

Abstract
Cisplatin is the first-line chemotherapy for the treatment of several cancers. However, the development of cisplatin resistance represents a major clinical problem, and the mechanisms of acquired resistance are not fully understood. Here we show that degradation of the Bcl-2 homology 3-only proapoptotic protein Bim plays an important role in cisplatin resistance in ovarian cancer. Specifically, we show that treatment of ovarian cancer cells with cisplatin caused Bim phosphorylation and subsequent degradation and that its degradation is associated with cisplatin resistance. We also show that cisplatin treatment caused the activation of ERK, which correlated with Bim phosphorylation and degradation. By inhibiting ERK phosphorylation with the MEK inhibitor and knocking down ERK expression with siRNA, we show that Bim phosphorylation and degradation were blocked, which suggests that Bim is phosphorylated by ERK and that such phosphorylation is responsible for cisplatin-induced Bim degradation. We show that ERK was activated in cisplatin-resistant OV433 cells as compared with their counterpart parental OV433 cells. We also show that Bim was phosphorylated and degraded in cisplatin-resistant OV433 cells but not in the parental OV433 cells. Importantly, we show that inhibition of Bim degradation by the proteasome inhibitor MG132 sensitized resistant OV433 cells to cisplatin-induced death. Taken together, our data indicate that degradation of Bim via ERK-mediated phosphorylation can lead to cisplatin resistance. Therefore, these findings suggest that cisplatin resistance can be overcome by the combination of cisplatin and the proteasome inhibitors in ovarian cancer cells.
Keywords: Cancer Therapy, Cell Death, Drug Resistance, MAP Kinases (MAPKs), Signal Transduction, Bim, ERK, Apoptosis, Cisplatin Sensitivity, Protein Degradation
Introduction
Ovarian cancer is the fifth leading cause of cancer-related deaths in women in the United States. Currently available therapeutic options include tumor debulking surgery and chemotherapy. Standard first-line chemotherapy is cisplatin-based treatment. However, the majority of ovarian cancer patients who are initially sensitive to cisplatin will eventually relapse, and in many cases acquired resistance will leave no curative treatments (1). The exact mechanisms of cisplatin resistance are not fully understood.
There are many molecules and pathways that have been shown to contribute to cisplatin resistance (2–10). Deregulation of these proteins results in a variety of consequences, including insufficient DNA binding, increased DNA repair, and altered expression of genes involved in the cell death and survival pathways (9, 11). Among survival pathways, activation of ERK contributes to cisplatin resistance (12). ERK is a member of the MAPK family that can be activated by growth signals (13). ERK is activated by MEK through a cascade of upstream kinases involving ras and raf (14). Once it is activated, ERK translocates into the nucleus to influence cellular responses via phosphorylation, leading to a number of cell processes, including cell proliferation and cell survival (14). There are many substrates that can be phosphorylated by ERK, including transcription factors and proteins that regulate apoptosis. One of its substrates is Bim (Bcl-2-interacting mediator of cell death)2 (15–17).
Bim is a member of the Bcl-2 homology 3 (BH3)-only subgroup of the Bcl-2 family that also includes Bid, Bad, Bik, Noxa, and Puma (18). Bim was previously identified as a BH3-only protein that induces apoptosis by interacting with and inhibiting the anti-apoptotic members of the Bcl-2 family (19). There are three splice forms of Bim: Bim-S, Bim-L, and Bim-EL (19, 20). Bim expression can be regulated through multiple mechanisms involving both transcriptional and posttranslational pathways. One of the mechanisms is phosphorylation (21). Bim phosphorylation can decrease its stability or alter its interaction with other proteins to regulate apoptosis (21, 22). A number of studies have shown that Bim can be phosphorylated by ERK and that such phosphorylation leads to ubiquitination and subsequent degradation in the proteasome (16, 17, 21). Importantly, ERK-mediated Bim phosphorylation increases resistance to some anticancer drugs, including paclitaxel (23, 24). Although cisplatin can induce ERK (25, 26) and ERK can phosphorylate Bim to target it for degradation, it is unclear thus far whether Bim degradation via cisplatin-induced ERK-mediated phosphorylation plays a role in cisplatin resistance, which is the focus of this study.
In this study, we showed that cisplatin induces ERK activation, and ERK in turn phosphorylates Bim. Phosphorylation targets Bim to the proteasome for degradation. Inhibition of ERK activation by a MEK inhibitor or down-regulation of ERK expression by siRNA inhibits Bim phosphorylation and degradation, which sensitizes ovarian cancer cells to cisplatin-induced apoptosis. Further, blockage of Bim degradation with a proteasome inhibitor sensitizes ovarian cancer cells to cisplatin. Importantly, we show that ovarian cancer cells with acquired cisplatin resistance express a lower level of Bim as compared with their parental counterparts. In addition, we show that proteasome inhibitor treatment increases the levels of Bim protein and renders cisplatin-resistant cells sensitive to cisplatin, further highlighting the importance of Bim degradation in cisplatin resistance. Taken together, our results suggest that ERK-mediated Bim degradation plays an important role in cisplatin resistance, which can be overcome by the combination treatment of cisplatin with proteasome inhibitors in ovarian cancer cells.
EXPERIMENTAL PROCEDURES
Reagents
Cisplatin and anti-actin antibody were purchased from Sigma. The MEK inhibitor U0126 and p38 inhibitor SB203580 were purchased from Promega (Madison, WI). The JNK inhibitor SP600125 was purchased from Calbiochem. Rabbit antibodies against ERK, phospho-ERK, p38, phospho-p38, JNK, phospho-JNK, CREB, phospho-CREB, c-Jun, phospho-c-Jun, Bim, and PARP were purchased from Cell Signaling Technology (Beverly, MA). MG132 was purchased from Enzo Life Sciences (Plymouth Meeting, PA). λ protein phosphatase was purchased from New England Biolabs (Ipswich, MA).
