

Abstract
Craniofacial dysmorphologies are some of the most variable and common defects affecting the population. Herein we examine a group of craniofacial disorders that are the result of defects in primary cilia; ubiquitous, microtubule based organelles that transduce molecular signals and facilitate the interactions between the cell and its environment. Based on the frequent appearance of craniofacial phenotypes in diseases born from defective primary cilia (ciliopathies) we propose a new class of craniofacial disorders referred to as craniofacial ciliopathies. We explore the most frequent phenotypes associated with ciliopathic conditions and the ciliary gene mutations responsible for craniofacial defects. Finally, we propose that some non-classified disorders may now be classified as craniofacial ciliopathies.
Keywords: primary cilia, craniofacial, Bardet-Biedl, Meckel-Gruber, oral-facial-digital, Joubert syndrome, Ellis-van Creveld, cranioectodermal dysplasia, frontonasal dysplasia
INTRODUCTION
Craniofacial dysmorphologies can be difficult to classify because of the complex anatomy of the craniofacial complex. The renewed interest in the primary cilium as an organelle that coordinates and transduces signals in the molecular environment has ushered in a fresh view for understanding the etiology of many craniofacial diseases. This review will discuss how the study of primary cilia has impacted our understanding of craniofacial disorders and how these minute organelles contribute in an enormous way to craniofacial development.
STRUCTURE AND FUNCTION OF THE PRIMARY CILIA
A primary cilium (Fig. 1A) is a finger-like projection from the cell surface that is composed of three parts: the axoneme, a microtubule based extension; the basal bodies that connect the microtubules to the cell body; and the specialized ciliary membrane that covers the axoneme and is separated from the cytoplasm by transition fibers [Sorokin 1962]. Receptors for a number of signaling pathways are preferentially localized to this specialized membrane [Rohatgi et al., 2007; Schneider et al., 2005; Vieira et al., 2006], making primary cilia responsible for coordinating signals from the Hedgehog, Platelet-derived growth factor (PDGF) alpha, Polycystin, and Wnt pathways (reviewed in [Pedersen and Rosenbaum 2008]).
Figure 1.

Structure of primary cilia. (A) Schematic diagram of primary cilia. The axoneme of the primary cilium is a receptor (pink and black) laden, membrane (green) bound, microtubule (light grey rods) extensions, anchored to the cell body by basal bodies/centrioles (gray barrels). Intraflagellar transport proteins (IFT88 or Kif3a; blue ovals) are important in the formation of cilia and carry molecular cargo up the axoneme. (B–D) Primary cilia are present in tissues of the craniofacial complex. (B) Arl13b expression (green) marks the primary cilia in the neural ectoderm. (C) Arl13b expression in cranial neural crest. (D) Arl13b expression in surface ectoderm. ne, neuroectoderm; v, ventricle; nc, neural crest; se, surface ectoderm.
The extension and retraction of primary cilia is a dynamic process dictated in part by the phase of the cell cycle [Santos and Reiter 2008]. When extended, the ciliary axoneme extends from a modified centriole, the basal body (Fig. 1A). Basal bodies are essential to the extension of the primary cilia as they provide the scaffold upon which the axoneme of the cilium can be built. The position of the basal body dictates the position and orientation of the cilium and serves as the point of entry for proteins that enter the cilium.
The extension of the axoneme from the basal body involves a process known as intraflagellar transport (IFT). During IFT large particles or “rafts” that contain protein cargo are transported bi-directionally along the microtubule tracks of the ciliary axoneme [Rosenbaum and Witman, 2002]. IFT proteins that travel to the distal end of the microtubules are referred to as anterograde IFT proteins whereas those that transport cargo proximally towards the cell body are referred to as retrograde IFT proteins. Mutations in any of the proteins involved in the assembly, function, or maintenance of primary cilia can cause human disease.
CILIOPATHIES AS A CLASS OF DISEASES
A ciliopathy is classified as a disorder that results from aberrant form or function of primary cilia. As a class of diseases, ciliopathies have an extraordinarily broad range of clinical manifestations [Badano et al., 2006]. The spectrum of phenotypes has been attributed to the purported ubiquitous nature of primary cilia. It is now known that a number of ciliopathies result in malformations of the craniofacial complex (Table I). Ciliopathies with craniofacial defects include Bardet-Biedl syndrome (BBS), Oro-facial-digital syndrome (OFD1), Meckel- or Meckel-Gruber syndrome (MKS), Joubert syndrome and Ellis-van Creveld syndrome. Whereas these syndromes are established ciliopathies, the underlying molecular and cellular disruptions responsible for the clinical manifestations remain somewhat elusive. In the following section, the genetic defects underlying these ciliopathies and their associated craniofacial phenotypes will be discussed.
Table I.
Craniofacial ciliopathies. Tabulation of known craniofacial ciliopathies and their accompanying phenotypes, associated genes, protein localization, ciliary defect and references.
Ciliopathies with craniofacial phenotypes.
| Syndrome | Craniofacial phenotype | OMIM | Genes | References |
|---|---|---|---|---|
| Bardet-Biedl syndrome | deep set eyes, hypertelorism, downward slanting palpebral fissures, flat nasal bridge anteverted nares, prominent nasolabial folds, long philtrum, thin upper lip, prominent forehead | 209900 | BBS1-14 | Beales et al., 1999; Lorda-Sanchez et al., 2001 |
| Joubert syndrome | large head and frontal prominence, prominent forehead and nasal bridge, bitemporal narrowing, epicanthal folds, ptosis, prognathism, eyebrow abnormalities, thick ear lobes trapezoid shaped mouth, lower lip eversion, upturned nose | 213300 | JBTS1 | Maria et al., 1999 |
| Meckel-Gruber syndrome | microcephaly, sloping forehead, occipital meningoencephalocele, cleft lip/palate, micrognathia, macrostomia, various glossal malformations | 249000 | MKS1-4 | Fraser and Lytwyn, 1981 |
| Orofaciodigital syndrome | malformations of the face, oral cavity, thickened alveolar ridges, abnormal dentition, absent lateral incisors, clefts of the jaw and tongue | 311200 | OFD1 | Gorlin et al., 1961; Ferrante et al., 2001 |
| Ellis-van Creveld syndrome | cleft lip and palate gingivo, labial musculofibrous fraenula, premature eruption of teeth, hypodontia, small cranial base, micrognathia, increased gonial angle, malocclusion | 225500 | EVC1-2 | Susami et al., 1999; Cahuana et al., 2004 z |
Asterisk indicates gene that has been associated with more that one syndrome (see text).
Bardet-Biedl syndrome (BBS)
BBS (OMIM 209900) is an autosomal, genetically heterogeneous disorder that is characterized by obesity, polydactyly, renal anomalies, retinal degeneration and mental retardation (Fig. 2). Although phenotypically variable, a subgroup of BBS patients present with characteristic facies including deep set eyes, hypertelorism, downward slanting palpebral fissures, a flat nasal bridge with anteverted nares and prominent nasolabial folds, a long philtrum, and a thin upper lip. In addition, BBS patients tend to have a wide, prominent forehead and a small mouth with a slightlyeverted lower lip and retrognathia [Beales et al., 1999].
Figure 2.
