. 1996;26:559–567.Benvegnu TJ, Toupet LJ, Gree RL. Tetrahedron. 1996;52:11811–11820.Benvegnu TJ, Gree RL. Tetrahedron. 1996;52:11821–11826.Gonzalez A, Aiguada J, Urpi F, Vilarrasa J. Tetrahedron Lett. 1996;49:8949–8952.Li S, Xu R, Bai D. Tetrahedron Lett. 2000;41:3463–3466.Li SK, Rui X, Xiao XS, Bai DL. Chin J Chem. 2000;18:910–923.Bonini C, Chiummiento L, Pullez M, Solladie G, Colobert F. J Org Chem. 2004;69:5015–5022. doi: 10.1021/jo049556l.Xiao X, Li S, Yan X, Liu X, Xu R, Bai D. Chem Lett. 2005;34:906–907.Bonini C, Chiummiento L, Videtta V, Colobert F, Solladie G. Synlett. 2006:2427–2430.Georgy M, Lesot P, Campagne JM. J Org Chem. 2007;72:3543. doi: 10.1021/jo0701942.Yadav JS, Gupta MK, Prathap I. Synthesis. 2007:1343–1348.Yadav JS, Kumar MR, Sabitha G. Tetrahedron Lett. 2008;49:463–466.
- 27.(a) Boyce RJ, Pattenden G. Tetrahedron Lett. 1996;37:3501–3504. [Google Scholar]; (b) Smith AB, III, Ott GR. J Am Chem Soc. 1998;120:3935–3948. [Google Scholar]; (c) Kim Y, Singer RA, Carreira EM. Angew Chem Int Ed. 1998;37:1261–1263. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1261::AID-ANIE1261>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]; (d) Marino JP, McClure MS, Holub DP, Comasseto JV, Tucci FC. J Am Chem Soc. 2002;124:1664–1668. doi: 10.1021/ja017177t. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda A, Kobayashi Y, Kimachi T, Takemoto Y. Tetrahedron. 2003;59:9305–9313. [Google Scholar]
- 29.Li S, Donaldson WA. Synthesis. 2003:2064–2068. [Google Scholar]
- 30.Godula K, Barmann H, Donaldson WA. J Org Chem. 2001;66:3590–3592. doi: 10.1021/jo001182i. [DOI] [PubMed] [Google Scholar]
- 31.Mukaiyama T, Hoshino T. J Am Chem Soc. 1960;82:5339–5342. [Google Scholar]
- 32.For an earlier synthesis of the C14–C24 segment of macrolactin A via organoiron chemistry see: Barmann H, Prahlad V, Tao C, Yun YK, Wang Z, Donaldson WA. Tetrahedron. 2000;56:2289–2295.
- 33.Le Gall T, Lellouche JP, Toupet L, Beaucourt JP. Tetrahedron Lett. 1989;47:6517–6520. [Google Scholar]
- 34.Franck-Neumann, et al. have reported that reduction of (diene)Fe(CO)3 complexes using freshly prepared Raney-nickel results in decomplexation. Franck-Neumann M, Geoffroy P, Bissinger P, Adelaide S. Tetrahedron Lett. 2001;42:6401–6404.