Cell Lines, Culture Conditions and Treatment
The human ovarian cancer cell lines RMG-1, OV433, OVCA432, OVCA420, and TOV112D were described previously (25). Cisplatin-resistant ovarian cancer cell line OV433-CR was established by chronically exposing the parental OV433 (OV433-P) cells to gradually increased concentrations of cisplatin starting from 0.1 μg/ml to 0.8 μg/ml for over 6 months (26). All cell lines were maintained in 1:1 MCDB105:M199 (Sigma) medium supplemented with 10% FBS and antibiotics at 37 °C in a humidified atmosphere consisting of 5% CO2 and 95% air. Cells were treated with various concentrations of cisplatin and/or inhibitors for different intervals of time as indicated in each figure legend.
siRNA Transfection for Knockdown of ERK and Bim
On-TARGETplus SMARTpool siRNAs for ERK, Bim, and corresponding control siRNA were purchased from Dharmacon Research (Lafayette, CO). Transfections were performed as suggested by Dharmacon with slight modifications, as described previously (27). Briefly, RMG-1 cells were plated at 8 × 105 cells/well in 6-well plates. The next day, cells were transfected with ERK or Bim siRNA oligonucleotides or non-target control oligonucleotides using Lipofectamine 2000 or Lipofectamine RNAiMax (Invitrogen). After 3 days, transfected cells were left untreated or treated with cisplatin (10 μm) for 48 h and then harvested for examining the expression of ERK and Bim protein by Western blot analysis. To determine cisplatin sensitivity, transfected cells were plated at 5000 cells/well in 96-well plates and then treated with or without cisplatin (10 or 20 μm) for 48 or 72 h, and cell growth inhibition was determined by MTT assays.
MTT Assays
The MTT assay was described previously (26). Briefly, cells were left untreated or pretreated with 10 μm U0126, 10 μm SB203580, 10 μm SP600125, or 250 nm MG132 for 30 min and then treated or untreated with 10 μm or 20 μm cisplatin for 48 or 72 h. After incubation with MTT solution, isopropanol was added to dissolve the formazan crystals. Absorbance was measured using a Vmax microplate reader (Molecular Devices, Sunnyvale, CA) at 590 nm. The survival was calculated from the mean of pooled data from three separate experiments with five wells each (26).
Western Blot Analysis
Cell lysates were prepared as described previously (28), and protein concentration was determined using the protein assay kit (Bio-Rad). Cell lysates were electrophoresed through 12 or 15% denaturing polyacrylamide gels and transferred to a PVDF membrane (Millipore, Bedford, MA). The blots were probed or reprobed with the antibodies, and bound antibody was detected using ECL or the Odyssey infrared imaging system according to the manufacturer's protocol.
Clonogenic Assays
The clonogenic assay was described previously (29). Briefly, cells were plated (2 × 105 cells/well) in 6-well plates. The next day, cells were treated with vehicle, inhibitors, or cisplatin. After 3 h of incubation with drugs, cells were trypsinized, counted, and seeded (400 cells/well) in new 6-well plates. After 12 days, colonies were stained with 0.25% crystal violet in 10% formalin and 80% methanol for 30 min, washed with water, and counted. Plating efficiency was determined as the fraction of cells that attached to the support and grew into colonies larger than 1 mm in diameter. The results represented at least three independent experiments.
Statistical Analysis
Statistical analyses were performed using Student's t test. The data were presented as the mean ± S.D., and a p value of less than 0.001 was considered very significant.
RESULTS
Cisplatin Treatment Causes Bim Phosphorylation
It has been shown that Bim phosphorylation and subsequent degradation plays an important role in chemoresistance in several types of cancer cells. However, the role of Bim phosphorylation in cisplatin resistance has not been determined thus far. To this end, a panel of ovarian cancer cell lines including RMG-1, OV433, OVCA420, OVCA432, and TOV112D were treated with 10 μm cisplatin for 24, 48, and 72 h, and the levels of Bim protein were assessed by Western blot analysis. As shown in Fig. 1A, cisplatin treatment caused a Bim protein mobility shift in all cell lines tested with different kinetics even though these cell lines exhibited differential cisplatin sensitivity (Fig. 1B). It is known that phosphorylation can cause mobility shifts. To determine whether the observed Bim mobility shift is due to phosphorylation, cell lysates from cisplatin-treated RMG-1 cells were incubated with and without λ phosphatase in the presence and absence of the phosphatase inhibitor vanadate. Fig. 1C shows that treatment of cell lysates with λ protein phosphatase inhibited the Bim mobility shift and that the phosphatase inhibitor vanadate abolished the effect of λ phosphatase on the mobility of the shift Bim band. These data suggest that the Bim mobility shift in cells treated with cisplatin is due to phosphorylation (Fig. 1C). We showed that different exposure times in RMG-1 cells clearly indicate three isoforms of Bim proteins (Fig. 1A) and that only Bim-EL is phosphorylated, which is consistent with previous reports (16, 17). Thus, these data suggest that an increase in Bim-EL (hereafter referred to as Bim) phosphorylation is a general event in cisplatin-treated human ovarian cancer cells and that this event may play an important role in cisplatin resistance.
FIGURE 1.

Cisplatin treatment causes Bim phosphorylation, leading to Bim-EL degradation in several ovarian cancer cell lines. A, Western blot analyses of Bim-EL expression in several ovarian cancer cell lines treated with 10 μm cisplatin for the indicated time periods. Actin was used as a loading control. In RMG-1 cells, two different exposures times were shown. B, MTT analyses of cell growth inhibition after 48 h of cisplatin treatment at the indicated concentrations. C, Western blot analyses of Bim-EL phosphorylation. RMG-1 cells were left untreated or treated with 10 μm cisplatin for 72 h. The whole cell lysates were collected and incubated with or without λ phosphatase in the presence (+) or absence (−) of the phosphatase inhibitor vanadate. The slower-migrating forms of Bim-EL are the result of phosphorylation. Actin was used as a loading control.