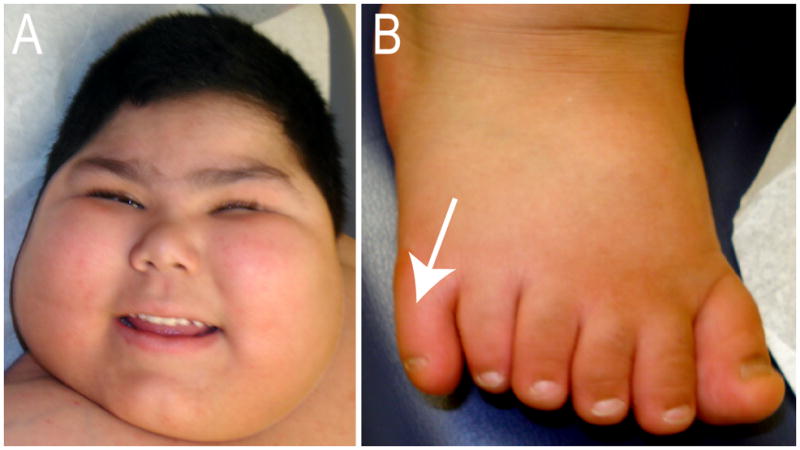
Clinical manifestations in a patient with Bardet-Biedl Syndrome type 8. (A) Face of child with BBS showing obesity, deep set eyes, flat nasal bridge, anteverted nares, long philtrum and thin upper lip. (B) Right foot with postaxial polydactyly (arrow).
Phenotypic variability has been attributed to the 14 different loci linked to BBS including BBS1-12, meckel syndrome1 (MKS1) and centrosomal protein 290kDa/nephronophthisis 6 (CEP290/NPHP6) [Leitch et al., 2008; Zaghloul and Katsanis, 2009]. All of the known BBS proteins localize to the centrosome/basal bodies or ciliary axoneme. Although it has been established that BBS proteins can associate with co-factors to promote the trafficking of vesicles to the cilium, a process necessary for the elongation of the axoneme [Beales 2005; Nachury et al., 2007; Zaghloul and Katsanis, 2009], the exact function of this protein family remains debatable. Two hypotheses exist regarding the function of BBS proteins during ciliogenesis and ciliary function. The first suggests BBS proteins function to bring additional proteins to the centrosome and ciliary dysfunction is an indirect consequence of centrosome/basal body abnormalities. The second suggests that BBS proteins promote cohesion between IFT subcomplexes inside the cilium and ciliary dysfunction is directly linked to the role of BBS proteins within the cilium [Nachury et al., 2007].
Animal models for BBS have shed some light on the function of BBS proteins. BBS2 and 4 knockout mice exhibit major components of the human BBS phenotype [Nishimura et al., 2004], and as such, have been used to elucidate BBS function. Although BBS proteins are not required for ciliogenesis (mice lacking Bbs4 do not appear to have aberrant cilia or basal body structure), the dynamics of cilia assembly is altered in Bbs4−/− cells, suggesting a role for Bbs4 in the regulation of ciliogenesis [Mokrzan et al., 2007].
Zebrafish models have also been used to explore the consequence of mutations in Bbs genes on craniofacial development. For example, defects in zebrafish Bbs genes cause craniofacial defects including shortening of the anterior neurocranium, partial cyclopia, and micrognathia [Tobin et al., 2008]. These defects were attributed to lack of cranial neural crest cell migration into the facial prominences [Tobin et al., 2008]. Molecular analysis of these fish indicate that down-regulation of Bbs proteins perturbs non-canonical Wnt signaling essential for the initiation of neural crest migration, and when coupled to the Shh insensitivity observed in Bbs-deficient neural crest, characteristic BBS craniofacial anomalies are produced. Whether this represents a unique function of primary cilia in teleosts, or is a consequence of the method of BBS gene inactivation (i.e., a morpholino approach) is not yet clear. How mutations in BBS genes specifically affect the cells and tissues that make up the craniofacial complex requires more in depth analysis.
Oro-facial-digital syndrome Type 1 (OFDS1)
OFDS1 (OMIM 311200) is an X-linked dominant disorder caused by mutations in OFD1. This male lethal disorder is characterized by digit abnormalities, polycystic kidney disease, central nervous system (CNS) malformations and facial dysmorphologies [Ferrante et al., 2001; Thauvin-Robinet et al., 2006] (Fig. 3). Craniofacial dysmorphologies occur in over 87% of reported cases [Macca and Franco 2009]. The most common craniofacial abnormalities of OFDS1 include hypertelorism, a broad nasal bridge, facial asymmetry, cleft palate, lingual hamartomas, hypodontia and hyperplastic buccal frenula which may lead to clefting [Anneren et al., 1984; Gorlin et al., 1961; Salinas et al., 1991; Thauvin-Robinet et al., 2006]. CNS malformations frequently observed in this disorder include agenesis of the corpus callosum, abnormal gyration, gray matter heterotopia, cerebellar and brain stem abnormalities and mental retardation [Holub et al., 2005]. The wide array of phenotypic variability often seen even among closely related females has been attributed to X inactivation [Franco and Ballabio 2006; Morleo and Franco 2008].
Figure 3.
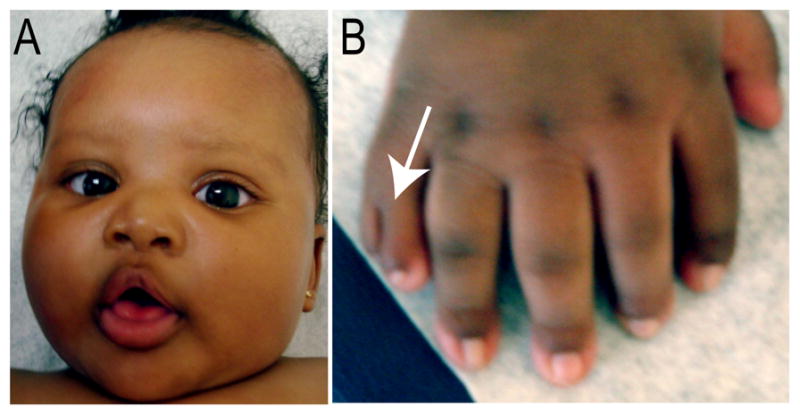
Clinical manifestations in a patient with Oro-facial-digital Syndrome type 1. (A) Face of a female child with OFDS type 1 showing mild facial asymmetry, frontal bossing, hypertelorism and broad nasal bridge. (B) Right hand with brachydactyly, postaxial polysyndactyly (arrow) with ulnar deviation involving the second digit.
To date the genetic basis for OFDS1 has been associated with only one gene, OFD1. OFD1 encodes for a centrosomal protein localized to the basal bodies at the base of ciliary axoneme [Romio et al., 2004; Macca and Franco, 2009]. Numerous mutations and genomic deletions in OFD1 have been identified in OFDS patients [Thauvin-Robinet et al., 2009]. OFD1 mutations usually result in truncations which produce non-functional gene products [Ferrante et al., 2001; Thauvin-Robinet et al., 2006]. Ofd1 knockout mice have absent or defective cilia in various tissues [Ferrante et al., 2003] and the OFDS phenotype has been attributed to abnormal cilia in multiple tissues [Stenram et al., 2007]. Additional forms of OFDS have been described and numbered OFDS type 2–11. Two additional types: OFDS12 and OFDS13, both of which are characterized by additional brain and cardiac abnormalities, have also been clinically diagnosed [Gurrieri et al., 2007; Moran-Barroso et al., 1998].
In a murine model system, knockout of Ofd1 generates phenotypes highly reminiscent of the OFDS in humans, yet more severe. The craniofacial complex in Ofd1 mutant mice is characterized by a shortened skull and facial region, cleft palate and exencephaly [Ferrante et al., 2006]. Researchers working with these mice attribute the increased phenotypic severity to differences of X-inactivation patterns observed between humans and mice [Ferrante et al., 2003; Franco and Ballabio 2006; Morleo and Franco 2008].
