

Abstract
HIV cell entry and infection are driven by binding events to the CD4 and Chemokine receptors with associated conformational change of the viral glycoprotein, gp120. Scyllatoxin mini-protein CD4 mimetics and a small molecule inhibitor of CD4 binding, NBD-556, also effectively induce gp120 conformational change. In this study we examine the fluctuation profile of gp120 in context of CD4, a mini-protein mimetic and NBD-556 with the aim of understanding the effect of ligand binding on gp120 conformational dynamics. Analysis of Molecular Dynamics trajectories indicate that NBD-556 binding in the Phe 43 cavity enhances the overall mobility of gp120 especially in the outer-domain in comparison to CD4 or mini-protein bound complex. Interactions with the more flexible bridging sheet strengthen upon NBD-556 binding and may contribute to gp120 restructuring. The enhanced mobility of D368, E370 and I371 with NBD-556 bound in the Phe 43 cavity suggests that interactions with α3-helix in the outer-domain are not optimal, providing further insights into gp120-small molecule interactions that may impact small molecule designs.
Keywords: HIV, gp120, Cd4 binding, NBD-566, Chemokine Receptor, Molecular Dynamics, small molecule inhibitor, protein-ligand interactions, HIV entry inhibitor
HIV infection is mediated by a series of attachment events initiated by the HIV viral coat glycoprotein, gp160 which is cleaved into its two components, gp120 and gp41 (1). The two HIV glycoproteins, gp120 and gp41 are assembled as a trimer (2, 3). HIV infection in human T-cell lymphocytes occurs via binding of gp120, to the host T-cell CD4 receptor (4, 5) followed by gp120 restructuring (6, 7). This conformational change exposes on gp120 the binding site for the chemokine receptor, either CCR5 or CXCR4, thus permitting, the second obligatory binding event for viral entry (8–11). Chemokine receptor binding is followed by insertion of the gp41 fusion-peptide in the host cell membrane allowing fusion and viral entry (12–15). A number of x-ray structures of CD4 bound to gp120 have been described revealing the inner, outer and bridging sheet domains-that form a large binding cavity (16–20). Two key CD4 residues, Phe43 and Arg59, bind in the gp120 cavity (21, 22) and to Asp 368 on an adjacent α-helix, respectively. Moreover, when Phe43 and Arg59 are mutated, to Alanine, CD4 no longer binds gp120 (23). Crystal structures of several mini-protein CD4 mimetics bound to gp120 have also been solved (16) revealing a biphenyl group bound deep within the CD4 cavity. The structure of unbound form SIV gp120, which has 35% sequence identity with HIV gp120, indicates an invariant outer domain, with conformational changes occurring in both the bridging sheet and inner domain (24). Although a crystal structure of the unbound form of HIV gp120 has not been reported, the plasticity of the inner domain and bridging sheet is revealed in the structure of gp120 bound to the CD4-binding site antibody, F105 (18).
The thermodynamic signature of gp120 conformational change upon CD4 binding has been ) described (25, 26 showing a highly favorable binding enthalpy (ΔH = −63 kcal/mol) balanced with a highly unfavorable molecular ordering (−TΔS = 52 kcal/mol). A similar thermodynamic signature is exhibited by the small molecule inhibitor, NBD-556 (27, 28) (Figure 1), which induces the CD4 stimulated conformational change in a manner similar to CD4 binding and is capable of enhancing viral infection on CD4 deficient target cells (27, 28). Second generation compounds (29, 30) with improved affinity have shown to cause a rapid inactivation of the virus. Furthermore, modification of the Region III piperidine modulates the magnitude of viral infection on CD4 deficient target cells and the entropic contribution to binding affinity (31), establishing this class of compounds as a potential entry inhibitor and antiviral therapeutic agent.
Figure 1.
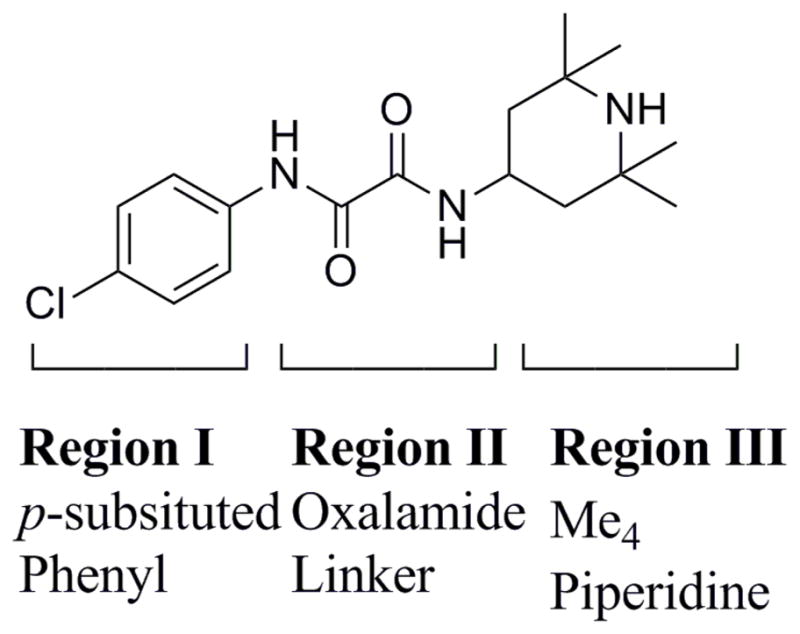
The three regions of NBD-556.