Cisplatin Treatment Activates MAPKs and Induces Bim Phosphorylation and Degradation
It has been shown that Bim can be phosphorylated by MAPKs, including ERK and JNK (16, 22, 30). Our previous study showed that cisplatin treatment can activate ERK, p38, and JNK (25). To determine whether cisplatin-induced ERK activation is responsible for Bim phosphorylation, we first determined the status of MAPK activation. RMG-1 cells were treated with cisplatin for 0, 24, 48, and 72 h, and activation of MAPK pathways was then determined. We chose RMG-1 cells because this cell line is already somewhat resistant to cisplatin. As shown in Fig. 2A, cisplatin treatment caused phosphorylation of ERK, p38, and JNK and their downstream targets including ATF-1, CREB, and c-Jun, indicating that cisplatin activates all three major MAPK pathways. To determine which pathway acts as survival signaling to counteract cisplatin-induced cell death, RMG-1 cells were left untreated or treated with the MEK1/2 inhibitor U0126 (10 μm), the p38 inhibitor SB203580 (10 μm), or the JNK inhibitor SP600125 (10 μm) for 30 min and then treated with 10 μm cisplatin for 48 and 72 h, and growth inhibition was determined by MTT assays. As shown in Fig. 2B, cisplatin alone slightly inhibited growth of RMG-1 cells, and U0126 alone had a minimal effect on cell growth. In contrast, inhibition of ERK signaling by U0126 enhanced cisplatin-induced inhibition, whereas such effects were minimal in combinations of cisplatin with SB203580 or SP600125. Similar results were obtained with several other ovarian cancer cell lines (data not shown). To further confirm the role of ERK signaling in counteracting cisplatin-induced killing, we performed colony formation assays to determine the role of ERK inhibition in cell survival. As shown in Fig. 2C, the combination of U0126 and cisplatin significantly inhibited colony formation, whereas a single agent alone had little effect. Thus, these data indicate that ERK but not JNK or p38 signaling contributes to cisplatin resistance in RMG-1 cells.
FIGURE 2.

Activation of the ERK pathway confers cisplatin resistance in RMG-1 cells. A, Western blot analyses of the levels of total and phosphorylated ERK, JNK, p38, c-Jun, and CREB protein levels in RMG-1 cells treated with 10 μm cisplatin for the indicated time periods. Phosphorylated ATF-1 is also shown. Actin was used as a loading control. B, MTT analyses of growth inhibition. RMG-1 cells left untreated or treated with 10 μm cisplatin (Cis) in the presence (+) or absence (−) of 10 μm U0126 (U), 10 μm SB203580 (SB), or 10 μm SP600125 (SP) for 48 or 72 h, respectively, as indicated. C, colony formation analyses. RMG-1 cells were left untreated or treated with 10 μm cisplatin in the presence or absence of 10 μm U0126 for 3 h. 400 cells/well were seeded in 6-well plates and grown for 12 days, followed by crystal violet staining. The lower panel shows quantification of survival colonies. The plating efficiencies of drug-treated wells were normalized to those of control wells. The plating efficiency of control wells was arbitrarily established as 100%.
Blockade of ERK Activation by U0126 Abrogates Bim Phosphorylation
It has been shown that ERK and JNK can phosphorylate Bim (21). We have shown that cisplatin treatment leads to the activation of MAPKs and Bim phosphorylation. To determine which MAPK is responsible for Bim phosphorylation, RMG-1 cells were treated with cisplatin in the absence and presence of U0126, SB203580, or SP600125, and Bim phosphorylation and the activation of MAPK pathways were determined. Fig. 3A shows that cisplatin caused Bim phosphorylation and activation of ERK, c-Jun, ATF-1, and CREB, which is consistent with the results obtained in Fig. 2A. Importantly, U0126 abolished cisplatin-induced Bim phosphorylation (Fig. 3A). Treatment with SB203580 followed by cisplatin was able to block CREB and ATF-1 phosphorylation, whereas SP600125 followed by cisplatin abolished c-Jun phosphorylation. SB203580 slightly blocked Bim phosphorylation, whereas SP600125 did not have any effect. Furthermore, blockage of Bim phosphorylation by U0126 but not SB203580 and SP600125 were also observed in the other two ovarian cancer cell lines, OVCA432 and OVCA420 (Fig. 3B). Taken together, these results strongly suggest that ERK is responsible for cisplatin-induced Bim phosphorylation.
FIGURE 3.

Activation of ERK by cisplatin phosphorylates Bim-EL, leading to Bim-EL degradation and cisplatin resistance in ovarian cancer cells. A, Western blot analyses of Bim-EL, ERK, c-Jun, and CREB phosphorylation. RMG-1 cells were left untreated or treated with 10 μm cisplatin in the presence (+) or absence (−) of 10 μm U0126, 10 μm SB203580, or 10 μm SP600125 for 48 h. Actin was used as a loading control. B, Western blot analyses of Bim-EL expression. OVCA432 and OVCA420 cells were left untreated or treated with 10 μm cisplatin in the presence or absence of 10 μm U0126, 10 μm SB203580, or 10 μm SP600125 for 48 h. Actin was used as a loading control.
Knockdown of ERK by siRNA Increases Cisplatin-induced Death, Whereas Knockdown of Bim Confers Cisplatin Resistance
To address the direct role of ERK in Bim phosphorylation, RMG-1 cells were transfected with either non-target or ERK siRNAs, and the effect of ERK knockdown on Bim phosphorylation and PARP cleavage were determined. Fig. 4A shows that total ERK in cells transfected with ERK siRNA was decreased significantly as compared with cells transfected with control siRNA. As expected, upon cisplatin treatment, Bim was rapidly phosphorylated and then degraded in cells transfected with control siRNA. By contrast, knockdown of ERK led to a significant decrease in Bim phosphorylation and degradation. Consistently, knockdown of ERK increased cisplatin-induced PARP cleavage, confirming a role for ERK in counteracting cisplatin-induced apoptosis.
FIGURE 4.