- 35.Evans DA, Chapman KJ, Carreira EM. J Am Chem Soc. 1988;110:3560–3578. [Google Scholar]
- 36.(a) Yun YK, Godula K, Cao Y, Donaldson WA. J Org Chem. 2003;68:901–910. doi: 10.1021/jo020421u. [DOI] [PubMed] [Google Scholar]; (b) Motiei L, Marek I, Gottlieb HE, Marks V, Lellouche JP. Tetrahedron Lett. 2003;44:5909–5012. [Google Scholar]
- 37.(a) Saberi SP, Slawin AMZ, Thomas SE, Williams DJ, Ward MF, Worthington PA. J Chem Soc, Chem Commun. 1994:2169–2170. [Google Scholar]; (b) Gibson SE, Saberi SP, Slawin AMZ, Stanley PD, Ward MF, Williams DJ, Worthington P. J Chem Soc, Perkin Trans 1. 1995:2147–2154. [Google Scholar]
- 38.(a) Shimamoto K, Ohfune Y. Synlett. 1993:919–920. [Google Scholar]; (b) Ohfune Y, Shimamoto K, Ishida M, Shinozaki H. Biorg Med Chem Lett. 1993;3:15–18. [Google Scholar]; (c) Pellicciari R, Marinozzi M, Natalini B, Constantino G, Lunei R, Giorgi Ge, Moroni F, Thomsen C. J Med Chem. 1996;39:2259–2269. doi: 10.1021/jm960059+. [DOI] [PubMed] [Google Scholar]; (d) Jullian N, Barbet I, Pin JP, Acher FC. J Med Chem. 1999;42:1546–1555. doi: 10.1021/jm980571q. [DOI] [PubMed] [Google Scholar]; (e) Mazon A, Pedregal C, Prowse W. Tetrahedron. 1999;55:7057–7064. [Google Scholar]; (f) Collado I, Pedregal C, Mazon A, Espinosa JF, Blanco-Urgoiti J, Schoeep DD, Wright RA, Johnson BG, Kingston AE. J Med Chem. 2002;45:3619–3629. doi: 10.1021/jm0110486. [DOI] [PubMed] [Google Scholar]; (g) Collado I, Pedregal C, Bueno AB, Marcos A, Gonzalez R, Blanco-Urgoiti J, Perez-Castells J, Schoeep DD, Wright RA, Johnson BG, Kingston AE, Moher ED, Hoard DW, Griffey KI, Tizzano JP. J Med Chem. 2004;47:456–466. doi: 10.1021/jm030967o. [DOI] [PubMed] [Google Scholar]
- 39.Sakai R, Suzuki K, Shimamoto K, Kamiya H. J Org Chem. 2004;69:1180–1185. doi: 10.1021/jo0355045. [DOI] [PubMed] [Google Scholar]
- 40.O’Donnell MJ. In: Encyclopedia of Reagents for Organic Synthesis. Paquette LA, editor. Vol. 1. John Wiley & Sons; New York: 1995. pp. 293–294. [Google Scholar]
- 41.Siddiquee TA, Lukesh JM, Lindeman S, Donaldson WA. J Org Chem. 2007;72:9802–9803. doi: 10.1021/jo7020604. [DOI] [PubMed] [Google Scholar]
- 42.Connor DT, Greenough RC, von Strandtmann M. J Org Chem. 1977;42:3664–3669. doi: 10.1021/jo00443a006. [DOI] [PubMed] [Google Scholar]; Just G, Potvin P. Can J Chem. 1980;58:2173–2175. [Google Scholar]
- 43.For an excellent review through 2004 see: Michelet V. Curr Org Chem. 2005;9:405–409.