The dynamics of gp120 glycoprotein have also been examined in computational studies (32–40). Molecular Dynamics (MD) simulations of wild-type gp120 (35) showed partial unfolding of some of the β-strands while the S375W mutant preferred the bound-like conformation. Non-equilibrium steered MD (36) showed that the bridging sheet strands β-2/3 had more flexibility than the β-20/21 strands which preferred interactions with the inner domain. Da et al. demonstrated that the bridging sheet refolds for the S375A mutant but not for the W112A or S375W/T257S mutants (38). Essential dynamics studies by Liu et al (34) of unbound gp120 homology models indicated that the S375W mutation favors the CD4 bound conformation, while the I423P mutation prefers the unliganded conformation. MD studies by Hsu et al. (32, 33) indicate that there are concerted loop motions in the vestibule of the CD4 cavity, stabilization of the bridging sheet and a coalescing of the bridging sheet and V3 loop to form the co-receptor binding site. Binding entropies extracted from these MD trajectories (33) suggest that the large entropy loss associated with CD4 binding is derived from hydrophobic interactions from CD4 Phe43 insertion into the cavity, the formation of a hydrogen-bond network and the restructuring of the bridging sheet. Large scale sampling of gp120 motions with temperature accelerated MD (39) predicted a counter rotation between the inner and outer domains and a disruption of the bridging sheet in the unbound form of gp120. A flexibility index, derived using the Floppy Inclusion and Rigid Substructure Topography (37), also been used to describe the extent and distribution of flexible and rigid regions in the inner, outer and bridging sheet domains of the twenty-two gp120 structures bound with various ligands. This analysis indicated that the inner domain and bridging sheet domains are more flexible while the outer domain is more rigid and that gp120 proteins bound with CD4 exhibited less flexibility in the inner domain than when bound by a mini-protein mimetic. We previously applied the coarse-grained Gaussian Network Model (GNM) (41–44) to gp120. The study of the fluctuation profiles from GNM resulted in the identification of critical residues in the outer domain with minima that line the Phe 43 cavity and may represent a core folding nucleus found in the pre-bound form of gp120 (40). Furthermore, W427, a highly conserved residue required for CD4 binding exhibited a GNM maximum, which is surrounded by two residues, H105 and M475, of GNM minima, purported to form a key structural element that stabilizes the formation of the CD4 cavity. Determination of residue pairs with efficient communication propensities from GNM and Molecular Dynamics (MD) simulations delineated an extensive residue network in the outer-domain that is topologically suited for signal propagation from the Phe 43 cavity throughout the gp120 outer domain.
In this study we focus on MD analysis of the gp120 Phe 43 cavity in the context of binding to the CD4 receptor, a CD4 mini-protein mimetic and a small molecule ligand (NBD-556). A docked conformation of NBD-556 bound to gp120 from strain HXBC2 has been previously described implicating cavity residues important in binding (30, 31). Here we examine CD4, scyllatoxin mini-protein and NBD-556 interactions with gp120 cavity residues and their influence on residue fluctuations in the inner, outer and bridging sheet domains. We aim to understand the similarities and differences in the gp120 fluctuation profile in the presence of these three ligands in the context of gp120 structuring and the potential implication on small molecule designs.
Materials and Methods
Small Molecule Modeling
NBD-556 was constructed in MOE (MOE Molecular Operating Environment Chemical Computing Group, version 2005.06 (Montreal, Canada) (http//www.chemcomp.com), ionized using MOE’s WashMDB function, and hydrogens were added. (45) The small molecule conformation was minimized to a gradient of 0.01 in the MMFF94x (46, 47) force field using a distance-dependent dielectric constant of 1. The minimized NBD-556 was used for docking with GOLD and then in MD simulations.
Protein modeling
Two X-ray crystal structures were prepared for docking and molecular dynamic calculations: CD4-bound HIV-1 gp120 core strain YU2: PDB code 1G9N (22) and the scyllatoxin mini-protein (CD4M47A) bound HIV-1 gp120 core strain YU2: PDB code 2I5Y (16). The deglycosylated core gp120 construction used in the two crystal structures, has a 19- and a 52- amino acid residue deletions from both the N- and C- termini respectively, is devoid of the V3 variable loop and contains a tripeptide substitution (Gly-Ala-Gly) for the 67 residues of the V1/V2 loop. Residue numbering in the crystal structures and in this study conforms to the numbering used in the full-length gp120 sequence. A gap in the numbering exists from residues (127 to 194 and 296 to 330) corresponding to the substitution of the V1/V2 loops with the tripeptide and the deletion of the V3 loops. For 2I5Y, the missing V4 loop was added from the 1G9N crystal structure and minimized. Four N-terminal residues in 2I5Y (S82, E83, V84, K85)) which were not present in the 1G9N structure were deleted from the starting coordinates for consistency. Hydrogen atoms were added and tautomeric states and orientations of Asn, Gln and His residues were determined with Molprobity (http://molprobity.biochem.duke.edu/) (48, 49). Hydrogens were added to crystallographic waters using MOE (45). The OPLS-AA force field (50) in MOE (45) was used and all hydrogens were minimized to a root mean square (rms) gradient of 0.01, holding the heavy atoms fixed. A stepwise minimization followed for all atoms, using a quadratic force constant (100) to tether the atoms to their starting geometries; for each subsequent minimization, the force constant was reduced by a half until 0.25. This was followed by a final cycle of unrestrained minimization. Water molecules, Asn-linked acetyl-D-glucosamine, 2-(acetylamino)-2-deoxy-a-d-glucopyranose and small molecules were removed prior to MD calculations.
NBD-556 docking
Docking calculations with GOLD (version 3.2) (51, 52) were performed without crystallographic water molecules in the cavity. One hundred genetic algorithm (GA) docking runs were performed with the following parameters: initial_virtual_pt_match_max=3.5, diverse_solutions=1, divsol_cluster_size=1, and divsol_rmsd=1.5. All other parameters were set as defaults. The best scoring pose was used as a starting conformation in MD simulations.
Cavity Analysis
The MOE (45) site finder utility was used for cavity analysis and calculation of volume for starting and simulated coordinates.
Molecular dynamics simulations
A preliminary energy minimization, followed by a short molecular dynamics simulation in an explicitely solvated environment, is expected to relax the x-ray structure, driving it closer to its true minimum. We thus performed MD simulations of solvated GPO3, GCD2, GCD3 and GPO3_NBD structures (Table 2), and analyzed the time evolution of their dynamics. Thirty nanosecond (ns) simulation trajectories were generated, using GROMACS (53, 54) in a fully solvated environment, at 310K and under constant number of particles, pressure and temperature (NPT) conditions.
Table 2.
Coordinate Sets and Ligands used in MD Studies
| Coordinate set | PDB Code | Ligand | Total Atoms | Production Run |
|---|---|---|---|---|
| GCD2 | 1G9N | CD4 | 94345 | 30ns |
| GCD3 | 2I5Y | CD4M47-biphenyl | 95047 | 30ns |
| GPO3_NBD | 2I5Y | NBD-556 docked | 71019 | 30ns |
| GPO3 | 2I5Y | None | 71007 | 30ns |
Molecular dynamics simulation trajectories were generated using Gromacs software package version 3.3.1(53, 54), using the GROMOS 43a1 force field and the SPC water model (55) for solvation. The solvated protein molecule was initially energy minimized with 1000 steps of steepest descent, followed by a short equilibration run of 200 picoseconds, during which the protein backbone atom were position restrained and the side chain atoms and water molecules were allowed to relax. During the production run of 30ns, the position restraints were removed.. A dielectric permittivity, ε=1 and timestep of 2 femtoseconds was used. The LINCS (56) algorithm was used to constrain all bond lengths and the Particle Mesh Ewald (PME) method (57) to compute the electrostatic term. In all simulations, the temperature was kept constant (T=310K) by coupling to an external temperature bath with a coupling constant of 0.1 ps, and isotropic pressure coupling was employed to maintain a constant pressure of 1bar.