Knockdown of Bim decreases cisplatin sensitivity, whereas knockdown of ERK decreases Bim-EL phosphorylation and degradation and increases cisplatin sensitivity. A, Western blot analyses of ERK, Bim-EL, and PARP expression. RMG-1 cells were transfected with control non-target siRNA or ERK siRNA. After 48 h, transfected cells were left untreated (−) or treated (+) with 10 μm cisplatin for 48 h. Actin was used as a loading control. B, Western blot analyses of Bim-EL and PARP levels. RMG-1 cells were transfected with control non-target siRNA or Bim siRNA. After 48 h, cells were left untreated (−) or treated (+) with 10 μm cisplatin for 48 h. Actin was used as a loading control. C, MTT analyses of growth inhibition. RMG-1 cells were transfected with control non-target siRNA, ERK siRNA, or Bim siRNA. 48 h later, cells were treated with cisplatin for 48 h.
We have shown a correlation between ERK activation and Bim phosphorylation/degradation (Figs. 3A and 4A). To address a direct role for Bim in cisplatin-induced cell death, we used siRNA to knock down Bim expression and then determined the effect of Bim knockdown on PARP cleavage. As shown in Fig. 4B, the levels of Bim were significantly decreased in cells transfected with Bim siRNA as compared with cells transfected with control siRNA in the absence and presence of cisplatin treatment. PARP cleavage was decreased in cells transfected with Bim siRNA as compared with cells transfected with control siRNA in response to cisplatin treatment. These data suggest that Bim is directly involved in cisplatin-induced apoptosis.
To determine the effect of knockdown of ERK and Bim on cisplatin sensitivity, RMG-1 cells transfected with ERK, Bim, or control siRNAs were treated with cisplatin, and growth inhibition was determined by MTT assays. Fig. 4C shows that 20 μm cisplatin treatment caused ∼5% growth inhibition, whereas 10 μm cisplatin had a minimal effect (∼2%) in cells transfected with Bim siRNA as compared with cells transfected with control siRNA (∼20 and ∼10%, respectively). In contrast, growth inhibition was ∼20 and ∼40% in cells transfected with ERK siRNA treated with 10 and 20 μm cisplatin, respectively (Fig. 4C). These data suggest that ERK contributes to cisplatin resistance in RMG-1 cells. Importantly, knockdown of Bim rendered cells resistant to cisplatin treatments at both concentrations (Fig. 4C). Thus, these data suggest that Bim plays a critical role in cisplatin-induced apoptosis and that ERK contributes to cisplatin resistance in ovarian cancer cells.
Bim Degradation by a Proteasome Pathway Plays a Critical Role in Cisplatin Resistance in Ovarian Cancer Cells
To determine whether cisplatin treatment affects the half-life of Bim protein, RMG-1 cells were left untreated or treated with cisplatin in the presence of the protein synthesis inhibitor cycloheximide, and the levels of Bim protein were determined. As shown in Fig. 5A, cycloheximide alone caused slight Bim degradation. In contrast, Bim was phosphorylated following cisplatin treatment, and the phosphorylated Bim protein was rapidly degraded in the presence of cycloheximide (Fig. 5A). It has been shown that ERK-mediated phosphorylation can target Bim for degradation (16). It is likely that cisplatin-induced Bim phosphorylation/degradation is mediated by a proteasome pathway. To this end, RMG-1 cells were left untreated or treated with cisplatin, and the effects of the treatment on Bim ubiquitination were determined. Cisplatin treatment led to Bim ubiquitination (Fig. 5B, upper panel) and subsequent degradation. Further, MG132 blocked Bim degradation, although it was phosphorylated (Fig. 5B, lower panel). Correspondingly, PARP cleavage was significantly increased in cells treated with both agents as compared with cells treated with either cisplatin or MG132 alone. These data suggest that inhibition of Bim degradation sensitizes ovarian cancer cells to cisplatin-induced apoptosis.
FIGURE 5.

Effects of cisplatin and proteasome inhibitors on Bim-EL degradation and cell death. A, Western blot analyses of Bim-EL expression. RMG-1 cells were treated with 2 μg/ml cycloheximide (CHX) for the indicated time periods in the presence and absence of 10 μm cisplatin for 48 h. Actin was used as a loading control. B, RMG-1 cells were left untreated or treated with 10 μm cisplatin (Cis) for 24 h (upper panel). Bim was immunoprecipitated, and Western blot analyses with anti-Bim and ubiquitin (Ub) antibodies were performed. The asterisk indicates ubiquitinated Bim appearing as a smear of bands with a higher molecular weight. RMG-1 cells were left untreated or treated with 10 μm cisplatin in the presence (+) or absence (−) of the proteasome inhibitor MG132 (250 nm) for 48 h (lower panel). Actin was used as a loading control (Con). IB, immunoblotting. C, MTT analyses of growth inhibition. RMG-1 cells were left untreated or treated with 10 μm cisplatin in the presence or absence of 250 nm MG132 for 48 or 72 h. D, colony formation analyses of cell growth. RMG-1 cells were left untreated or treated with 10 μm cisplatin in the presence or absence of 250 nm MG132 (MG) for 3 h. 400 cells/well were seeded in 6-well plates and grown for 12 days, followed by crystal violet staining. Quantification of survival colonies is shown (lower panel). The plating efficiencies of drug-treated wells were normalized to those of control wells. The plating efficiency of the control wells was arbitrarily established as 100%.
To determine whether blocking Bim degradation affects growth, RMG-1 cells were treated with cisplatin, MG132, or their combination, and growth inhibition was determined by MTT assays. As shown in Fig. 5C, cisplatin or MG132 alone had minimal effects on cell growth. In contrast, the combination treatments significantly inhibited growth. To further validate the effects of the combination on cell growth, a long-term colony formation assay was performed. Consistent with the results obtained with MTT assays, combination treatments significantly inhibited colony formation, whereas single treatment alone had a minimal effect. Collectively, these data strongly suggest that the combination of cisplatin and proteasome inhibitors can effectively kill cisplatin-resistant ovarian cancer cells.