Molecular analysis of Ofd1 null mice reveals that Shh expression is absent in the neural tube, but present in the notochord of Ofd1 mutants. Expression of downstream targets of the Hh pathway, including Ptc1 and Gli1, is markedly reduced in the ventral neural tube of the mutant embryo. The resulting abnormal patterning of the neural tube recapitulates the phenotype of some IFT mutants suggesting that Ofd1 may act in the same genetic cascade as these IFT genes [Ferrante et al., 2006].
Defects in the limb of Ofd1 mutant mice suggest an association between Ofd1 and Gli3. Although Ofd1 disruption was not shown to affect Gli3 transcription, the possibility that Gli3 protein function is affected remains a possibility [Ferrante et al., 2006]. IFT proteins have been shown to regulate Gli protein function [Liu et al., 2005]; therefore it is possible that Ofd1 also has a functional interaction with the Gli proteins. Whereas associations like this one remain circumstantial at best, given the current literature it would not be hard to hypothesize that the loss of Ofd1 could disrupt ciliary processing of Gli3 in such a manner that would resemble a loss of Gli3 phenotype [Toriello, 2009].
Work in zebrafish has also contributed to our understanding of Ofd1 function. Knockdown of Ofd1 via antisense morpholinos produces shorter cilia with disrupted axonemes and perturbed intravesicular fluid flow in Kupffer’s vesicle. The resultant phenotype includes a bent body axis, hydrocephalus, edema and randomized situs inversus. As in humans, zebrafish Ofd1 is localized to the centrosome/basal body. The molecular consequence of the loss of Ofd1 is attributed to a defect in Wnt signaling, as these defects are enhanced by loss of Slb/Wnt11 or Tri/Vangl2, two proteins functioning in the non-canonical Wnt/planar cell polarity pathway [Ferrante et al., 2009].
Meckel-Gruber Syndrome (MKS)
MKS (OMIM 249000) is an autosomal recessive, perinatal lethal disorder characterized by congenital heart defects, polycystic kidneys, postaxial polydactyly and numerous craniofacial defects including microcephaly, sloping forehead, occipital meningoencephalocele, cleft lip/palate, micrognathia, micropthalmia, macrostomia, and various malformations of the tongue (Fig. 4). Six subtypes of MKS, MKS type 1–6, exist and are phenotypically variable with linkages to five loci and four genes [Baala et al., 2007; Delous et al., 2007; Kyttala et al., 2006; Roume et al., 1998; Smith et al., 2006]. Phenotypic variation between the subtypes is attributed to unique mutations at the five distinct MKS loci. Unfortunately, only a minor proportion of reported MKS cases are attributable to these identified genes [Tallila et al., 2008].
Figure 4.

Clinical manifestations in a fetus with Meckel-Gruber Syndrome. (A) Lateral view of the head showing mild micrognathia, low set rotated ear and short neck. This exemplifies the variability in phenotypic expression, as this fetus does not have a sloping forehead or cleft lip/palate. (B) Posterior view of skull showing occipital encephalocele (arrow). (C) Right foot with postaxial supernumerary digit (arrow).
MKS1 is a cytosolic protein that is enriched at the centrosome and localized to the base of cilia [Dawe et al., 2007a; Williams et al., 2008]. MKS1 is required for the outgrowth of primary cilia and normal Shh signaling in both the neural tube and the developing limb. Like its human homolog, murine Mks1 also encodes a basal body protein associated with ciliary function[Dawe et al., 2007b].
Mks1 loss of function mice (krc mutants) provide an excellent model of human MKS type 1 because they phenocopy the structural abnormalities of the human disorder, including those of the neural tube, limb, kidney and liver [Weatherbee et al., 2009]. The craniofacial defects in krc mutants are also highly reminiscent of the human disorder. Exencephaly, hypomineralization and/or splitting of the supraoccipital bone are frequently seen in both krc mutants and human patients. Furthermore, krc mutants have cleft palate and hydrocephaly accompanied by reduced ossification of the frontal and parietal bones. Data obtained from the krc mutants suggest that aberrant Hedgehog signaling is likely to underlie many, but not all of the morphologic defects associated with MKS type 1 [Weatherbee et al., 2009].
Although the human gene product has not been identified for MKS type 2 (OMIM 603194), the human ortholog for the murine basal body protein C2cd3 lies in proximity to the critical regions of the locus [Hoover et al., 2008]. Work in mice suggests that C2cd3 is an essential regulator of intracellular transduction of Hedgehog signals and C2cd3 mutants exhibit disrupted proteolytic processing of Gli3. Further studies are required to identify the gene responsible for human MKS type 2.
The remaining types of MKS have been associated with various gene defects. MKS type 3 (OMIM 607361) is associated with mutations in TMEM67, which encodes Meckelin, an integral transmembrane protein found in the ciliary membrane of the axoneme. A mouse model for MKS type 3 provides a strong tool to study the role of TMEM67 [Cook et al., 2009; Dawe et al., 2007b]. MKS type 4 (OMIM 611134) has been linked to mutations in CEP290, which encodes the centrosomal protein Nephrocystin-6 [Baala et al., 2007; Frank et al., 2008]. MKS type 5 (OMIM 611561) results from mutations in RPGRIP1L that encodes RPGR-interacting protein 1-like protein. RPGRIP1L localizes to the basal bodies [Delous et al., 2007] and is allelic to JBTS7 in Joubert syndrome. MKS type 6 (OMIM 612284) results from mutations in CC2D2A. The function of CC2D2A remains unknown [Tallila et al., 2008, Mougou-Zerelli, 2009 #27764].
Joubert Syndrome (JBTS)
JBTS (OMIM 213300) is a group of disorders (JBTS type 1–10) characterized by variable dysmorphologies including the absence or underdevelopment of the cerebellar vermis and a malformed brain stem. The most common features of this disorder include developmental delay, ataxia, hyperpnea (abnormal breathing patterns), sleep apnea, abnormal eye and tongue movements, hypotonia, renal/liver anomalies, retinal anomalies and polydactyly. Craniofacial dysmorphologies associated with this group of disorders include a long face and frontonasal prominence, bitemporal narrowing, ptosis, prominent nasal bridge and tip, prognathism, eyebrow abnormalities, trapezoid shaped mouth, lower lip eversion, and thick ear lobes [Maria et al., 1999]. Despite being associated with several dysmorphic facial characteristics and distinct anthropometric facial patterns, current data prevents a recognizable pattern of malformation. Variability and overlap of features in JBTS and other hindbrain syndromes makes clinical diagnosis difficult and probably reflects genetic heterogeneity [Braddock et al., 2007].
Difficulty in diagnosis of JBTS may also be attributed to the numerous subtypes, the multiple genes associated with the syndrome and the extensive overlap with MKS. JBTS type 3 (OMIM 608629) is associated with mutations in the AHI1 gene, which encodes for the protein Jouberin. JBTS type 4 (OMIM 609583) is associated with deletions in NPHP1. JBTS type 5 (OMIM 610188), like MKS type 4, is associated with mutations in CEP290. JBTS type 6 (OMIM 610688) is caused by mutation in TMEM67 (MKS3), JBTS type 7 (OMIM 611560), as mentioned earlier is caused by mutations in RPGRIP1L and is allelic to MKS type 5. JBTS type 8 (OMIM 612291) is caused by mutations in Arl13b. JBTS type 9 (OMIM 612285), like MKS type 6, is caused by mutations in CC2D2A, and JBTS type 10 (OMIM 300804) is caused by mutations in the CXORF5 (OFD1). Recently, Edvardson suggested that JBTS type 2 in Ashkenazi Jews is caused by mutations in the TMEM216 gene in the centromeric region of chromosome 11 (11p12-q13.3) [Edvardson et al., 2010]. Of the identified genes, CC2D2A, NPHP1, AHI1, CEP290, RPGRIP1L, TMEM67, and ARL13B all localize to and function in the primary cilium/basal body organelle. The molecular mechanism of how these genes produce craniofacial defects is poorly understood, however of the above listed candidate genes, Arl13b has been extensively studied and closely linked to Hedgehog processing.
