

Abstract
How phospholipase D (PLD) is involved in myogenesis remains unclear. At the onset of myogenic differentiation of L6 cells induced by the PLD agonist vasopressin in the absence of serum, mTORC1 complex was rapidly activated, as reflected by phosphorylation of S6 kinase1 (S6K1). Both the long (p85) and short (p70) S6K1 isoforms were phosphorylated in a PLD1-dependent way. Short rapamycin treatment specifically inhibiting mTORC1 suppressed p70 but not p85 phosphorylation, suggesting that p85 might be directly activated by phosphatidic acid. Vasopressin stimulation also induced phosphorylation of Akt on Ser-473 through PLD1-dependent activation of mTORC2 complex. In this model of myogenesis, mTORC2 had a positive role mostly unrelated to Akt activation, whereas mTORC1 had a negative role, associated with S6K1-induced Rictor phosphorylation. The PLD requirement for differentiation can thus be attributed to its ability to trigger via mTORC2 activation the phosphorylation of an effector that could be PKCα. Moreover, PLD is involved in a counter-regulation loop expected to limit the response. This study thus brings new insights in the intricate way PLD and mTOR cooperate to control myogenesis.
Keywords: Cell Differentiation, mTOR Complex (mTORC), Phosphatidic Acid, Phospholipase D, Protein Kinase C (PKC), Skeletal Muscle, Myogenesis, Vasopressin
Introduction
The mammalian target of rapamycin (mTOR)2 is a serine/threonine protein kinase that integrates signals provided by growth factors, nutrient availability, energy levels, or redox status to adapt protein synthesis and major cell functions such as growth, proliferation, and survival to the physiological conditions (1). It exists in two complexes, mTORC1 and mTORC2, that are differentially regulated, have distinct effector substrates, and are differentially sensitive to rapamycin, a bacterial macrolide endowed with anti-proliferative and immunosuppressant activities. Whereas activity of mTORC1 complex is highly sensitive to acute treatment by nanomolar concentrations of rapamycin, only prolonged rapamycin treatment is able to induce mTORC2 disruption and inactivation (2–4).
A recently identified regulatory signal impacting on mTOR activity is the production of phosphatidic acid (PA) by phospholipase D (PLD)-mediated hydrolysis of phosphatidylcholine (5, 6). PLD, which can be activated by a variety of hormones, growth factors, and cytokines, is present in most tissues under two isoforms, PLD1 and PLD2, endowed with different properties, regulations, and functions (7). Phosphatidic acid has been shown to specifically bind to mTOR protein on the FRB domain, a regulatory site also responsible for the binding of rapamycin in complex with protein FKBP12. The protein/phospholipid interaction causes the activation of mTOR kinase, and rapamycin has been proposed to exert its inhibitory effects on mTOR by competing with PA and blocking PA-mediated activation (8–10).
Signaling by mTOR has become a topic of particular interest in the field of skeletal muscle biology due to the critical involvement of this kinase in muscle remodeling. Thus, muscle hypertrophy induced by exercise has been shown to involve mTOR signaling (11–13). Rapamycin inhibits IGF-1-induced myotube hypertrophy (14, 15) and re-growth of myofibers after denervation (11). Knocking out the mTOR effector S6K1 produces mice with smaller myofibers, suggesting that mTOR, via S6K1 activation, is required for muscle cell growth (16). Furthermore, mTOR regulates in vivo muscle regeneration after tissue damage (17), and the potent inhibition exerted by rapamycin on in vitro myogenic differentiation has been known for more than a decade (18–20), although the precise mechanism by which mTOR inhibition affects the myogenic process remains controversial (4, 20, 21).
We and others have reported that PLD, particularly the PLD1 isoform, is positively involved in the regulation of myogenic differentiation. A PLD agonist, the neurohypophysial hormone arginine-vasopressin (AVP), efficiently stimulates in vitro differentiation of myogenic L6 cells and primary human myoblasts in the presence of reduced serum concentration (22, 23). Using L6 myoblasts cultured in the presence of AVP, we have previously observed that AVP induces a stimulation of PLD signaling necessary for the myogenic response (24–26). Besides, PLD1-mediated mTOR activation has been proposed to support C2C12 cell differentiation through a kinase-independent enhancement of IGF-2 expression (27). However, which mTOR complex is involved and whether the regulation of IGF-2 production by PLD1 and mTOR intervenes in other myogenic models remained pending questions.