Bim Is Required for Proteasome Inhibitor-mediated Sensitization of Ovarian Cancer Cells to Cisplatin
We have shown that ERK-mediated Bim phosphorylation and subsequent degradation can be blocked by MG132. To determine the extent to which ERK plays a role in Bim-mediated cisplatin sensitivity, RMG-1 cells were transfected with either ERK or control siRNA, and the expression of ERK was then determined. Fig. 6A shows that ERK protein was effectively knocked down in cells transfected with ERK siRNA as compared with cells transfected with control siRNA. Cisplatin slightly caused growth inhibition (15%) in cells transfected with control siRNA as compared with cells transfected with ERK siRNA in which cisplatin caused about 30% growth inhibition (Fig. 6B), suggesting that the activation of ERK plays a role, but not a critical role, in cisplatin resistance. We also found that MG132 had a minimal effect on growth inhibition and that ERK knockdown did not increase the effect of MG132 on growth inhibition in ERK knockdown cells, indicating that ERK acts upstream of the Bim degradation pathway. Importantly, the combination of cisplatin and MG132 significantly inhibited growth (50%) as compared with untreated cells or cells treated with single agent alone, whereas ERK knockdown did not increase such effects. These results suggest that MG132 can sensitize resistant cells to cisplatin.
FIGURE 6.
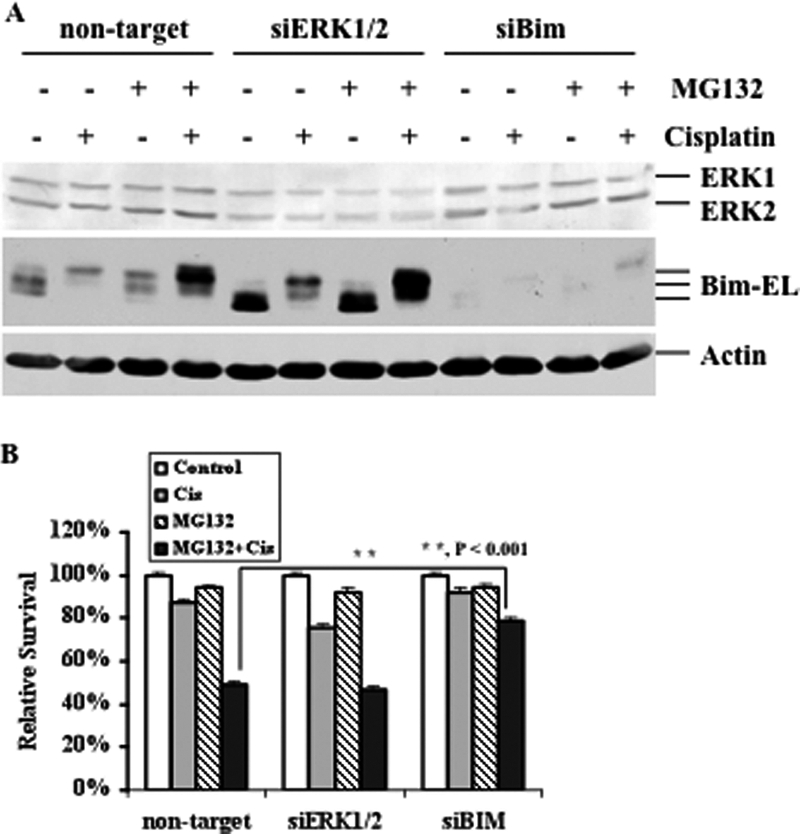
The Effects of ERK and Bim knockdown on proteasome inhibitor, cisplatin, and their combination-induced growth inhibition. A, Western blot analyses of Bim-EL and ERK expression. RMG-1 cells were transfected with control non-target siRNA, ERK siRNA, or Bim siRNA. After 48 h, cells were left untreated or treated with 10 μm cisplatin in the presence (+) or absence (−) of 250 nm MG132 for 48 h. Actin was used as a loading control. B, MTT analyses of growth inhibition. RMG-1 cells were transfected with control non-target siRNA, ERK siRNA, or Bim siRNA. 48 h later, cells were left untreated or treated with 10 μm cisplatin in the presence or absence of 250 nm MG132 for 48 h.
To determine whether MG132-induced sensitization is mediated by Bim, we first knocked down Bim in RMG-1 cells (Fig. 6A) and then treated these cells with cisplatin. As shown in Fig. 6B, knockdown of Bim led to cisplatin resistance, whereas a modest effect was observed in non-target siRNA-transfected cells. Importantly, although the combination treatments effectively inhibited growth in non-target siRNA transfected cells, knockdown of Bim conferred cells resistant to the combination treatment. Taken together, these data strongly suggest that Bim plays a critical role in cisplatin sensitivity, whereas ERK contributes to cisplatin resistance. Thus, the combination of cisplatin and proteasome inhibitor may effectively kill cisplatin resistant ovarian cancer cells.
The Combination Treatments Effectively Inhibit Growth of Ovarian Cancer Cells with Acquired Cisplatin Resistance
The major problem with the use of cisplatin is that cancer cells initially sensitive to cisplatin acquire resistance (12). Thus, acquired resistance is an urgent issue that is clinically relevant. To determine whether ERK-mediated Bim phosphorylation and subsequent degradation plays a role in acquired cisplatin resistance in ovarian cancer cells, we tested this mechanism in a pair of ovarian cancer cells: parental OV433 (OV433-P) cells and their derivative cisplatin-resistant OV433 cells (OV433-CR). OV433-CR cells were generated by chronically exposing cisplatin to parental OV433 cells for over 6 months (26). As shown in Fig. 7A, OV433-CR cells were much more resistant than OV433-P to cisplatin. As expected, cisplatin treatment caused PARP cleavage in OV433-P cells but not in OV433-CR cells. Importantly, OV433-CR had much higher levels of the basal and cisplatin-induced phosphorylated ERK as compared with OV433-P cells under the same treatment conditions (Fig. 7B). We found that Bim was phosphorylated and that cisplatin treatment increased its phosphorylation and degradation in OV433-CR cells, whereas such changes were not detected in OV433-P cells under the same treatment conditions. Further, the levels of Bim protein were much lower in OV433-CR cells than OV433-P cells (Fig. 7B). These results clearly show that the higher levels of phosphorylated ERK protein and Bim phosphorylation inversely correlated with the basal levels of Bim protein in both cell lines.