Arl13b is a small GTPase of the Arf/Arl family that localizes to the ciliary axoneme. An Arl13b null mutation was recently identified in the lethal hennin mouse mutant that displays a phenotype suggesting aberrant Shh signaling [Caspary et al., 2007]. Analysis of hennin mice indicates that Gli3 repressor activity is normal, but Gli activators are constitutively active at low levels. Studies in zebrafish confirm that Arl13b is localized to the cilium and is required for cilia formation in multiple regions outside of the brain. These studies have also confirmed that defects in Arl13b lead to phenotypes in multiple organs [Duldulao et al., 2009]. Together, the identification of Arl13b mutations in mouse, zebrafish and humans with JBTS indicates an extremely broad spectrum of phenotypes across species all related to defective cilia function. These results also suggest a role for cilia-mediated Shh signaling in the pathogenesis of JBTS in humans.
Ellis-van Creveld (EVC) and Acrofacial dysostosis (AFD)
EVC (OMIM 225500) is an autosomal recessive disorder characterized by craniofacial abnormalities with disproportionate short stature, polydactyly, nail dysplasia and cardiac malformations [Ellis and van Creveld 1940; McKusick et al., 1964; Ruiz-Perez and Goodship 2009]. The craniofacial anomalies include cleft lip and palate, multiple gingivolabial musculofibrous frenula, premature eruption of teeth, the presence of teeth at birth, missing primary or permanent teeth, hypodontia, small posterior cranial base, increased gonial angle (the angle formed by the junction of the posterior and lower borders of the mandible) and malocclusion [Cahuana et al., 2004; Susami et al., 1999]. The genetic basis for Ellis-van Creveld syndrome is attributable to loss of function mutations in two genes at 4p16; EVC1 and EVC2 [Galdzicka et al., 2002; Polymeropoulos et al., 1996; Ruiz-Perez et al., 2000]. These two genes lie in a head-to-head configuration that is conserved from fish to man. Defects in these genes may also cause Weyers AFD (OMIM 193530), an autosomal dominant disorder that has a milder phenotype compared to Ellis-van Creveld.
Critical analyses demonstrate that the craniofacial presentation of Evc1−/− mice phenocopy that of the human disorder. Evc1 is expressed in the craniofacial region throughout murine development. As early as e11.5 Evc1 expression is detected in regions of the developing face including the lateral nasal, maxillary and mandibular processes, the nasal septum and cartilages of the nasal capsule, the upper and lower lip mesenchyme, the mesenchyme outlining the growing bones of the maxilla and mandible and the developing bones of the orofacial region [Ruiz-Perez et al., 2007].
The murine Evc1 protein localizes to the distal end of the maternal centriole at the base of the axoneme. Although the loss of Evc1 does not interfere with ciliogenesis, molecular signaling that requires ciliary function is disrupted in Evc1 mutants. A loss of Evc1 causes a disruption in Indian hedgehog signaling [Ruiz-Perez et al., 2007]. It is not clear yet whether disruptions in Evc1 function in mice affect Shh signaling in the craniofacial complex, but in the appendicular skeleton Evc1 acts as a positive mediator of Hh signaling and is not essential for Gli processing [Ruiz-Perez et al., 2007].
CILIARY GENES AS CANDIDATES FOR CRANIOFACIAL DISORDERS
The increasing overlap between genes localized to and/or essential for ciliary function and those responsible craniofacial dysmorphologies has raised the possibility that some craniofacial syndromes with a yet to be determined etiology may be ciliopathies. In the following section we introduce genes that are both essential for ciliary function and maintenance, and when disrupted lead to craniofacial abnormalities in mouse models.
Intraflagellar transport proteins (IFTs)
IFT proteins are a highly conserved family of multimeric proteins present in all ciliated eukaryotic cells [Pan et al., 2005; Rosenbaum and Witman 2002; Scholey and Anderson 2006]. Properly functioning IFT proteins are essential for the full extension of the ciliary axoneme. In addition to their role in cilia formation and maintenance, IFT machinery is essential for Hh signaling downstream of Ptc1 and upstream of direct transcriptional targets of Hh [Fliegauf et al., 2007]. As such, mutations in many IFT proteins strongly resemble loss of Hedgehog phenotypes [Murcia et al., 2000; Zhang et al., 2003], including holoprosencephaly, polydactyly, craniofacial defects and skeletal malformations [McMahon et al., 2003]. We will explore how mutations in two distinct IFT proteins lead to craniofacial dysmorphologies.
Ift88, which encodes the protein Polaris, is concentrated just below the apical membrane in the region of the basal bodies and within axoneme. [Taulman et al., 2001]. Mice with null mutations in ift88, lack cilia on all cells and die mid-gestation with severe defects in neural tube patterning/closure, left-right axis determination, polydactyly, situs inversus and skeletal patterning abnormalities. Craniofacial defects in ift88 null mice include hydrocephalus and a hypoplastic maxilla and mandible [Murcia et al., 2000; Zhang et al., 2003]. Conditional mutations and hypomorphic variants (e.g., Tg737orpk embryos) of ift88 also exhibit polydactyly, hypoplastic maxilla and mandible and supernumerary teeth [Ohazama et al., 2009; Zhang et al., 2003] (Fig. 5). Humans with Ift88 mutations have polycystic kidney disease, which is observed in a number of uncharacterized craniofacial disorders.
Figure 5.
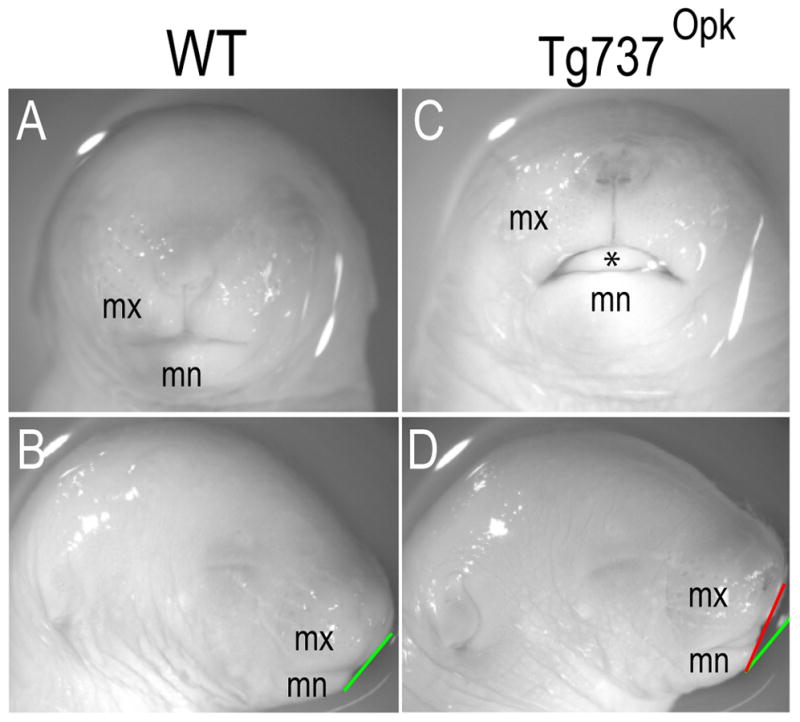
Loss of ciliary proteins produce facial dysmorphologies in murine embryos. (A,B) Frontal and lateral views of e17.5 wild-type embryos. (C, D) Frontal and lateral views of e17.5 Tg737Opk embryos. Gonial angle is indicated by white line in wild-type embryos (B) and black line in Tg737Opk embryos (D). Note that the hypoplastic maxilla produces an increased gonial angle in the mutant (compare black and white line in (D)).