Results and Discussion
Two crystal structure complexes from the HIV YU2 strain were used to form the basis of this study: the CD4, 17b anti-body, gp120 complex (1G9N) (22) and the CD4M47A scyllatoxin, 17b antibody, gp120 complex (2I5Y) (16), Figure 2, A–B. The gp120 coordinates from the CD4M47 scyllatoxin bound complex were used to dock NBD-556 to the Phe 43 cavity (Figure 2C). Previously published structure activity relationships combined with docking and mutagenesis experiments indicate that the NBD-556 chlorophenyl group binds at the bottom of the Phe 43 cavity while the tetramethylpiperidine is positioned in the Phe 43 cavity vestibule (30, 31). The docked model predicts cavity lining residues that are within interaction distance to NBD-556 (less than 0.5 nm), Table 1. Also listed in Table 1 are characteristics of cavity residues, such as whether they are conserved, display GNM slow mode minimum or maximum or have efficient communication propensities with nearby residues. The docked NBD-556–gp120 complex was used as a starting point in MD simulations and is abbreviated as GPO3_NBD. MD simulations were used to elucidate the fluctuation profiles of these residues in comparison to the three other coordinate sets, the CD4 -gp120 complex, with Phe 43 bound in the cavity (GCD2, pdb code 1G9N), the CD4M47 -gp120 complex, with biphenyl bound in the cavity (GCD3, pdb code 2I5Y) and gp120 in the CD4 bound form but without ligand bound in cavity during simulation. (GP03, pdb code 2I5Y), Table 2. MD simulation with GROMACS (53, 58) produced stable 30 nanosecond (ns) trajectories for the four coordinate sets (Figure 3). As we have previously reported, GNM and MD produced similar fluctuation profiles for receptor and mini-protein bound gp120 (40). Here, Root Mean Square Fluctuation (RMSF) from the average structure (averaged over 30ns simulation trajectory for each amino-acid residue (C-α) show similar profiles between the four coordinate sets for the majority of gp120 residues (Figure 4) However, in general, the fluctuations are enhanced, i.e. peak height, when the Phe 43 cavity is occupied by NBD-556, (Figure 4). Individual RMSF difference plots among the bound gp120 systems compared to the unbound GPO3 also indicate that the complexes with the deepest cavity binding ligands have dynamics that are enhanced compared to GPO3 Figure S1. When RMSF’s are mapped to the gp120 fold, a picture emerges that with NBD-556 bound in the cavity the dynamics of the gp120 are accentuated, Figure 5. The two regions of largest enhancement in backbone fluctuation when NBD-556 is bound are residues 364–367 (located adjacent to helix α-3 in the outer domain) and 459–463 (V5 loop in the outer domain), (Figure 5D) compared to bound CD4 (Figure 5B, Figure S1B) or the mini-protein (Figure 5C). Residues 364–367, adjacent to the cavity, are within van der Waals contact of the β-strand from CD4M47 mini-protein or CD4 residues and a hydrogen bond is formed between the backbone of D368 and the mini-protein or CD4. Residues 459–463 are quite distant from the cavity, do not form inter-protein contacts with CD4M47 and only sparingly with CD4. Enhanced mobility in loop V5 (residues 459–463) in GPO_NBD (Figure 5D) can be attributed to the inherent flexibility in this loop as has also been observed in various crystal structures (16, 22, 58), whereas enhanced RMSF for residues 364–367 appears ligand dependent.
Figure 2.

The four constructs used in MD simulation are shown in panels A – D with the three gp120 domains; inner, outer and bridging sheet colored as red, yellow and green ribbons, respectively. A) The gp120-CD4 complex (GCD2) with CD4 shown in blue with Phe 43 depicted as space-filling model. B) The gp120-CD4M47 complex (GCD3) with the scorpion toxin derived mini-protein shown in orange with the residue 23 biphenyl depicted as space-filling model. C) NBD-556 docked to gp120 (GPO3_NBD) with NBD-556 shown as purple space filling model. D) the unbound gp120 structure (GPO3) E) Comparison of the NBD-556 (purple), biphenyl (orange) and Phe-43 (cyan) as bound in the cavity gp120 for GPO3_NBD, GCD3 and GCD2, respectively.
Table 1.
gp120 Residues within 5.0 Å of Cavity Binding Ligands
| Domain | GPO3-NBD- 556 | GCD3 Biphenyl | GCD2 Phe 43 | GPO3 GNM Profile | GPO3 residue CP < 0.20 |
|---|---|---|---|---|---|
| Inner | H105 | Min. | |||
| Inner | W112 | W112 | |||
| Inner | V255 | V255 | Min. | ||
| Outer | S256 | S256 | Min. | L260, S375 I371, H374 S375, |
|
| Outer | T257 | T257 | T257 | Min. | F376 |
| Outer | D368 | D368 | D368 | ||
| Outer | E370 | E370 | E370 | Min. | |
| Outer | I371 | I371 | I371 | Min. | H374 F382,F383,Y384, |
| Outer | S375 | S375 | Min. | C385 | |
| Outer | F376 | F376 | Min. | F382, F383, Y384 | |
| Outer | N377 | N377 | Min. | F382, F383 | |
| Outer | F382 | F382 | |||
| Outer | Y384 | Y384 | |||
| Bridging Sheet | I424 | I424 | Max. | C418, I420 | |
| Bridging Sheet | N425 | N425 | N425 | Max. | |
| Bridging Sheet | M426 | M426 | M426 | Max. | |
| Bridging Sheet | W427 | W427 | W427 | Max. | |
| Bridging Sheet | N428 | Max. | |||
| Bridging Sheet | E429 | Max. | |||
| Bridging Sheet | V430 | Max. | |||
| Bridging Sheet | G431 | Max. | |||
| Outer Domain | G472 | Max. | |||
| Outer Domain | G473 | G473 | G473 | Min. | |
| Outer Domain | D474 | D474 | D474 | Min. | |
| Inner Doman | M475 | M475 | M475 | Min. | |
Residues conserved in all primates viruses, all human viruses and moderately in human viruses are indicated in bold, underlined and italic fonts, respectively. Residues with Gaussian Network Model calculated slow mode maximum and minimum as well as residues pairs with efficient communication propensity (CP <0.20) are also listed.
Figure 3.