FIGURE 7.

The role of ERK and Bim in acquired cisplatin resistance. A, MTT analyses of growth inhibition. Parental OV433 cells (OV433-P) and their derivative cisplatin-resistant OV433 cells (OV433-CR) were treated with cisplatin for 48 h. B, Western blot analyses of the levels of ERK, Bim-EL, and PARP proteins. OV433-P and OV433-CR cells were left untreated (−) or treated (+) with 10 μm cisplatin for 24 h. Actin was used as a loading control. C, Western blot analyses of the levels of Bim-EL protein. OV433-P and OV433-CR cells left untreated or treated with 10 μm cisplatin in the presence (+) or absence (−) of 250 nm MG132 for 24 h. Actin was used as a loading control. D, MTT analyses of growth inhibition. OV433-P and OV433-CR cells left untreated or treated with 10 μm cisplatin (Cis) in the presence or absence of 250 nm MG132 for 48 h.
To determine whether increased ERK-mediated Bim phosphorylation is in fact directly associated with acquired cisplatin resistance, we treated both cell lines with MG132 to inhibit Bim degradation and then tested the effect of such treatments on cisplatin sensitivity. As shown in Fig. 7B, cisplatin treatment caused Bim phosphorylation in OV433-CR cells, whereas such change was not obvious in OV433-P cells. MG132 treatment increased the total and phosphorylated levels of Bim protein in OV433-CR cells but not in OV433-P cells (Fig. 7C). Furthermore, we found that the combination of cisplatin and MG132 significantly increased Bim phosphorylation in OV433-CR but not in OV433-P cells (Fig. 7C). Further, OV433-CR cells were more resistant than OV433-P cells to cisplatin, whereas both cell lines were resistant to MG132 despite the fact that OV433-P cells were slightly more sensitive than OV433-CR cells to MG132. Importantly, the combination of cisplatin with MG132 significantly inhibited growth of OV433-P cells, and such synergistic effects were also observed in OV433-CR cells, although its effect was not as prominent as that in OV433-P cells (Fig. 7D). Considering the fact that OV433-CR cells are very resistant to cisplatin, our data strongly suggest that degradation of Bim plays a critical role in cisplatin resistance and that the combination of cisplatin with MG132 may be a new strategy to reverse cisplatin resistance in ovarian cancer cells.
DISCUSSION
Our previous studies showed that cisplatin treatment led to the activation of MAPKs, including ERK (25, 26). It is generally accepted that activation of ERK can lead to cell survival and apoptosis resistance, but the mechanism by which activation of ERK contributes to cisplatin resistance is not understood. In this study, we showed that cisplatin treatment not only caused ERK activation but also increased Bim phosphorylation. Because Bim phosphorylation was observed in all cells treated with cisplatin regardless of their cisplatin sensitivities (Fig. 1B), this consideration suggests that Bim phosphorylation may be a general event in cisplatin-treated ovarian cancer cells.
It has been shown that ERK is an important player in mediating Bim phosphorylation to confer cell survival and apoptosis resistance. However, the role of ERK-mediated Bim phosphorylation and degradation in cisplatin resistance was not previously recognized. In this study, we observed a correlation between ERK activation and Bim phosphorylation, which suggests that cisplatin-induced ERK activation may be responsible for Bim phosphorylation and subsequent degradation, leading to cisplatin resistance. It has been shown that ERK-mediated Bim phosphorylation plays an important role in growth factor-induced cell survival (15). Subsequent studies showed that ERK phosphorylates Bim at Ser-65 and Ser-69 and that such phosphorylation targets Bim to the proteasome for degradation (17, 31). Because binding of Bim to Bcl-Xl and MCl-1 inhibits their anti-apoptotic function, degradation of Bim by ERK-mediated phosphorylation can release Bcl-Xl and MCL-1, allowing them to block apoptosis (31). Furthermore, it has been shown that the Bcr-ABL inhibitor imatinib can inhibit MEK-mediated Bim phosphorylation, leading to apoptosis in K562 leukemia cells (17). Importantly, accumulation of Bim protein has been shown to play a critical role for paclitaxel-induced chemosensitivity, and activation of the H-ras/MAPK pathway decreases the levels of Bim protein via a phosphorylation/degradation mechanism, leading to chemoresistance (23). In agreement, this study showed that cisplatin treatment led to ERK activation and Bim phosphorylation.
We showed that cisplatin not only induced the activation of ERK but also activated p38 and JNK (Fig. 2). Several kinases, including ERK and JNK, have been shown to phosphorylate Bim (16, 22, 32, 33). It is possible that Bim can be phosphorylated by other kinases rather than ERK. However, we found that U0126, but not SP600125 or SB203580, blocked Bim phosphorylation (Fig. 3). Furthermore, we showed that knockdown of ERK by siRNA inhibited cisplatin-induced Bim phosphorylation, whereas Bim phosphorylation by cisplatin treatment is intact in cells transfected with control siRNA (Fig. 4). Therefore, these data strongly suggest that ERK is responsible for Bim phosphorylation in response to cisplatin treatment.
It is known that cisplatin treatment can activate a number of pathways, including both cell death and survival pathways. Although we showed that ERK is activated by cisplatin, it is possible that the activation of ERK may have nothing to do with cisplatin-induced apoptosis. To exclude this possibility, we treated cells with cisplatin in the presence and absence of U0126, SP600125, and SB203580. Our data clearly showed that neither SP600125 nor SB203580 increased cisplatin-induced growth inhibition (Fig. 2B). By contrast, U0126 was not only able to block Bim phosphorylation but also sensitized cells to cisplatin-induced growth inhibition (Figs. 2B and 3). Importantly, we showed that the combination of cisplatin with U0126 significantly inhibited colony formation as compared with either U0126 or cisplatin alone (Fig. 2C). Taken together, these data clearly suggest that the activation of ERK plays a role in counteracting cisplatin-induced cell death, leading to cell survival.