In contrast to Ift88, Ift172 is not required for the induction of cilia, yet the loss of Ift172 impairs proper cilia extension. Loss of Ift172 results in severely truncated axonemes without visible microtubules. Ift172 plays a crucial role in forebrain growth and patterning and in the maintenance of the isthmic organizer, a signaling center essential for the anterior–posterior patterning of the mid- and hindbrain regions. Ift172 is required for proper signaling of the head organizing center, the anterior mesendoderm [Gorivodsky et al., 2009].
Many of the defects related to mutations in Ift172 have been attributed to aberrant Hh signaling as Ift172 acts downstream of Smo and is required for proteolytic processing of Gli3 to its repressor form. Loss of function Ift172 mutants (Slb mutants) die at approximately e12.5, and exhibit severe craniofacial malformations including holoprosencephaly, open cranial neural tube and exencephaly [Huangfu and Anderson 2005]. A second Ift172 mutant, the wimple mutant, carries a point mutation in Ift172 that causes one amino acid substitution at the C-terminus of the protein [Huangfu et al., 2003]. As in Slb mutants, expression of both Hh ligands and targets is profoundly reduced in the developing forebrain of wimple mutants [Huangfu and Anderson 2005] [Huangfu et al., 2003]. Defects associated with IFT172 in humans have yet to be identified.
One caveat exists when using mutations in IFT genes as indicators of ciliopathies. IFT proteins have been identified in non-ciliated cells associated with the process of exocytosis. Specifically, Ift20 was found in T lymphocytes, a cell population that never extends a primary cilium, and was shown to be vital to the redistribution of proteins and lipids on the plasma membrane [Baldari and Rosenbaum 2010; Finetti et al., 2009]. Furthermore, RNAi studies suggest that Ift27 plays an important role during cytokinesis [Qin et al., 2007]. In light of these recent findings, we must use caution when classifying all IFT mutations as ciliopathies.
Fibroblast growth factors (FGF)
Members of the FGF family have long been considered key factors in craniofacial development and disease. Many FGF ligands have been implicated in the growth and development of facial prominences as well as development of the facial skeleton [Creuzet et al., 2004; Macatee et al., 2003; Szabo-Rogers et al., 2008; Trumpp et al., 1999]. Furthermore, FGF receptors have been implicated in suture development and gain of function mutations in these receptors are frequently reported causes for craniosynostosis (reviewed in [Lenton et al., 2005]).
Recent studies have shown that FGF signaling regulates cilia length and function [Neugebauer et al., 2009]. When FGF signaling via Fgf receptor 1 (Fgfr1) is disrupted, expression of ift88 and two transcription factors important in controlling ciliary gene expression, foxj1 and rfx2, is reduced. Phenotypes produced from mutations in fgfr1 are consistent with those seen in ift88 mutants, including curved body axis, kidney cysts and shortened cilia. The acting hypothesis suggests ligands that signal through Ffgr1 including Fgf8 and Fgf2/4 activate the receptor cell-autonomously to maintain a transcriptional network that allows normal expression of IFT proteins required for normal length cilia [Neugebauer et al., 2009]. The findings in this report allow for the postulation that some syndromes caused by mutations in members of the FGF family may actually be ciliopathic in nature.
POTENTIAL CILIOPATHIES WITH CRANIOFACIAL PHENOTYPES
On average 90,000 children per year are born with some type of craniofacial malformation [Nabarra and Papiernik 1988; Nicaise et al., 1998; NIDCR, 2005]. Many of these anomalies are of an unknown etiology. We raise the possibility that some identified human disorders, like cranioectodermal dysplasia and frontonasal dysplasia are among a number of craniofacial disorders that may be ciliopathies.
Cranioectodermal dysplasia (CED)
CED (OMIM 218330), also known as Sensenbrenner syndrome, is a rare autosomal recessive disorder characterized by typical craniofacial, skeletal, ectodermal and renal defects. The craniofacial signature includes dolichocephaly (long, narrow head with or without sagittal suture synostosis), sparse hair, epicanthal folds, ptosis, hypodontia and/or microdontia, retrognathia, high arched palate and anteverted lower lip with protruding tongue [Konstantinidou et al., 2009; Young 1989].
Ectodermal dysplasias, including CED, have been extensively studied and systematically distinguished from other conditions with defects in skeletal and ectodermal systems including Ellis-van Creveld syndrome, GAPO syndrome (Growth retardation, Alopecia, Pseudoanodontia, and Optic atrophy), cartilage-hair hypoplasia, and the tricho-dento-osseous syndrome [Young, 1989]. The low number of documented cases has made it difficult to determine the genetic etiology of CED in particular, however patients with CED often present with characteristics associated with ciliopathies. The phenotypic similarities between CED and other established ciliopathies will most likely aide in the gene identification studies for CED [Konstantinidou et al., 2009].
Frontonasal Dysplasia (FND)
FND (OMIM 136760), also referred to as median cleft syndrome, is a rare disorder affecting the face and head. FND is characterized by malformations of the central portion of the face especially the forehead, the nose, and the philtrum. Phenotypic hallmarks of FND include a broad nasal root and, in extreme cases, a bifid nasal septum, hypertelorism, frontal bossing, anterior cranium occultum, agenesis of the corpus callosum and clefting that traverses the upper lip [Gorlin et al., 1990; Guion-Almeida and Richieri-Costa 2001]. Current studies have associated distinct presentations of FND in humans to compound mutations in Alx3/Alx4 and Six2 [Beverdam et al., 2001; Fogelgren et al., 2008; Twigg et al., 2009]. Studies from our lab have revealed a murine mutant that phenocopies human FND; however this mutant was produced via a conditional deletion of an IFT protein, Kif3a, and subsequent loss of primary cilia on neural crest cells.
Kif3a is an IFT protein essential for ciliary function. Kif3a is a heterotrimeric member of the kinesin super-family of microtubule-associated motors involved in anterograde transport of membrane bound organelles. This protein motor mediates transport between the endoplasmic reticulum and the Golgi, and transports protein complexes within cilia and flagella required for their extension and morphogenesis [Kondo et al., 1994]. Kif3a null mice have a host of embryonic defects including severe developmental abnormalities characterized by neural tube degeneration, mesodermal and caudal dysgenesis and cardiovascular insufficiency that culminate in embryonic lethality during mid-gestation [Takeda et al., 1999]. A deeper understanding for the role of Kif3a during development has been achieved by creating conditional knockouts in multiple tissues including the neuroectoderm [Huangfu et al., 2003] and the neural crest [Brugmann et al., 2010].