In the present work we took advantage of the model of AVP-induced L6 cell differentiation, which avoids the use of serum and thus allows close control of the stimuli supplied to the cells, to investigate the role of regulation by PLD of both mTOR complexes. We observed that mTORC1 and mTORC2 are both activated in a PLD-dependent way and play opposite roles in regulating the myogenic response.
EXPERIMENTAL PROCEDURES
Materials and Reagents
ECL immunodetection reagent was from Pierce. Bradford protein assay was from Bio-Rad. Rapamycin was purchased from Coger (Paris, France). Dioctanoyl-PA and egg yolk phosphatidic acid (sodium salts), propranolol, AVP, wortmannin, mTOR inhibitor PP242, insulin, and mouse recombinant IGF-2 were from Sigma. ZSTK474 compound was supplied by LC Laboratories. Negative control siRNA was from Eurogentec (Angers, France). The following antibodies were used: anti-phospho-Thr-1135-Rictor anti-phospho-Thr-389/Thr-412-S6K1, anti-phospho-Ser-473-Akt, anti-S6K1, anti-Akt, anti-Raptor, anti-Rictor (Cell Signaling Technology); anti-HA tag and anti-α-tubulin monoclonal antibody (Sigma); PLD1-specific polyclonal antibody (kindly provided by Dr. S. Bourgoin, Université Laval, Canada); anti-phospho-Ser-657-PKCα (Millipore); anti-PKCα antibody (Abgent); anti-IGF2 monoclonal antibody clone S1F2 (Upstate Biotechnology), F5D anti-myogenin monoclonal antibody (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA); HRP-conjugated anti-mouse- or anti-rabbit-IgG antibodies (Jackson ImmunoResearch Laboratories (Soham, UK)). PLD1, PLD2, PKCα, Raptor, and Rictor siRNAs were synthesized by Sigma. 5-Fluoro-2-indolyldeschlorohalopemide, a potent inhibitor of both PLD isoforms (28, 29), was obtained from Sigma. Selective inhibitors of PLD1 (CAY10593) and PLD2 (CAY10594) (30) were supplied by Cayman Chemical Co.
Cell Culture and Transfection
L6 myoblasts of the C5 subclone (22) were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/liter glucose and 10% fetal bovine serum at 37 °C with 5% CO2. To induce differentiation, cells were grown to 100% confluence and switched to differentiation medium (DMEM containing 10−7 m AVP without serum). L6 cells grown on 6-well plates to 40–50% confluence were transfected with 2 μg of plasmidic DNA per well using Exgene transfection reagent (from Euromedex) for 48 h. Medium was changed after 24 h of transfection. The hS6K1-pMT2 plasmid expressing both p85- and p70-S6K1 was a generous gift of Dr. P. J. Coffer (Utrecht, The Netherlands). The Myc-tagged-Akt1-pCDNA3 vector and mutant p110-PI3K α-pEGFP vectors have been described previously (31).
Determination of PLD Activity
Cells were labeled with 2 μCi/ml [3H]palmitic acid for 2 h at 37 °C in serum-free medium. Butan-1-ol (1% final concentration) was added 15 min before cell treatment by 10−7 m AVP for 10 min. Cells were then collected, lipids were extracted, and phosphatidylbutanol was separated by bidimensional TLC as described in Zeiller et al. (32). TLC plates were then stained with Coomassie Brilliant Blue R, phosphatidylbutanol spots were scraped off, and radioactivity was determined by liquid scintillation counting. Radioactivity associated with phosphatidylbutanol was expressed as a percentage of total phospholipid radioactivity.
Western Blots
Cells were lysed in ice-cold lysis buffer containing 20 mm Tris/HCl, 100 mm NaCl, 10 mm sodium pyrophosphate, 10 mm glycerophosphate, 50 mm NaF, 1.5 mm Na3VO4, 1% Triton, and protease inhibitor mixture (pH 7.6). Lysates were kept on ice for 15 min and cleared by centrifugation at 13,000 × g for 15 min. Protein concentrations were determined by the Bio-Rad protein assay. Cell lysates were separated by SDS/PAGE. Particular sample treatment and electrophoresis conditions for PLD analysis have already been described (25). Proteins were transferred onto PVDF membranes blocked with 5% BSA in Tris-buffered saline, 0.1% Tween 20 and incubated with the various antibodies following the manufacturer's recommendations. Immunoblots were revealed with the ECL detection system (Pierce) and quantified with Image J software.