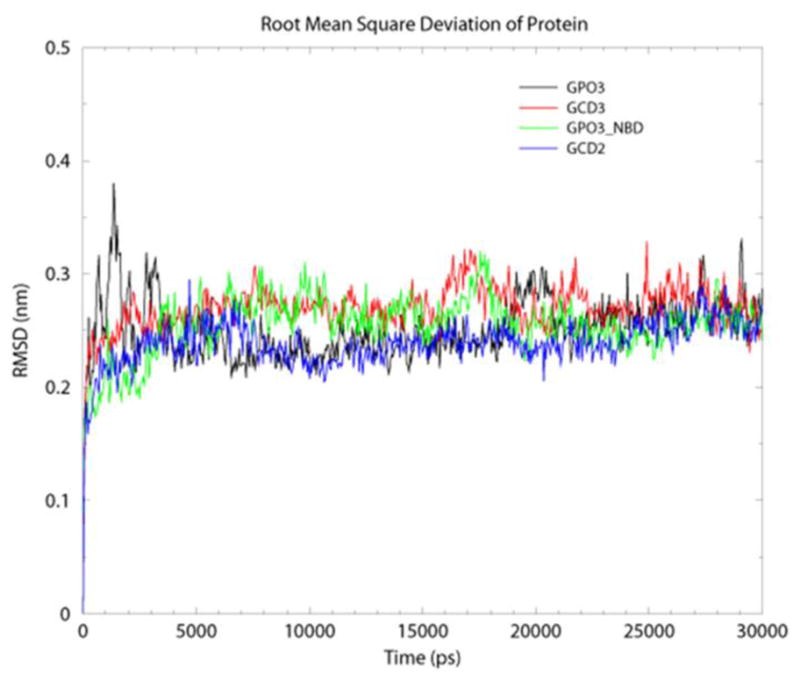
Calculated RMSDs for backbone C-α atoms from the starting structure for the four constructs, GPO3 (black), GCD2 (red), GCD3 (blue) and GPO3_NBD (green). The Mean RMSDs and standard deviations in nm for the four complexes, GPO3, GCD2, GCD3 and GPO3_NBD are; 0.25 (0.03), 0.24 (0.02), 27 (0.02) and 0.25 (0.02), respectively.
Figure 4.
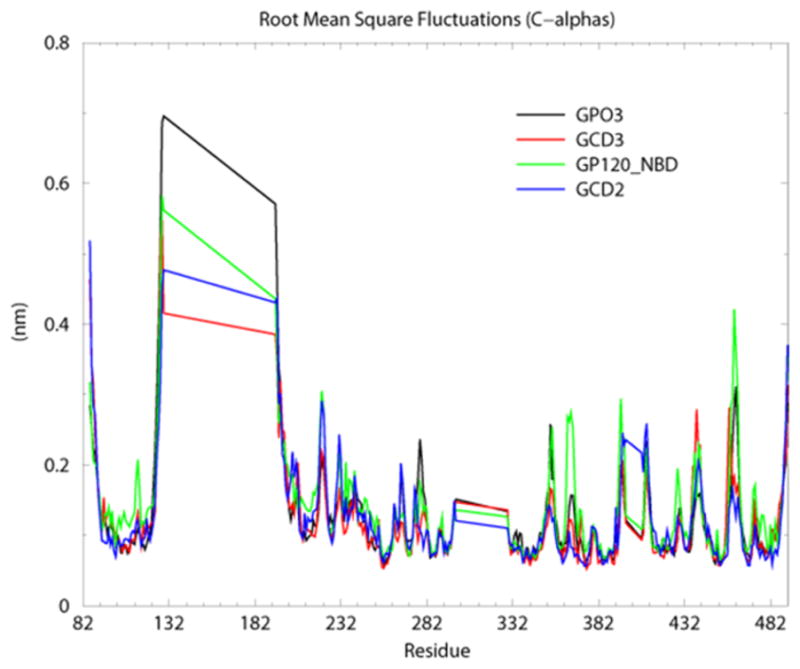
Calculated root mean square fluctuations (RMSFs) from the average, for gp120 residue backbone C-α atoms for the four constructs, GPO3 (black), GCD2 (red), GCD3 (blue) and GPO3_NBD (green). Residue numbering for the core gp120 crystal structures maintains the numbering from the full-length gp120 sequence (residues 82–492). A gap in the numbering exists from residues (127 to 194 and 296 to 330) corresponding to the substitution of the V1/V2 loops with the tripeptide and the deletion the V3 loops. The peaks at residue 127 and 296 correspond to these gaps.
Figure 5.

The calculated root mean square fluctuations (RMSFs) for gp120 residue backbone C-α atoms mapped to ribbon diagram for the four constructs: A) GPO3, B) GCD2, C) GCD3 and D) GPO3_NBD. High to low RMSFs are colored from blue, red, orange, yellow, green, respectively.
To better understand overall fluctuation patterns in the Phe 43 cavity we choose to first analyze the behavior of several residues of moderate to high sequence conservation in Phe 43 cavity (I109, P124, D368, W427, V430) and their interacting residue partners for the four simulation trajectories. The inter-residue distances over the course of the simulation (Supplementary, Figure S2) indicate that there is a tight interaction (the distance between the center of mass of the interacting pairs of residues is less than ~0.3 nm and exhibits low fluctuation) between F210-W112, W427-I109, D113-K117 and K432-L122 residue pairs while V430-T123, V430-P124, K432-D368 are loosely associated (the distance greater than 0.3nm and exhibits high fluctuation). We observe that for the gp120-CD4 complex (GCD2) V430-T123 (Figure S2C black curve) is a rather loose association (distance increases to greater than 0.5nm from 5ns to 15ns), while in the presence of NBD-556 (GPO3_NBD) this association (Figure S2D, black curve) strengthens (distance decreases to less than 0.25nm from 15ns onwards) over the course of the simulation. Conversely, W427-I109 is stable (distance less than 0.3nm) in the CD4, CD4M47 and unliganded constructs GCD2 (Figure S2C, green curve). GCD3 (Figure S2B green curve), GPO3 (Figure S2A green curve), respectively, while in GPO3_NBD (Figure S2D, green curve) this interaction weakens (distance greater than 0.3 nm from 20ns onwards) during the course of the simulation. This weakening of the interactions is characterized by movement of W427 away from the Phe 43 cavity with an accompanying shift in neighboring inner domain residues H105, M475, W112 (Supplementary Figure S3).