The disruption of ciliary function due to the loss of Kif3a in cranial neural crest cells produces a severe craniofacial phenotype in mice (Fig. 6A,B). Mice that lack Kif3a in neural crest cells exhibit extreme hypertelorism, bifid nasal septum, cranium occultum, aglossia, dental abnormalities and agenesis of the corpus callosum [Brugmann et al., 2010]. When viewed as a whole, this phenotype was strikingly similar to that of human FND (Fig. 6C).
Figure 6.
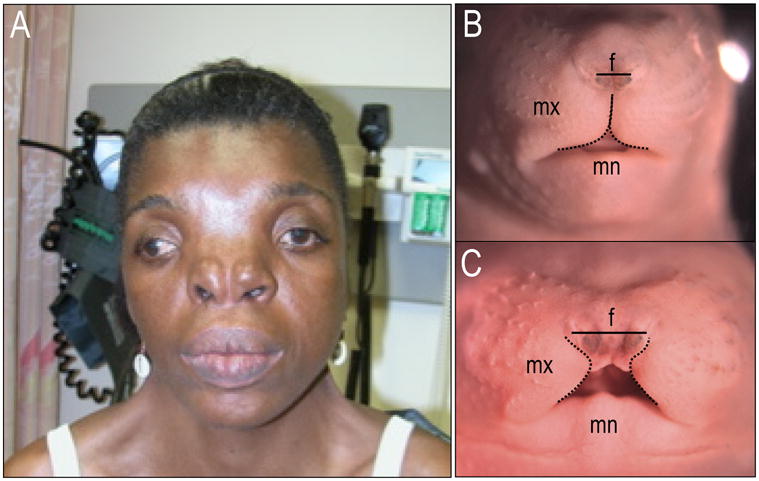
Phenotypic similarities between the Kif3a knockout mouse and human frontonasal dysplasia. (A) Frontal view of a wild-type e16.5 mouse embryo. (B) Frontal view of a Kif3a conditional knockout at e16.5. Black solid line indicates width of frontonasal prominence, dotted black line indicates maxillary boundary. (C) Human patient with frontonasal dysplasia showing hypertelorism, a broad nasal root, nose with median cleft and absent tip of the nose. mx, maxillary prominence; mn, mandibular prominence; f, frontonasal prominence.
FUTURE DIRECTIONS
Normal structure and function of primary cilia are required for a cell to transduce molecular signals from the environment. To date no distinct craniofacial gestalt has been associated with ciliopathies. Several lines of evidence show that primary cilia are involved in the transduction of both Hedgehog and Wnt signals. This has significant implications on both normal and abnormal facial development given the importance of these signaling molecules during development of the face. Furthermore, the relationship between primary cilia and these signaling molecules raises the possibility that a number of craniofacial dysmorphologies may arise as a consequence of abnormal Hedgehog or Wnt signaling secondary to ciliary dysfunction. In the near future it is likely that a number of craniofacial disorders, in which a genetic cause is not presently known, will be shown to involve impaired ciliogenesis or ciliary function. Recent advances in the understanding of how primary cilia contribute to craniofacial development have introduced a new class of genetic candidates for craniofacial syndromes of unknown etiology. The continued engineering of transgenic, ciliopathic animal models will allow for a deeper comprehension of how each tissue involved in the development of the craniofacial complex utilizes primary cilia.
Acknowledgments
We would like to thank Paul Sharpe and Atsushi Ohazama for sharing images of Tg737orpk mice, Alan Shanske for sharing three images of BBS and FND patients, and Edmond Lemire for sharing images of Meckel-Gruber patients. This work was supported by the National Institute of Health (NIH) grants K99 DE019853-01 to S.A.B, The Eunice Kennedy Shriver National Institute of Child Health and Human Development to D.R.C, Women’s Reproductive Health Research Career Development grant K12HD01255 to D.R.C and The Eleanor and Miles Shore Scholars in Medicine, Brigham and Women’s Hospital, Department of Obstetrics, Gynecology and Reproductive Medicine and Harvard Medical School.
References
- Anneren G, Arvidson B, Gustavson KH, Jorulf H, Carlsson G. Oro-facio-digital syndromes I and II: radiological methods for diagnosis and the clinical variations. Clin Genet. 1984;26(3):178–86. doi: 10.1111/j.1399-0004.1984.tb04365.x. [DOI] [PubMed] [Google Scholar]
- Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, Esculpavit C, Toutain A, Moraine C, Parent P, Marcorelles P, Dauge MC, Roume J, Le Merrer M, Meiner V, Meir K, Menez F, Beaufrère AM, Francannet C, Tantau J, Sinico M, Dumez Y, MacDonald F, Munnich A, Lyonnet S, Gubler MC, Génin E, Johnson CA, Vekemans M, Encha-Razavi F, Attié-Bitach T. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007;81(1):170–9. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–48. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Baldari CT, Rosenbaum J. Intraflagellar transport: it’s not just for cilia anymore. Curr Opin Cell Biol. 2010;22(1):75–80. doi: 10.1016/j.ceb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales PL. Lifting the lid on Pandora’s box: the Bardet-Biedl syndrome. Curr Opin Genet Dev. 2005;15(3):315–23. doi: 10.1016/j.gde.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–46. [PMC free article] [PubMed] [Google Scholar]
- Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development. 2001;128(20):3975–86. doi: 10.1242/dev.128.20.3975. [DOI] [PubMed] [Google Scholar]
- Braddock SR, Henley KM, Maria BL. The face of Joubert syndrome: a study of dysmorphology and anthropometry. Am J Med Genet A. 2007;143A(24):3235–42. doi: 10.1002/ajmg.a.32099. [DOI] [PubMed] [Google Scholar]
- Brugmann SA, Allen NC, James AW, Mekonnen Z, Madan E, Helms JA. A primary cilia dependent etiology for midline facial disorders. Human Molecular Genetics. 2010:19. doi: 10.1093/hmg/ddq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahuana A, Palma C, Gonzales W, Gean E. Oral manifestations in Ellis-van Creveld syndrome: report of five cases. Pediatr Dent. 2004;26(3):277–82. [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007;12(5):767–78. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Cook SA, Collin GB, Bronson RT, Naggert JK, Liu DP, Akeson EC, Davisson MT. A mouse model for Meckel syndrome type 3. J Am Soc Nephrol. 2009;20(4):753–64. doi: 10.1681/ASN.2008040412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creuzet S, Schuler B, Couly G, Le Douarin NM. Reciprocal relationships between Fgf8 and neural crest cells in facial and forebrain development. Proc Natl Acad Sci U S A. 2004;101(14):4843–7. doi: 10.1073/pnas.0400869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe HR, Farr H, Gull K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J Cell Sci. 2007a;120(Pt 1):7–15. doi: 10.1242/jcs.03305. [DOI] [PubMed] [Google Scholar]
- Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, Blair-Reid S, Sriram N, Katsanis N, Attie-Bitach T, Afford SC, Copp AJ, Kelly DA, Gull K, Johnson CA. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007b;16(2):173–86. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbemann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Berthélémé JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Rüther U, Schneider-Maunoury S, Attié-Bitach T, Saunier S. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39(7):875–81. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Duldulao NA, Lee S, Sun Z. Cilia localization is essential for in vivo functions of the Joubert syndrome protein Arl13b/Scorpion. Development. 2009;136(23):4033–42. doi: 10.1242/dev.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Shaag A, Zenvirt S, Erlich Y, Hannon GJ, Shanske AL, Gomori JM, Ekstein J, Elpeleg O. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet. 2010;86(1):93–7. doi: 10.1016/j.ajhg.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RWB, van Creveld S. A syndrome characterized by ectodermal dysplasia, polydactyly, chondro-dysplasia and congenital morbus cordis: report of three cases. Arch Dis Child. 1940;15:65–84. doi: 10.1136/adc.15.82.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Barra A, Truong JP, Banfi S, Disteche CM, Franco B. Characterization of the OFD1/Ofd1 genes on the human and mouse sex chromosomes and exclusion of Ofd1 for the Xpl mouse mutant. Genomics. 2003;81(6):560–9. doi: 10.1016/s0888-7543(03)00091-0. [DOI] [PubMed] [Google Scholar]
- Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, Sylvie O, Bernard L, Malcolm S, Winter R, Ballabio A, Franco B. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68(3):569–76. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL, Woolf AS, Wilson SW. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum Mol Genet. 2009;18(2):289–303. doi: 10.1093/hmg/ddn356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dolle P, Franco B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38(1):112–7. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, Baldari CT. Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol. 2009;11(11):1332–9. doi: 10.1038/ncb1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8(11):880–93. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Fogelgren B, Kuroyama MC, McBratney-Owen B, Spence AA, Malahn LE, Anawati MK, Cabatbat C, Alarcon VB, Marikawa Y, Lozanoff S. Misexpression of Six2 is associated with heritable frontonasal dysplasia and renal hypoplasia in 3H1 Br mice. Dev Dyn. 2008;237(7):1767–79. doi: 10.1002/dvdy.21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco B, Ballabio A. X-inactivation and human disease: X-linked dominant male-lethal disorders. Curr Opin Genet Dev. 2006;16(3):254–9. doi: 10.1016/j.gde.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Frank V, den Hollander AI, Bruchle NO, Zonneveld MN, Nurnberg G, Becker C, Du Bois G, Kendziorra H, Roosing S, Senderek J, Nürnberg P, Cremers FP, Zerres K, Bergmann C. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum Mutat. 2008;29(1):45–52. doi: 10.1002/humu.20614. [DOI] [PubMed] [Google Scholar]
- Galdzicka M, Patnala S, Hirshman MG, Cai JF, Nitowsky H, Egeland JA, Ginns EI. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Mol Genet Metab. 2002;77(4):291–5. doi: 10.1016/s1096-7192(02)00178-6. [DOI] [PubMed] [Google Scholar]
- Gorivodsky M, Mukhopadhyay M, Wilsch-Braeuninger M, Phillips M, Teufel A, Kim C, Malik N, Huttner W, Westphal H. Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev Biol. 2009;325(1):24–32. doi: 10.1016/j.ydbio.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Anderson VE, Scott CR. Hypertrophied frenuli, oligophrenia, famflial trembling and anomalies of the hand. Report of four casesin one family and a forme fruste in another. N Engl J Med. 1961;264:486–9. doi: 10.1056/NEJM196103092641004. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, Cohen MM, Levin LS. Syndromes of the Head and Neck. New York: Oxford University Press; 1990. [Google Scholar]
- Guion-Almeida ML, Richieri-Costa A. Frontonasal dysplasia, macroblepharon, eyelid colobomas, ear anomalies, macrostomia, mental retardation and CNS structural anomalies: defining the phenotype. Clin Dysmorphol. 2001;10(2):81–6. doi: 10.1097/00019605-200104000-00002. [DOI] [PubMed] [Google Scholar]
- Gurrieri F, Franco B, Toriello H, Neri G. Oral-facial-digital syndromes: review and diagnostic guidelines. Am J Med Genet A. 2007;143A(24):3314–23. doi: 10.1002/ajmg.a.32032. [DOI] [PubMed] [Google Scholar]
- Holub M, Potocki L, Bodamer OA. Central nervous system malformations in oral-facial-digital syndrome, type 1. Am J Med Genet A. 2005;136(2):218. doi: 10.1002/ajmg.a.30751. [DOI] [PubMed] [Google Scholar]
- Hoover AN, Wynkoop A, Zeng H, Jia J, Niswander LA, Liu A. C2cd3 is required for cilia formation and Hedgehog signaling in mouse. Development. 2008;135(24):4049–58. doi: 10.1242/dev.029835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102(32):11325–30. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426(6962):83–7. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Kondo S, Sato-Yoshitake R, Noda Y, Aizawa H, Nakata T, Matsuura Y, Hirokawa N. KIF3A is a new microtubule-based anterograde motor in the nerve axon. J Cell Biol. 1994;125(5):1095–107. doi: 10.1083/jcb.125.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidou AE, Fryssira H, Sifakis S, Karadimas C, Kaminopetros P, Agrogiannis G, Velonis S, Nikkels PG, Patsouris E. Cranioectodermal dysplasia: a probable ciliopathy. Am J Med Genet A. 2009;149A(10):2206–11. doi: 10.1002/ajmg.a.33013. [DOI] [PubMed] [Google Scholar]
- Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestila M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38(2):155–7. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40(4):443–8. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- Lenton KA, Nacamuli RP, Wan DC, Helms JA, Longaker MT. Cranial suture biology. Curr Top Dev Biol. 2005;66:287–328. doi: 10.1016/S0070-2153(05)66009-7. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132(13):3103–11. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Macatee TL, Hammond BP, Arenkiel BR, Francis L, Frank DU, Moon AM. Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development. 2003;130(25):6361–74. doi: 10.1242/dev.00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macca M, Franco B. The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet C Semin Med Genet. 2009;151C(4):318–25. doi: 10.1002/ajmg.c.30224. [DOI] [PubMed] [Google Scholar]
- Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999;14(9):583–90. doi: 10.1177/088307389901400906. discussion 590–1. [DOI] [PubMed] [Google Scholar]
- McKusick VA, Egeland JA, Eldridge R, Krusen DE. Dwarfism in the Amish I. the Ellis-Van Creveld Syndrome. Bull Johns Hopkins Hosp. 1964;115:306–36. [PubMed] [Google Scholar]
- McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- Mokrzan EM, Lewis JS, Mykytyn K. Differences in renal tubule primary cilia length in a mouse model of Bardet-Biedl syndrome. Nephron Exp Nephrol. 2007;106(3):e88–96. doi: 10.1159/000103021. [DOI] [PubMed] [Google Scholar]
- Moran-Barroso V, Valdes Flores M, Garcia-Cavazos R, Kofman-Alfaro S, Saavedra-Ontiveros D. Oral-facial-digital (OFD) syndrome with associated features: a new syndrome or genetic heterogeneity and variability? Clin Dysmorphol. 1998;7(1):55–7. [PubMed] [Google Scholar]
- Morleo M, Franco B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J Med Genet. 2008;45(7):401–8. doi: 10.1136/jmg.2008.058305. [DOI] [PubMed] [Google Scholar]
- Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127(11):2347–55. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- Nabarra B, Papiernik M. Phenotype of thymic stromal cells. An immunoelectron microscopic study with anti-IA, anti-MAC-1, and anti-MAC-2 antibodies. Lab Invest. 1988;58(5):524–31. [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–13. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- Neugebauer JM, Amack JD, Peterson AG, Bisgrove BW, Yost HJ. FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature. 2009;458(7238):651–4. doi: 10.1038/nature07753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicaise P, Gleizes A, Sandre C, Forestier F, Kergot R, Quero AM, Labarre C. Influence of intestinal microflora on murine bone marrow and spleen macrophage precursors. Scand J Immunol. 1998;48(6):585–91. doi: 10.1046/j.1365-3083.1998.00487.x. [DOI] [PubMed] [Google Scholar]
- NIDCR. 2005 http://www.nidcr.nih.gov/HealthInformation/OralHealthInformationIndex/SpectrumSeries/CraniofacialResearch.htm.
- Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, Carmi R, Stone EM, Sheffield VC. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A. 2004;101(47):16588–93. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohazama A, Haycraft CJ, Seppala M, Blackburn J, Ghafoor S, Cobourne M, Martinelli DC, Fan CM, Peterkova R, Lesot H, Yoder BK, Sharpe PT. Primary cilia regulate Shh activity in the control of molar tooth number. Development. 2009;136(6):897–903. doi: 10.1242/dev.027979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Wang Q, Snell WJ. Cilium-generated signaling and cilia-related disorders. Lab Invest. 2005;85(4):452–63. doi: 10.1038/labinvest.3700253. [DOI] [PubMed] [Google Scholar]
- Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Ide SE, Wright M, Goodship J, Weissenbach J, Pyeritz RE, Da Silva EO, Ortiz De Luna RI, Francomano CA. The gene for the Ellis-van Creveld syndrome is located on chromosome 4p16. Genomics. 1996;35(1):1–5. doi: 10.1006/geno.1996.0315. [DOI] [PubMed] [Google Scholar]
- Qin H, Wang Z, Diener D, Rosenbaum J. Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr Biol. 2007;17(3):193–202. doi: 10.1016/j.cub.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–6. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J Am Soc Nephrol. 2004;15(10):2556–68. doi: 10.1097/01.ASN.0000140220.46477.5C. [DOI] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3(11):813–25. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Roume J, Genin E, Cormier-Daire V, Ma HW, Mehaye B, Attie T, Razavi-Encha F, Fallet-Bianco C, Buenerd A, Clerget-Darpoux F, Munnich A, Le Merrer M. A gene for Meckel syndrome maps to chromosome 11q13. Am J Hum Genet. 1998;63(4):1095–101. doi: 10.1086/302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, Blanco MJ, Wilson A, Liu YN, Miles C, Peters H, Goodship JA. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development. 2007;134(16):2903–12. doi: 10.1242/dev.007542. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Goodship JA. Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to hedgehog ligands. Am J Med Genet C Semin Med Genet. 2009;151C(4):341–51. doi: 10.1002/ajmg.c.30226. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, King L, Francomano C, Freisinger P, Spranger S, Marino B, Dallapiccola B, Wright M, Meitinger T, Polymeropoulos MH, Goodship J. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. 2000;24(3):283–6. doi: 10.1038/73508. [DOI] [PubMed] [Google Scholar]
- Salinas CF, Pai GS, Vera CL, Milutinovic J, Hagerty R, Cooper JD, Cagna DR. Variability of expression of the orofaciodigital syndrome type I in black females: six cases. Am J Med Genet. 1991;38(4):574–82. doi: 10.1002/ajmg.1320380416. [DOI] [PubMed] [Google Scholar]
- Santos N, Reiter JF. Building it up and taking it down: the regulation of vertebrate ciliogenesis. Dev Dyn. 2008;237(8):1972–81. doi: 10.1002/dvdy.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15(20):1861–6. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Scholey JM, Anderson KV. Intraflagellar transport and cilium-based signaling. Cell. 2006;125(3):439–42. doi: 10.1016/j.cell.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Malik Sharif S, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, Miller CA, Cox P, Algazali L, Trembath RC, Torres VE, Attie-Bitach T, Kelly DA, Maher ER, Gattone VH, 2nd, Harris PC, Johnson CA. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38(2):191–6. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol. 1962;15:363–77. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenram U, Cramnert C, Axfors-Olsson H. Oralfacialdigital-like syndrome with respiratory tract symptoms from birth and ultrastructural centriole/basal body disarray. Acta Paediatr. 2007;96(7):1101–4. doi: 10.1111/j.1651-2227.2007.00326.x. [DOI] [PubMed] [Google Scholar]
- Susami T, Kuroda T, Yoshimasu H, Suzuki R. Ellis-van Creveld syndrome: craniofacial morphology and multidisciplinary treatment. Cleft Palate Craniofac J. 1999;36(4):345–52. doi: 10.1597/1545-1569_1999_036_0345_evcscm_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Geetha-Loganathan P, Nimmagadda S, Fu KK, Richman JM. FGF signals from the nasal pit are necessary for normal facial morphogenesis. Dev Biol. 2008;318(2):289–302. doi: 10.1016/j.ydbio.2008.03.027. [DOI] [PubMed] [Google Scholar]
- Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A−/− mice analysis. J Cell Biol. 1999;145(4):825–36. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallila J, Jakkula E, Peltonen L, Salonen R, Kestila M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet. 2008;82(6):1361–7. doi: 10.1016/j.ajhg.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12(3):589–99. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Cossee M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, Bieth E, Layet V, Parent P, David A, et al. Goldenberg A, Mortier G, Héron D, Sagot P, Bouvier AM, Huet F, Cusin V, Donzel A, Devys D, Teyssier JR, Faivre L. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43(1):54–61. doi: 10.1136/jmg.2004.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Franco B, Saugier-Veber P, Aral B, Gigot N, Donzel A, Van Maldergem L, Bieth E, Layet V, Mathieu M, Teebi A, Lespinasse J, Callier P, Mugneret F, Masurel-Paulet A, Gautier E, Huet F, Teyssier JR, Tosi M, Frébourg T, Faivre L. Genomic deletions of OFD1 account for 23% of oral-facial-digital type 1 syndrome after negative DNA sequencing. Hum Mutat. 2009;30(2):E320–9. doi: 10.1002/humu.20888. [DOI] [PubMed] [Google Scholar]
- Tobin JL, Di Franco M, Eichers E, May-Simera H, Garcia M, Yan J, Quinlan R, Justice MJ, Hennekam RC, Briscoe J, Tada M, Mayor R, Burns AJ, Lupski JR, Hammond P, Beales PL. Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung’s disease in Bardet-Biedl syndrome. Proc Natl Acad Sci U S A. 2008;105(18):6714–9. doi: 10.1073/pnas.0707057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriello HV. Are the oral-facial-digital syndromes ciliopathies? Am J Med Genet A. 2009;149A(5):1089–95. doi: 10.1002/ajmg.a.32799. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes and Development. 1999;13(23):3136–48. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Versnel SL, Nurnberg G, Lees MM, Bhat M, Hammond P, Hennekam RC, Hoogeboom AJ, Hurst JA, Johnson D, Robinson AA, Scambler PJ, Gerrelli D, Nürnberg P, Mathijssen IM, Wilkie AO. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am J Hum Genet. 2009;84(5):698–705. doi: 10.1016/j.ajhg.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Gaus K, Verkade P, Fullekrug J, Vaz WL, Simons K. FAPP2, cilium formation, and compartmentalization of the apical membrane in polarized Madin-Darby canine kidney (MDCK) cells. Proc Natl Acad Sci U S A. 2006;103(49):18556–61. doi: 10.1073/pnas.0608291103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet. 2009;18(23):4565–75. doi: 10.1093/hmg/ddp422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Winkelbauer ME, Schafer JC, Michaud EJ, Yoder BK. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol Biol Cell. 2008;19(5):2154–68. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ID. Cranioectodermal dysplasia (Sensenbrenner’s syndrome) J Med Genet. 1989;26(6):393–6. [Google Scholar]
- Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119(3):428–37. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Murcia NS, Chittenden LR, Richards WG, Michaud EJ, Woychik RP, Yoder BK. Loss of the Tg737 protein results in skeletal patterning defects. Dev Dyn. 2003;227(1):78–90. doi: 10.1002/dvdy.10289. [DOI] [PubMed] [Google Scholar]