Immunofluorescence Microscopy
Myogenin nuclear accumulation was detected by immunofluorescence. The cells were fixed by 3.7% formaldehyde for 20 min at room temperature and permeabilized with 0.1% Triton for 10 min at room temperature, and aspecific labeling was blocked in 1% BSA for 20 min. Anti-myogenin F5D monoclonal antibody was added undiluted and incubated overnight at room temperature. After washing with 1% BSA in PBS, fluorescein- or rhodamine-conjugated anti-mouse IgG antibody was added, diluted 1:1000 in 1% BSA, for 1 h. Nuclei were stained with 1 μg/ml 4.5-diamidino-2-phenylindole (DAPI) for 3 min. The cells were examined by fluorescence microscopy with an Axiovert 200 microscope, an objective LD A-plan, 20×/0.30 PHI ∞/40, an Axiocam MRm camera, and Axiovision 4.1 image acquisition software (Carl Zeiss, Göttingen, Germany). The total number of nuclei in the considered fields was assessed on phase contrast images or after nuclei staining with DAPI.
Short Interfering RNA (siRNA) Transfection
The siRNA used were targeted to rat PLD1 sequence 5′-AAGTTAAGAGGAAATTCAAGC-3′, rat PLD2 sequence 5′-GACACAAAGTCTTGATGAG-3′, rat PKCα sequence 5′-GAAGCAAGCACAAGTTCAA-3′, rat Raptor sequence 5′-GACAGTGGGCCTCTCAGGA-3′, and rat Rictor sequence 5′-GTTCGTTCCGACACTATAA-3′. Transfection of siRNAs was performed using Hiperfect reagent (Qiagene) with 50 nm siRNA for 48 h, and medium was changed after 24 h of transfection.
Reverse Transcriptase and Real-time PCR
Total RNA was isolated from L6 cells using TRIzol reagent (Invitrogen). After quantification, 1 μg of total RNA was reverse-transcribed in the presence of 100 units of Superscript II (Invitrogen) using random hexamers and oligo(dT). Real-time PCR was performed with the Fast Start DNA Master SYBR Green kit using Rotor-Gene 6000 (Corbett Research, Mortlake, Australia). Data were analyzed using LightCycler software (Roche Diagnostics) and normalized to the TATA-binding protein (TBP) housekeeping gene transcripts. Alternatively, for RT-PCR of IGF-2 mRNA, 1 μg total RNA was processed with Access RT-PCR kit (Promega). Specific Sense and Antisense Primers Used for Amplification Were: myogenin sense (CAATGCACTGGAGTTTGGTC) and myogenin antisense (CATATCCTCCACCGTGATGC); PLD1 sense (GGTCAGAAAGATAACCCAGG) and PLD1 antisense (GAAGCGAGACAGCGAAATGG); PLD2 sense (TTGCTGGCTGTGTGTCTGGC) and PLD2 antisense (GGACCTCCAGAGACACAAAG); troponin I skeletal slow 1 sense (TGAGGAGCGCTATGATATCG) and troponin I skeletal slow 1 antisense (TTCACAGACTTGAGGTTGGC); IGF-2 sense (GGAAGTCGATGTTGGTGCTT) and IGF-2 antisense (CGAGATCTTCATGAGGTAGTC).
Adenoviral Constructions and Virus Production
Recombinant adenoviral genomes carrying the HA-tagged cDNA of interest (hPLD1b, hPLD2, or GFP) were generated as previously described (32). Infections were performed at a multiplicity of infection of 100 in complete medium. After 12 h of incubation in the presence of viral particles, the medium was changed, and cells were cultured for 24–72 h. Under these conditions, >75% of the cells expressed GFP.
Statistical Analysis
Data expressed as the means ± S.E. were analyzed by one-way analysis of variance, and means were compared by a protected t test. p ≤ 0.05 was considered significant.
RESULTS
In Differentiating Conditions, mTORC1 and S6K1 Are Activated in a Phospholipase D-dependent Way
We first investigated the effects of AVP, a PLD agonist that strongly enhances the differentiation of L6 cells in low serum culture conditions, on the phosphorylation status of protein substrates of the mTORC1 complex. AVP markedly stimulated the phosphorylation of both p70 and p85 isoforms of S6K1 on the mTORC1 target residues (Thr-389 on p70-S6K1 and the homologous Thr-412 on p85-S6K1) (Fig. 1A). In addition, the mTORC1 substrate 4E-BP1 and S6 ribosomal protein, a substrate of S6K1, were phosphorylated in response to AVP (not shown). The induction of differentiation was thus accompanied by mTORC1 and S6K1 activation. As a confirmation of mTORC1 involvement in the response to AVP, we assessed the effects on S6K1 phosphorylation of depletion of Raptor, a specific component of mTORC1 complex. siRNA-induced Raptor silencing (i.e. inactivation of mTORC1), but not Rictor silencing (i.e. inactivation of mTORC2), markedly decreased AVP-induced phosphorylation of p70 and p85 S6K1 isoforms (Fig. 1B).