We also monitored the distances between the loops surrounding the Phe 43 cavity to ascertain cavity dynamics over the course of the simulation. Nodes were defined by the center of mass of residues G473-M475, G367-P369, G379-E381, and V127-A129 corresponding to turns or loops between β-strands, β-24/β-25, β-14/β-15, β-16/β-17 and the V1/V2 stem, respectively, Figure 6. The inter-node distances to the residue pair W427-E429 on β-20/21 was chosen since W427 is a highly conserved residue and is predicted to interact with the NBD-566 p-chloro-phenyl group. These distances for the four gp120 coordinate sets are plotted in Figure 6. Significant fluctuation of the V1/V2 stem in GPO3, GCD3 and GPO3_NBD is observed (Figure 6A, C and D) while more overall cavity instability is seen in GCD3 (Figure 6C) and GPO3_NBD (Figure 6D) compared to GPO3 and GCD2 (Figure 6A–B). Of note, GCD3-biphenyl and GPO3_NBD ligands are more deeply bound in the cavity compared to GCD2 (Figure 2E). This cavity instability is consistent with the conclusion drawn from root mean square fluctuations in Figure 4 that NBD-556 bound in the gp120 cavity enhances over all gp120 mobility. Although dynamics were enhanced overall, significant changes in Phe 43 cavity shape and volume did not occur when comparing GPO3 (790 Å3) and GPO3_NBD (809Å3) coordinates after 30 ns of MD simulation.
Figure 6.

The inter-node distance to the residue pair W427-E429 (purple) on β-20/21 was measured (nm) for the four constructs: A) GPO3, B) GCD2, C) GCD3 and D) GPO3_NBD for the residues (E) G473-M475(red), G367-P369 (blue), G379-E381 (green), and V127-A129 (black) corresponding to turns or loops between β-strands, β-24/β-25, β-14/β-15, β-16/β-17 and the V1/V2 stem, respectively.
To follow-up the observation of enhanced cavity mobility we examined the extent of NBD-556 fluctuation as predicted to bind in the cavity of GPO3_NBD during the simulation. The RMSDs of the three functional regions of NBD-556, as defined in Figure 1, do not fluctuate significantly over the 30 ns time frame (Supplementary Figure S4). However, it is apparent that the Region II oxalamide stem displays larger RMSDs during the simulation (we note that only one tautomer for the Region II oxalamide in Figure 1 was considered here as there is no experimental evidence suggesting another active tautomer). The residue RMSDs are the deviation from the starting conformation of all residue atoms as a function of time. Such an analysis at the residue level, allows examination of the stability (or flexibility) of residues in a localized region. We thus examined residue RMSDs to delineate the dynamics of those residues which are predicted to be within interacting distance (less than 0.5 nm) (Table 1) of the bound NBD-556 as indicated in GPO3_NBD (Figure 7) and in GPO3, GCD2 and GCD3 (Supplementary Figure S5, S6 and S7 respectively). Inspection of Figure 7 indicates that cavity residues, S256, N377, I424, N425, G431, G472 and G473 have low mean RMSD’s (less than 0.05 nm). Of these, the RMSD’s of residues N377, G431, G472, and G473 also have low standard deviations (less than 0.02 nm) (Supplementary Table 1). We can attribute these residues with low RMSDs to maintaining the structural integrity of the Phe 43 cavity in the presence of NBD-556. Two other residues T257, S375, transitioned to a different conformation from the starting structure, as evidenced from the jump in the RMSD (Figure 7A) and remained stable at the new conformation for the duration of the simulation with a low standard deviation of 0.019 and 0.004 respectively (Supplementary Table S1). An inspection of the starting and final structure revealed that the change in the residue conformation, facilitated interaction of these two residues with the NBD-556 aromatic group, simultaneously. This phenomenon is not observed in GPO3 and GCD2 for residues T257 and S375 which display stable trajectories, while in GCD3, T257 and S375 exhibit highly fluctuating positions. Several other residues I371 (Figure 7A), W427 and Q428 (Figure 7B) exhibit distinct substates throughout the GPO3_NBD simulation. Residues W427 and Q428 reside on the turn of β-20/β-21 on the bridging sheet. Furthermore, W427 is predicted to form an aromatic-aromatic stacking interaction with the p-chloro-phenyl group of Region I. Inspection of the first frame and last frame indicates that at later stages of simulation the NBD-556 p-chloro-phenyl group rotates and that W427 may adjust its position to maintain the stacking interaction. Inspection of I371, W427, and Q428 RMSDs calculated from the simulations trajectories (Supplementary Figures S5, S6 and S7 ) for GPO3, GCD2 and GCD3 does not show distinct states for these residues and only in the case of GCD2 is an overall increase in fluctuation observed for W427 and Q428.
Figure 7.

RMSDs for the 25 residues that form the Phe 43 cavity of GPO3_NBD and are within 4.5 Å of NBD-556 as predicted by docking. A) Residues are: H105, W112, V255, S256, T257, D368, E370, I371, S375 B) Residues are: F376, N377, F382, Y384, I424, N425, M426, W427, Q428, E429 C) Residues are: V430, G431, G472, G473, D474, M475 D) Residues surrounding NBD-556 in the Phe 43 cavity vestibule that exhibit larger fluctuations during simulation.
The remaining residues, D368, E370, M426, E429, V430, D474 and M475, exhibit the highest degree of fluctuation (RMSD standard deviation > 0.02 (Supplementary Table 1). These residues are located at the rim of cavity vestibule (Figure 7D) within 4.5 Å of region III tetra-methyl piperidine. Three of these, D368, E370, and D474 are from the outer domain and demonstrated slow mode minimia in GNM analysis of GCD2, GCD3 and GPO3 (40). (The GNM studies did not include a gp120-NBD-556 complex because the small molecule is inadequately represented in the Gaussian Network Model.) Thus three dynamics states, stable, transitional and highly mobile, are exhibited by residues that form the Phe 43 cavity when bound by the NBD-556 ligand.
We also analyzed the extent for which these twenty-five residues have coordinated fluctuations with NBD-556 by measuring the pair wise residue distances with NBD region I, II and III as shown in Supplementary Figures S8, S9 and S10. Interactions distances of less than 0.40 nm (minimum distance between the center of mass of the respective NBD region and the residue) and with low standard deviations reflect tight interactions. Residues with tight interaction for Region I are V255, S256, T257, S375, F376, F382, I424, N425, W427; for Region II are N425, M426, W427, G473, D474 and Region III are M426, W427, E429, V430, G473, D474 and M475. Those residues with the tightest NBD-556 interactions and lowest residue RMSDs are S256, N377, I424, N425 and G473, all from the outer domain suggesting that these residues-NBD-556 interactions provide the maximum stabilization to the protein-ligand complex. Those residues that have higher RMSDs but tight NBD-556 interactions are F376, F382, M426, W427, Q428, E429, V430, D474 and M475, suggesting these residues have relaxed away from the corresponding crystal structure positions, allowing a more favorable interaction with the bound NBD-556 molecule. With the exception of F376 and F382, all reside in the bridging sheet domain. Given that the bridging sheet region is highly flexible and displays alternate conformations in several gp120-antibody crystal structures (59), tight NBD-556 interactions with these same residues can be postulated to be in important in driving NBD-556 structuring of gp120.