- 44.(a) Kende AS, Mendoza JS, Fujii Y. J Am Chem Soc. 1990;112:9645–9646. [Google Scholar]; (b) Liu P, Jacobsen EN. J Am Chem Soc. 2001;123:10772–10773. doi: 10.1021/ja016893s. [DOI] [PubMed] [Google Scholar]; (c) Kirkland TA, Colucci J, Geraci LS, Marx MA, Schneider M, Kaelin DE, Jr, Martin SF. J Am Chem Soc. 2001;123:12432–12433. doi: 10.1021/ja011867f. [DOI] [PubMed] [Google Scholar]; (d) Lee E, Choi SJ, Kim H, Han HO, Kim YK, Min SJ, Son SH, Lim SM, Jang WS. Angew Chem Int Ed. 2002;41:176–178. doi: 10.1002/1521-3773(20020104)41:1<176::aid-anie176>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 45.Lukesh JM, Donaldson WA. Chem Commun. 2005:110–112. doi: 10.1039/b413129k. [DOI] [PubMed] [Google Scholar]
- 47.(a) Wallock NJ, Donaldson WA. Org Lett. 2005;7:2047–2049. doi: 10.1021/ol050637t. [DOI] [PubMed] [Google Scholar]; (b) Wallock NJ, Bennett DW, Siddiquee T, Haworth DT, Donaldson WA. Synthesis. 2006:3639–3646. [Google Scholar]; (c) Pandey RK, Wang L, Wallock NJ, Lindeman S, Donaldson WA. J Org Chem. 2008;73:7236–7245. doi: 10.1021/jo801446q. [DOI] [PubMed] [Google Scholar]
- 48.(a) Hudlicky T, Fan R, Reed JW, Gadamasetti KG. Org React. 1992;41:1–133. [Google Scholar]; (b) Hudlicky R, Kutchan TM, Naqvi SM. Org React. 1985;33:247–335. [Google Scholar]
- 49.Fischer NH, Olivier EJ, Fischer HD. Progress in the Chemistry of Organic Natural Products. 1979:38.Breitmaier E. Terpenes Wiley-VCH. Germany: 2006. pp. 37–39.For a recent review on the synthesis of guianolides with a trans-lactone moiety see: Schall A, Reiser O. Eur J Org Chem. 2008:2353–2364.Zhang S, Wang J, Xue H, Deng Q, Xing F, Ando M. J Nat Prod. 2002;65:1927–1929. doi: 10.1021/np0202632.Samek Z, Holub M, Drozdz B, Iommi G, Carbella A, Gariboldi P. Tetrahedron Lett. 1971:4475–4478.Daniewski WM, Danikiewics W, Gumulka M, Pankowska E, Krajewski J, Grabarczyk H, Wichlacz M. Phytochem. 1993;34:1639–1641.
- 50.Gone JR, Wallock NJ, Lindeman S, Donaldson WA. Tetrahedron Lett. 2009;50:1023–1025. [Google Scholar]
- 51.Yong W, Vandewalle M. Synlett. 1996:911–912. [Google Scholar]
- 52.Gree R, Laabassi M, Moset P, Carrie R. Tetrahedron Lett. 1985;26:2317–2318. [Google Scholar]
- 53.(a) Cotton JD, Crisp GT, Daly VA. Inorg Chim Acta. 1981;47:165–169. [Google Scholar]; (b) Cotton JD, Crisp GT, Latif L. Inorg Chim Acta. 1981;47:171–176. [Google Scholar]
- 54.(a) McDaniel KF, Kracker LR, II, Thamburaj PK. Tetrahedron Lett. 1990;31:2373–2376. [Google Scholar]; (b) Donaldson WA, Shang L, Tao C, Yun YK, Ramaswamy M, Young VG., Jr J Organomet Chem. 1997;539:87–98. [Google Scholar]; (c) Chaudhury S, Donaldson WA. J Am Chem Soc. 2006;128:5984–5985. doi: 10.1021/ja055668y. [DOI] [PubMed] [Google Scholar]; (d) Chaudhury S, Li S, Bennett DW, Siddiquee T, Haworth DT, Donaldson WA. Organometallics. 2007;26:5295–5303. [Google Scholar]
- 55.(a) Aumann R. J Am Chem Soc. 1974;96:2361–2362. [Google Scholar]; (b) Schulze MM, Gockel U. J Organomet Chem. 1996;525:155–158. [Google Scholar]; (c) Taber DF, Kanai K, Jiang Q, Bui G. J Am Chem Soc. 2000;122:6807–6808. [Google Scholar]; (d) Taber DF, Bui G, Chen B. J Org Chem. 2001;66:3423–3426. doi: 10.1021/jo001737+. [DOI] [PubMed] [Google Scholar]; (e) Taber DF, Joshi PV, Kanai K. J Org Chem. 2004;69:2268–2271. doi: 10.1021/jo0302760. [DOI] [PubMed] [Google Scholar]; (f) Taber DF, Sheth RB. J Org Chem. 2008;73:8030–8032. doi: 10.1021/jo801767n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Christie SDR, Cummins J, Elsegood MRJ, Dawson G. Synlett. 2009:257–259. [Google Scholar]