FIGURE 1.


PLD is required for S6K1 phosphorylation. A, left panel, as a control for the immunodetection of S6K1 isoforms, L6 myoblasts were transfected with pMT2-S6K1, which expresses both p85- and p70-S6K1 (52), or empty pMT2 vector for 48 h, lysed, and analyzed by Western blotting for total and Thr-389/Thr-412-phosphorylated S6K1. The blots show that both p70 and p85 isoforms can be detected by the anti-protein antibody and by the anti-phosphoprotein antibody. Right panel, L6 myoblasts were serum-starved overnight and treated with 10−7 m AVP for the indicated times, and cell lysates were analyzed as above. B, shown is a Western blot of cells transfected with 50 nm control siRNA, Raptor-siRNA, or Rictor-siRNA and stimulated for 40 min with 10–7 M AVP. C and D, cells were serum-starved overnight and pretreated or not with 0.5% 1-butanol (BuOH1) or 2-butanol (BuOH2) for 30 min before 40 min of stimulation by 10−7 m AVP or 100 μm dioctanoyl-PA (PA-diC8) or 100 μm propranolol (propra). E, cells were transfected with 50 nm control siRNA or PLD1-siRNA or PLD2-siRNA for 48 h, serum-starved overnight, and stimulated by AVP for 40 min. Quantitative RT-PCR was performed to assess the efficiency of PLD1 and PLD2 knockdown (left diagram, means ± S.E. of at least four determinations). The cell lysates were analyzed by Western blot (right panel). F, cells were infected with GFP-adenovirus or PLD1-adenovirus or PLD2-adenovirus at a multiplicity of infection of 100 for 48 h, serum-starved overnight, and stimulated by AVP for 40 min. The cell lysates were analyzed by Western blot (right panel). The efficiency of PLD overexpression was assessed in control Western blots of cells infected at various multiplicities of infection and probed with either anti-PLD1 antibody or anti-HA tag antibody for recombinant PLD2 detection (left panel). G, cells were shifted to serum-free medium and pretreated for 40 min with 100 nm rapamycin (rapa) before 40 min of treatment with 10−7 m AVP or 100 μm propranolol (propra). The cell lysates were analyzed by Western blotting for total and Thr-389/Thr-412-phosphorylated S6K1. The diagrams show the quantification of Rictor, Raptor, or phospho-S6K1 isoforms in the Western blot above after normalization by either tubulin or the S6K1 proteins (mean ± S.E. of three determinations except diagram G, right : data of one experiment representative of two performed). *, significantly different from control cells, p < 0.05; **, p < 0.01.
To determine whether the activation of the mTORC1 pathway by AVP involved PLD, 1-butanol, which specifically prevents the formation of PA, the normal product of PLD, was added to the cells. We observed that 1-butanol more efficiently inhibited S6K1 phosphorylation than 2-butanol, an isomer not recognized by PLD used as a control for aspecific alcohol effects, showing that AVP-induced S6K1 phosphorylation requires PLD activity (Fig. 1C). This was confirmed by using compounds that mimic PLD activation. The addition to the cells of either exogenous PA or propranolol, an inhibitor of PA phosphatase that induces an accumulation of endogenous PA, induced S6K1 phosphorylation (Fig. 1D). To identify the PLD isoform(s) that is responsible for the regulation of S6K1, selective silencing of either PLD1 or PLD2 isoform was performed by using siRNAs. Depletion of PLD1 strongly decreased S6K1 phosphorylation, whereas PLD2 silencing was less efficient. In agreement, the adenovirus-induced overexpression of PLD1 strongly enhanced S6K1 phosphorylation, PLD2 overexpression being less effective. (Fig. 1, E and F). These results strongly suggest that PLD, and especially the PLD1 isoform, regulates S6K1 phosphorylation on critical positions, and as a consequence, S6K1 activity in L6 cells induced to differentiate.