We showed that ERK plays a critical role in Bim phosphorylation because inhibition of ERK activation by U0126 blocked Bim phosphorylation (Fig. 3). Importantly, we showed that knockdown of ERK by siRNA significantly increases the basal level of Bim protein in the absence of cisplatin treatment, which is consistent with a role for ERK in Bim phosphorylation and degradation (Fig. 4A). However, although the total level of Bim protein was increased in ERK siRNA-transfected cells as compared with cells transfected with control siRNA, a small amount of Bim was still phosphorylated in response to cisplatin treatment (Fig. 4A). This result suggests that cisplatin treatment may activate other kinases that are involved in Bim phosphorylation. It has been shown that JNK can phosphorylate Bim (22). It has also been shown that there is interaction among MAPKs. Although inhibition of JNK by its pharmacological inhibitor SP600125 did not affect cisplatin-induced Bim phosphorylation, it is possible that when ERK is inhibited, JNK may compensate for ERK knockdown to phosphorylate Bim (21). As a result, Bim can be phosphorylated in ERK knockdown (Fig. 4). We speculate that inhibition of both ERK and JNK may completely block cisplatin-induced Bim phosphorylation. This possibility is under investigation. Furthermore, it has been shown that Akt can phosphorylate Bim (33). We showed previously that cisplatin treatment can activate Akt to counteract cisplatin-induced cell death (34). It is possible that Akt may be also involved in Bim phosphorylation in cells when ERK is knocked down. We are currently testing whether inhibition of ERK and Akt can completely block Bim phosphorylation and subsequent degradation. Because Bim plays an important role in regulating cell death, as expected, blocking ERK-mediated Bim phosphorylation and degradation can increase cisplatin-induced apoptosis. Consistent with this, we showed that knockdown of ERK increased cisplatin-induced PARP cleavage and growth inhibition (Fig. 4, A and C). Conversely, we showed that knockdown of Bim increased cell survival (Fig. 4B). Collectively, these data suggest that ERK activation plays an important role in Bim degradation and cell survival.
It has been shown that Bim degradation is required for paclitaxel resistance and that inhibition of Bim degradation by Velcade can restore Bim expression and thereby abrogate ras-dependent paclitaxel resistance (23). Consistent with this, we showed that MG132 can inhibit Bim degradation (Figs. 5B, 6A, and 7C). We also showed that although Bim is phosphorylated by cisplatin treatment, MG132 is still able to sensitize cells to cisplatin-induced apoptosis (Fig. 5). Despite the fact that MG132 can increase Bim protein accumulation in the absence of cisplatin treatment, MG132 alone did not significantly affect apoptosis and cell survival (Fig. 5). These data suggest two possibilities. The first one is that RMG-1 cells are very resistant to proteasome inhibitors at the concentration used in this experiment and that blockage of Bim degradation alone is not sufficient to cause apoptosis and inhibit cell growth and colony formation. Another one is that simultaneous activation of apoptotic pathways and inhibition of Bim degradation can significantly increase the cisplatin-induced killing effect. Consistent with this, we showed that knockdown of Bim did not increase the effects of combination of cisplatin and MG132 on growth inhibition, whereas knockdown of ERK modestly increased cisplatin-induced growth inhibition (Fig. 6B), which is consistent with our previous study showing that ERK plays a role in cisplatin resistance in ovarian cancer (25). Our data also showed that in Bim knockdown cells, the combination of cisplatin with MG132 still caused about 20% growth inhibition as compared with cisplatin or MG132 alone (Fig. 6B), further confirming that inhibition of Bim degradation does not solely account for cisplatin-induced growth inhibition.
Although we showed that ERK-mediated Bim phosphorylation and subsequent degradation is important for cisplatin resistance, the question becomes whether this mechanism is clinically relevant. Because the majority of ovarian cancer patients who are initially sensitive to cisplatin will eventually relapse and in many cases acquire cisplatin resistance (1), we asked if Bim degradation via ERK-mediated phosphorylation plays a role in acquired cisplatin resistance. We showed that OV433-CR cells express higher levels of phosphorylated ERK and lower levels of Bim as compared with their counterpart OV433-P cells (Fig. 7). As expected, cisplatin treatment caused apoptosis in OV433-P cells but not in OV433-CR cells (Fig. 7B). Thus, we conclude that combination of cisplatin with MG132 can effectively inhibit growth inhibition in cisplatin-resistant OV433-CR cells.
In conclusion, we demonstrate that Bim degradation plays a critical role in cisplatin resistance. We show that cisplatin treatment activates the ERK survival pathway and in turn phosphorylates Bim, leading to Bim degradation and cisplatin resistance (Fig. 8). We also show that inhibition of Bim degradation by MG132 increases Bim expression, leading to Bim-mediated apoptotic cell death. In addition, we believe that cisplatin can also activate apoptotic pathways, including JNK (35), leading to cell death. Blockage of Bim degradation and activation of cisplatin-induced apoptosis pathways simultaneously (e.g. JNK) can effectively induce ovarian cancer cell death. Thus, this study suggests that the combination of the proteasome inhibitors (e.g. Velcade) with cisplatin may overcome cisplatin resistance in ovarian cancer.
FIGURE 8.

A model for the possible mechanism of cisplatin resistance involving ERK and Bim-EL in ovarian cancer cells. Activation of JNK can lead to apoptosis as shown previously (35).
This work was supported by the Gail Purtan Ovarian Cancer Research Fund.
- Bim
- Bcl-2-interacting mediator of cell death
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- CREB
- cAMP response element-binding protein
- CHX
- cycloheximide
- MAPK
- mitogen-activated protein kinase
- PARP
- poly (ADP-ribose) polymerase.