While some residues which were initially within interacting distance of the bound NBD-556 (distance < 0.45 nm) maintained their interactions (V255, S256, T257, S375, F376, F382, I424, N425, W427, M426, G473, D474, E429 and V430 (Supplementary Table S1), other residues moved away from NBD-556, during the course of the simulation. Of note are residues with large pair-wise distances (> 0.6 nm) to NBD-556 during MD simulation when docking predicted interactions less than 0.45 nm. Residue D368 exhibits an average pair-wise distance of 0.7 nm with Region III with significant fluctuation during the simulation. Nearby residues I371, E370 also display large mean pair-wise distance to NBD-556 Region III, 0.78 nm and 1.07 nm, respectively. Thus, Region III contact with outer domain residues in the vicinity of D368, E370 and I371 are not stabilized by NBD-556 binding. Most of the residues interacting with NBD-556 regions I and II also have a low standard deviation (<= 0.05 nm), while those interaction with region III, have a higher standard deviation (>= 0.05 nm). This suggests that the residues interacting with the NBD-556 molecule are not rigid, but have local flexibility, allowing the bound NBD-556 molecule some flexibility as well.
To verify NBD-556 enhancement of cavity dynamics when docked in the Phe 43 cavity, residue RMSDs are compared to GPO3, GCD2 and GCD3, Figure 8 and Supplementary Figures S5–S7. Overall, residues with the largest RMSDs in GPO3_NBD also exhibit larger RMSDs in GPO3, GCD2, and GCD3 throughout the simulations. However, for residues D368, E370, I371 the magnitude and type of fluctuation differs among the four simulation sets. GCD3 and GPO3_NBD have similar RMSDs for D368 and E370 and I371, while only GPO3_NBD exhibits distinct dynamic states for I371. Moreover, in GCD2, D368 dynamics are accentuated (mean RMSD 0.12 nm) while E370 and I371 fluctuations are substantially reduced (mean RMSDs 0.04). A second area of differing dynamics is W427 and Q428 (mean RMSD of 0.13 nm and 0.10 nm, respectively) on the bridging sheet. This contrasts with moderate dynamics with transitional states for W427 and Q428 in GPO3_NBD and very stable dynamics for these residues in GPO3 and GCD2. Greater fluctuations in the bridging sheet for M426, E429, V430 and D474 as exhibited in the GPO3_NBD simulation are similarly observed for GPO3, GCD2, and GCD3. Thus, MD simulations of GPO3_NBD in comparison with GPO3, GCD2 and GCD3 show differential dynamics for both outer domain (D368, E370, I317) and bridging sheet residues (W427 and Q428) in the Phe 43 cavity.
Figure 8.

The mean residue RMSDs for the twenty residues in the Phe 43 cavity within 5.0 Å of the docking binding mode of NBD-556 for each simulation complex, GPO3 (blue), GCD2 (yellow), GCD3 (green), GPO3_NBD (red).
Conclusions
In this study we compare the fluctuation dynamics of the gp120 core (residues 86–492) when bound to its cognate ligand (CD4 receptor), a CD4 mini-protein mimetic derived from the scyllatoxin scaffold (CD4M47), a small molecule ligand (NBD-556) and without ligand bound, but in the CD4 bound structured form. The docked binding mode of NBD-556 with p-chloro-phenyl group bound most deeply in the Phe 43 cavity is consistent with mutagenesis experiments (30) and SAR studies (31) thus providing the rationale for further computational studies using this model.
As a docked model of NBD-556 bound to gp120 is used rather than a crystal structure, trends in weakening and strengthening of specific protein-ligand interactions can be interpreted as an adaptation of the MD relaxed complex to a form closer to the native state. Nonetheless, the overall dynamics are revealing and consistent with previous studies. Molecular dynamics studies have shown that the bridging sheet is highly mobile and that two strands β-2/3 and β-20/21 have distinct dynamics (35). In the context of NBD-556, interactions with W427 are tight and intra-bridging sheet contacts as measured between V123 and W427 strengthen, while interactions between the bridging sheet and inner domain weaken consistent with the counter rotation between the inner and outer domain and disruption of the bridging sheet observed in computations by Abrams (39) and Tan (37).
While NBD-556, has a stable fluctuation profile the simulations indicate that its presence in the Phe 43 cavity enhances the overall mobility of the gp120 envelop protein in comparison to the CD4, mini-protein bound and uncomplexed forms. Evaluation of gp120 intra-protein interactions and cavity stability indicated that in response to NBD-556 binding the bridging sheet β-2/β-3 interaction strengthens and the interface between inner domain and bridging sheet weakens and is less stable. Overall, individual RMSDs reveal three dynamic states for cavity residues; stable, transitional and highly mobile depending on the cavity filling capacity of the ligand. At the cavity bottom, residues T257 and S375 have notably increased dynamics with biphenyl (GCD3) and NBD-556 (GPO3_NBD) bound, compared to GCD2 with Phe43 in the cavity. At the cavity vestibule the largest enhancement is observed for in residues 364–368 and 370-and 371. Enhancement of residues 364–368 dynamics in this region is not surprising as there is no compensating D368- CD4-R59 interaction predicted to form in the NBD-556-gp120 complex to stabilize the α3-helix. Indeed, the lack of gp120 binding for CD4-R59A (23) mutant indicates that this is an important region of protein-ligand interaction. The concurrent enhancement of E370 and I371 dynamics however, was also unexpected, as both residues are well conserved and were thought to form a stable surface on the outer domain as assessed by GNM studies (40). Moreover, in the absence of the D368- CD4 R59 stabilization several dynamics states are observed for these two residues, in the case of GPO3_NBD. In summary, residues with low RMSD and tight interactions with Regions I, II, III in the outer domain (S256, I424, N425, and G473) may provide the most stabilization to the protein-ligand complex. Conversely, those residues with larger RMSDs but tight NBD-556 interactions (S375, F382, M426, W427, Q428, E429, V430, D474 and M474) may represent residues and regions that contribute to NBD-556 induced gp120 structuring.