REFERENCES
- 1. Weinberg L. E., Rodriguez G., Hurteau J. A. (2010) J. Surg. Oncol. 101, 334–343 [DOI] [PubMed] [Google Scholar]
- 2. Johnson S. W., Shen D., Pastan I., Gottesman M. M., Hamilton T. C. (1996) Exp. Cell Res. 226, 133–139 [DOI] [PubMed] [Google Scholar]
- 3. Cullen K. J., Newkirk K. A., Schumaker L. M., Aldosari N., Rone J. D., Haddad B. R. (2003) Cancer Res. 63, 8097–8102 [PubMed] [Google Scholar]
- 4. Turchi J. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 4337–4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dabholkar M., Vionnet J., Bostick-Bruton F., Yu J. J., Reed E. (1994) J. Clin. Invest. 94, 703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perego P., Giarola M., Righetti S. C., Supino R., Caserini C., Delia D., Pierotti M. A., Miyashita T., Reed J. C., Zunino F. (1996) Cancer Res. 56, 556–562 [PubMed] [Google Scholar]
- 7. Beale P. J., Rogers P., Boxall F., Sharp S. Y., Kelland L. R. (2000) Br. J. Cancer 82, 436–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Altomare D. A., Wang H. Q., Skele K. L., De Rienzo A., Klein-Szanto A. J., Godwin A. K., Testa J. R. (2004) Oncogene 23, 5853–5857 [DOI] [PubMed] [Google Scholar]
- 9. Kelland L. (2007) Nat. Rev. Cancer 7, 573–584 [DOI] [PubMed] [Google Scholar]
- 10. Kuo M. T., Chen H. H., Song I. S., Savaraj N., Ishikawa T. (2007) Cancer Metastasis Rev. 26, 71–83 [DOI] [PubMed] [Google Scholar]
- 11. Wu G. S. (2007) Cancer Metastasis Rev. 26, 579–585 [DOI] [PubMed] [Google Scholar]
- 12. Wang D., Lippard S. J. (2005) Nat. Rev. Drug Discov. 4, 307–320 [DOI] [PubMed] [Google Scholar]
- 13. Johnson G. L., Lapadat R. (2002) Science 298, 1911–1912 [DOI] [PubMed] [Google Scholar]
- 14. Chang L., Karin M. (2001) Nature 410, 37–40 [DOI] [PubMed] [Google Scholar]
- 15. Weston C. R., Balmanno K., Chalmers C., Hadfield K., Molton S. A., Ley R., Wagner E. F., Cook S. J. (2003) Oncogene 22, 1281–1293 [DOI] [PubMed] [Google Scholar]
- 16. Ley R., Balmanno K., Hadfield K., Weston C., Cook S. J. (2003) J. Biol. Chem. 278, 18811–18816 [DOI] [PubMed] [Google Scholar]
- 17. Luciano F., Jacquel A., Colosetti P., Herrant M., Cagnol S., Pages G., Auberger P. (2003) Oncogene 22, 6785–6793 [DOI] [PubMed] [Google Scholar]
- 18. Cory S., Adams J. M. (2002) Nat. Rev. Cancer 2, 647–656 [DOI] [PubMed] [Google Scholar]
- 19. O'Connor L., Strasser A., O'Reilly L. A., Hausmann G., Adams J. M., Cory S., Huang D. C. (1998) EMBO J. 17, 384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsu S. Y., Lin P., Hsueh A. J. (1998) Mol. Endocrinol. 12, 1432–1440 [DOI] [PubMed] [Google Scholar]
- 21. Ley R., Ewings K. E., Hadfield K., Cook S. J. (2005) Cell Death Differ. 12, 1008–1014 [DOI] [PubMed] [Google Scholar]
- 22. Lei K., Davis R. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tan T. T., Degenhardt K., Nelson D. A., Beaudoin B., Nieves-Neira W., Bouillet P., Villunger A., Adams J. M., White E. (2005) Cancer Cell 7, 227–238 [DOI] [PubMed] [Google Scholar]
- 24. Pei X. Y., Dai Y., Tenorio S., Lu J., Harada H., Dent P., Grant S. (2007) Blood 110, 2092–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J., Zhou J. Y., Wu G. S. (2007) Cancer Res. 67, 11933–11941 [DOI] [PubMed] [Google Scholar]
- 26. Wang J., Zhou J. Y., Zhang L., Wu G. S. (2009) Cell Cycle 8, 3191–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu J., Zhou J. Y., Tainsky M. A., Wu G. S. (2007) Cancer Res. 67, 1203–1211 [DOI] [PubMed] [Google Scholar]
- 28. Zhou J. Y., Liu Y., Wu G. S. (2006) Cancer Res. 66, 4888–4894 [DOI] [PubMed] [Google Scholar]
- 29. Wu G. S., Burns T. F., McDonald E. R., 3rd, Jiang W., Meng R., Krantz I. D., Kao G., Gan D. D., Zhou J. Y., Muschel R., Hamilton S. R., Spinner N. B., Markowitz S., Wu G., el-Deiry W. S. (1997) Nat. Genet. 17, 141–143 [DOI] [PubMed] [Google Scholar]
- 30. Cai B., Chang S. H., Becker E. B., Bonni A., Xia Z. (2006) J. Biol. Chem. 281, 25215–25222 [DOI] [PubMed] [Google Scholar]
- 31. Ewings K. E., Hadfield-Moorhouse K., Wiggins C. M., Wickenden J. A., Balmanno K., Gilley R., Degenhardt K., White E., Cook S. J. (2007) EMBO J. 26, 2856–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ley R., Ewings K. E., Hadfield K., Howes E., Balmanno K., Cook S. J. (2004) J. Biol. Chem. 279, 8837–8847 [DOI] [PubMed] [Google Scholar]
- 33. Qi X. J., Wildey G. M., Howe P. H. (2006) J. Biol. Chem. 281, 813–823 [DOI] [PubMed] [Google Scholar]
- 34. Peng D. J., Wang J., Zhou J. Y., Wu G. S. (2010) Biochem. Biophys. Res. Commun. 394, 600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Z., Xu J., Zhou J. Y., Liu Y., Wu G. S. (2006) Cancer Res. 66, 8870–8877 [DOI] [PubMed] [Google Scholar]