The delineation of cavity dynamics in the context of NBD-556 binding suggests that tight interactions with the bridging sheet are important and drive structuring. Interactions in the outer-domain with residues 472–475 between β-24 and the α5-helix in the inner domain may dominate binding rather than interactions with α3-helix and D368. Overall enhancement of gp120 dynamics may result from the small molecule ligand binding more deeply in the Phe 43 cavity and/or a lack of stabilizing protein-protein interactions in the absence of protein ligand (cf CD4 or CD4 mini-protein mimetic). The thermodynamic signature of NBD-556 while similar to CD4 has diminished enthalpic and entropic terms (ΔH= −24.5 kcal/mol and TΔS = −57.4 kcal/mol) when compared to CD4 (ΔH= −34.5 kcal/mol and TΔS = −79 kcal/mol) (27, 28). The enhanced dynamics observed in this study provides an indication for which gp120 residues may be contributing to less efficient binding of NBD-556 in comparison to the CD4 receptor. A previous report has noted that several NBD analogues have differential effects on gp120 structuring and viral enhancement when the NBD Region III is modified (31) and that efficient inhibition of CD4-gp120 binding may be obtained when unwanted triggering of gp120 conformational change is eliminated. Results from this MD study, in the absence of protein-ligand crystal structure, provide insights on which residues and interactions may be responsible for this inducing gp120 structuring and suggests which interactions may have the most impact on future small molecule designs.
Supplementary Material
Acknowledgments
FUNDING: This work was supported by a grant from the National Institutes of Health GM56550 (JL) and support from NIH grant 1U54GM087519-01A1 to Ivet Bahar’s lab is gratefully acknowledged by IS.
The authors appreciate the useful suggestions and discussions with Navid Madani and Ivet Bahar. Support from NIH grant 1U54GM087519-01A1 to Ivet Bahar’s lab is gratefully acknowledged by IS.
This work was supported by a grant from the National Institutes of Health GM56550 (JL).
ABREVIATIONS
- MD
Molecular Dynamics
- HIV
Human immunodeficiency virus
- GNM
Gaussian Network Model
- RMSF
root mean square fluctuations
- RMSD
root mean square deviations
- SAR
structure activity relationships
Footnotes
Supporting Information Available
Supplementary Figures S1-9 and table 1S are available free of charge via the internet at http://pubs.acs.org.
References
- 1.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987;237:1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 2.Blacklow SC, Lu M, Kim PS. A trimeric subdomain of the simian immunodeficiency virus envelope glycoprotein. Biochemistry. 1995;34:14955–14962. doi: 10.1021/bi00046a001. [DOI] [PubMed] [Google Scholar]
- 3.Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 4.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 5.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 6.Sattentau QJ, Moore JP. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J Exp Med. 1991;174:407–415. doi: 10.1084/jem.174.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sattentau QJ, Moore JP, Vignaux F, Traincard F, Poignard P. Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J Virol. 1993;67:7383–7393. doi: 10.1128/jvi.67.12.7383-7393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 9.Deng WP, Zhong M, Guo XC, Kende AS. Total Synthesis and Structure Revision of Stachybotrys Spirolactams. J Org Chem. 2003;68:7422–7427. doi: 10.1021/jo030202o. [DOI] [PubMed] [Google Scholar]
- 10.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 11.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 12.Bosch ML, Earl PL, Fargnoli K, Picciafuoco S, Giombini F, Wong-Staal F, Franchini G. Identification of the fusion peptide of primate immunodeficiency viruses. Science. 1989;244:694–697. doi: 10.1126/science.2541505. [DOI] [PubMed] [Google Scholar]
- 13.Brasseur R, Cornet B, Burny A, Vandenbranden M, Ruysschaert JM. Mode of insertion into a lipid membrane of the N-terminal HIV gp41 peptide segment. AIDS Res Hum Retroviruses. 1988;4:83–90. doi: 10.1089/aid.1988.4.83. [DOI] [PubMed] [Google Scholar]
- 14.Helseth E, Olshevsky U, Gabuzda D, Ardman B, Haseltine W, Sodroski J. Changes in the transmembrane region of the human immunodeficiency virus type 1 gp41 envelope glycoprotein affect membrane fusion. J Virol. 1990;64:6314–6318. doi: 10.1128/jvi.64.12.6314-6318.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins fusogens antigens and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 16.Huang CC, Stricher F, Martin L, Decker JM, Majeed S, Barthe P, Hendrickson WA, Robinson J, Roumestand C, Sodroski J, Wyatt R, Shaw GM, Vita C, Kwong PD. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure. 2005;13:755–768. doi: 10.1016/j.str.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Kwon YD, Zhou T, Wu X, O’Dell S, Cavacini L, Hessell AJ, Pancera M, Tang M, Xu L, Yang Z-Y, Zhang M-Y, Arthos J, Burton DR, Dimitrov DS, Nabel GJ, Posner MR, Sodroski J, Wyatt R, Mascola JR, Kwong PD. Structural Basis of Immune Evasion at the Site of CD4 Attachment on HIV-1 gp120. Science. 2009;326:1123–1127. doi: 10.1126/science.1175868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diskin R, Marcovecchio PM, Bjorkman PJ. Structure of a clade C HIV-1 gp120 bound to CD4 and CD4-induced antibody reveals anti-CD4 polyreactivity. Nat Struct Mol Biol. 2010;17:608–613. doi: 10.1038/nsmb.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, Kwon YD, Scheid JF, Shi W, Xu L, Yang Y, Zhu J, Nussenzweig MC, Sodroski J, Shapiro L, Nabel GJ, Mascola JR, Kwong PD. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwong PD, Wyatt R, Majeed S, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure Fold Des. 2000;8:1329–1339. doi: 10.1016/s0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 23.Moebius U, Clayton LK, Abraham S, Harrison SC, Reinherz EL. The human immunodeficiency virus gp120 binding site on CD4: delineation by quantitative equilibrium and kinetic binding studies of mutants in conjunction with a high-resolution CD4 atomic structure. J Exp Med. 1992;176:507–517. doi: 10.1084/jem.176.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 25.Dey B, Pancera M, Svehla K, Shu Y, Xiang SH, Vainshtein J, Li Y, Sodroski J, Kwong PD, Mascola JR, Wyatt R. Characterization of human immunodeficiency virus type 1 monomeric and trimeric gp120 glycoproteins stabilized in the CD4-bound state: antigenicity, biophysics, and immunogenicity. J Virol. 2007;81:5579–5593. doi: 10.1128/JVI.02500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myszka DG, Sweet RW, Hensley P, Brigham-Burke M, Kwong PD, Hendrickson WA, Wyatt R, Sodroski J, Doyle ML. Energetics of the HIV gp120-CD4 binding reaction. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9026–9031. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schon A, Madani N, Klein JC, Hubicki A, Ng D, Yang X, Smith AB, 3rd, Sodroski J, Freire E. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry. 2006;45:10973–10980. doi: 10.1021/bi061193r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339:213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Haim H, Si Z, Madani N, Wang L, Courter JR, Princiotto A, Kassa A, DeGrace M, McGee-Estrada K, Mefford M, Gabuzda D, Smith AB, 3rd, Sodroski J. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog. 2009;5:e1000360. doi: 10.1371/journal.ppat.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madani N, Schon A, Princiotto AM, Lalonde JM, Courter JR, Soeta T, Ng D, Wang L, Brower ET, Xiang SH, Kwon YD, Huang CC, Wyatt R, Kwong PD, Freire E, Smith AB, 3rd, Sodroski J. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure. 2008;16:1689–1701. doi: 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaLonde JM, Elban MA, Courter JR, Sugawara A, Soeta T, Madani N, Princiotto A, Kwon YD, Kwong PD, Schön A, Freire E, Sodroski J, Smith AB., III Design, synthesis and biological evaluation of small molecule inhibitors of CD4-gp120 binding based on virtual screening. Bioorganic & Medicinal Chemistry. 2011;19(1):91–101. doi: 10.1016/j.bmc.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu STD, Bonvin AMJJ. Atomic insight into the CD4 binding-induced conformational changes in HIV-1 gp120. Proteins: Struc Funct & Bioinformatics. 2004;55:582–593. doi: 10.1002/prot.20061. [DOI] [PubMed] [Google Scholar]
- 33.Hsu S-TD, Peter D, van Gunsteren WF, Bonvin AMJJ. Entropy Calculation of HIV-1 Env gp120, its Receptor CD4, and their Complex: An Analysis of Configurational Entropy Changes upon Complexation. Biophysical Journal. 2005;88:14–24. doi: 10.1529/biophysj.104.044933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu SQ, Liu SX, Fu YX. Molecular motions of human HIV-1 gp120 envelope glycoproteins. J Mol Model. 2008;14:857–870. doi: 10.1007/s00894-008-0327-7. [DOI] [PubMed] [Google Scholar]
- 35.Pan Y, Ma B, Keskin O, Nussinov R. Characterization of the conformational state and flexibility of HIV-1 glycoprotein gp120 core domain. The Journal of biological chemistry. 2004;279:30523–30530. doi: 10.1074/jbc.M404364200. [DOI] [PubMed] [Google Scholar]
- 36.Pan Y, Ma B, Nussinov R. CD4 binding partially locks the bridging sheet in gp120 but leaves the beta2/3 strands flexible. Journal of molecular biology. 2005;350:514–527. doi: 10.1016/j.jmb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Tan H, Rader AJ. Identification of putative, stable binding regions through flexibility analysis of HIV-1 gp120. Proteins. 2009 doi: 10.1002/prot.22196. [DOI] [PubMed] [Google Scholar]
- 38.Da LT, Quan JM, Wu YD. Understanding of the Brdiging Sheet Formation of HIV-1 Glycoprotein gp120. J Phys Chem B. 2009;113:14536–14543. doi: 10.1021/jp9081239. [DOI] [PubMed] [Google Scholar]
- 39.Abrams CF, Vanden-Eijnden E. Large-scale conformational sampling of proteins using temperature-accelerated molecular dynamics. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4961–4966. doi: 10.1073/pnas.0914540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shrivastava I, LaLonde JM. Fluctuation dynamics analysis of gp120 envelope protein reveals a topologically based communication network. Proteins: Structure, Function, and Bioinformatics. 2010;78:2935–2949. doi: 10.1002/prot.22816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bahar I, Atilgan AR, Demirel MC, Erman B. Vibrational dynamics of folded proteins: significance of slow and fast modes in relation to function and stability. Phys Rev Letters. 1998;80:2733–2736. [Google Scholar]
- 42.Bahar I, Jernigan RL. Vibrational dynamics of transfer RNAs: a comparison of the free synthetase bound forms. J Mol Biol. 1998;281:871–884. doi: 10.1006/jmbi.1998.1978. [DOI] [PubMed] [Google Scholar]
- 43.Bahar I, Wallqvist DG, Covell DG, Jernigan RL. Correlation between native-state hydrogen exchange and cooperative residue fluctuations from a simple model. Biochemistry. 1998;37:1067–1075. doi: 10.1021/bi9720641. [DOI] [PubMed] [Google Scholar]
- 44.Haliloglu T, Bahar I, Erman B. Gaussian dynamics of folded proteins. Phys Rev Letters. 1997;79:3090–3093. [Google Scholar]
- 45.MOE, Molecular Operating Environment. 2006.08. Chemical Computing Group; Montreal Canada: 2006. [Google Scholar]
- 46.Halgren TA. MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem. 1999;20:720–729. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 47.Halgren TA. MMFF MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J Comput Chem. 1999;20:740–774. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 48.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 49.Word J, Lovell S, Richardson J, Richardson D. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 50.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;117:11225–11236. [Google Scholar]
- 51.Kirton SB, Murray CW, Verdonk ML, Taylor RD. Prediction of binding modes for ligands in the cytochromes P450 and other heme-containing proteins. Proteins. 2005;58:836–844. doi: 10.1002/prot.20389. [DOI] [PubMed] [Google Scholar]
- 52.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved Protein-Ligand Docking Using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 53.VanDerSpoel E, Lindahl B, Hess G, Groenhof AM, Berendsen H. GROMACS Fast Flexible and Free. J Comp Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 54.Scott WRP, Hunenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, Torda AE, Huber T, Kruger P, van Gunsteren WF. The GROMOS biomolecular simulation program package. J Phys Chem A. 1999;103:3596–3607. [Google Scholar]
- 55.Berendsen HJC, Grigera JR, Straatsma PR. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- 56.Hess B, Bekker H, Berendsen HJC, Fraaije J. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 57.Darden T, York D, Pedersen L. Particle mesh Ewald - An N.log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 58.Zhou T, Xu L, Dey B, Hessell AJ, Van Ryk D, Xiang SH, Yang X, Zhang MY, Zwick MB, Arthos J, Burton DR, Dimitrov DS, Sodroski J, Wyatt R, Nabel GJ, Kwong PD. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen L, Kwon YD, Zhou T, Wu X, O’Dell S, Cavacini L, Hessell AJ, Pancera M, Tang M, Xu L, Yang ZY, Zhang MY, Arthos J, Burton DR, Dimitrov DS, Nabel GJ, Posner MR, Sodroski J, Wyatt R, Mascola JR, Kwong PD. Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science. 2009;326:1123–1127. doi: 10.1126/science.1175868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.