Because the activity of mTOR kinase is regulated by PA binding (5, 6), we asked whether the effects of the PLD and PA level changes on S6K1 were solely mediated by mTORC1 action. To this end, the cells were submitted to a short treatment by rapamycin (40 min) in conditions known to suppress only mTORC1 activity (Fig. 1G). This treatment totally suppressed the activation of p70 phosphorylation by either AVP or propranolol, showing that PA-dependent p70-S6K1 phosphorylation was entirely mediated by mTORC1. Instead, p85-S6K1 phosphorylation was incompletely inhibited by rapamycin, showing that p85-S6K1 can be in part activated by PA in a mTORC1/rapamycin-independent way.
Under Differentiating Conditions, mTORC2 and Akt Are Activated in a Phospholipase D-dependent Way
Upon AVP stimulation, the phosphorylation of Ser-473 on Akt was rapidly increased, the peak of phosphorylation being reached in 6–8 min. This Akt activation also involved the participation of PLD, as it was reproduced by treatment of the cells by exogenous PA or propranolol (Fig. 2A). PLD implication was further supported by the effect of PLD isoforms overexpression, PLD1 inducing a marked increase in Akt phosphorylation, whereas PLD2 was less effective (Fig. 2B). In agreement, siRNA-mediated depletion of PLD, especially PLD1, decreased Ser-473-Phospho-Akt in the presence of AVP (Fig. 2C).
FIGURE 2.

mTORC2 and Akt are activated in a phospholipase D-dependent way. A, cells were serum-starved overnight and stimulated for the indicated times with 10−7 m AVP or 100 μm dioctanoyl-PA (PA-diC8) or 100 μm propranolol (propra). Cell lysates were subjected to Western blotting analysis for total Akt protein and Ser-473-phospho-Akt. As a positive control, the effect of insulin is shown in the upper blot, the lane on the right. B, cells were infected with GFP, PLD1, or PLD2 adenovirus for 48 h, serum-starved overnight, treated with 10−7 m AVP for 7 min, and analyzed by Western blotting. C and D, cells were transfected with 50 nm control siRNA or PLD1-siRNA or PLD2-siRNA or Raptor-siRNA or Rictor-siRNA for 48 h, serum-starved overnight, treated with 10−7 m AVP for 7 min, and analyzed by Western blotting. E, cells treated with 100 nm rapamycin (rapa) for 24 h were stimulated with 10−7 m AVP or 100 μm propranolol (propra) for 7 and 30 min, respectively, and subjected to Western blotting analysis. The diagrams show the quantification of phospho-Ser-473-Akt in the Western blot above after normalization by Akt protein amount (means ± S.E. of three determinations; *, significantly different from control cells, p < 0.05).
It is well known that phosphorylation of Ser-473 position on Akt can be mediated by the mTORC2 complex. To verify the participation of mTORC2 in PA-stimulated Akt phosphorylation, siRNA silencing experiments were performed. Depletion of Rictor, but not of Raptor, inhibited Akt phosphorylation induced by AVP (Fig. 2D) or propranolol (not shown), showing that it was mTORC2-dependent and mTORC1-independent.
Besides siRNA silencing, another means to deplete cells in mTORC2 complex is the use of longer term treatment by rapamycin (3). 24-h rapamycin inhibited Akt phosphorylation induced by AVP or propranolol, in agreement with the conclusion that it was mediated by mTORC2 activation (Fig. 2E).
Regulation of mTOR by PLD and Myogenic Differentiation
We had previously proposed that PLD activity is required for in vitro myogenic differentiation (24–26). In further support to this conclusion, we observed in the present work that fluoro-2-indolyldeschlorohalopemide, a potent synthetic inhibitor of both PLD isoforms (28, 29), almost totally suppressed AVP-induced PA accumulation at 250 nm (Fig. 3A) and dose-dependently inhibited myogenin expression with an approximate EC50 of 100 nm (Fig. 3B). Isoform-specific PLD inhibitors were also studied (30). Each of the PLD1- and PLD2-specific inhibitors used at 50 nm roughly inhibited by 50% AVP-induced PA accumulation (Fig. 3A). However, they had clearly different effects on myogenin expression. The PLD1 inhibitor dose-dependently inhibited this response with an approximate EC50 of 5 nm, whereas the PLD2 inhibitor had no significant effect up to 500 nm (Fig. 3C). The observation that adenovirus-mediated overexpression of the PLD1 isoform increased the percentage of myogenin-positive nuclei in cells after 48 h in differentiation medium, further supported the positive involvement of PLD1 in the myogenic response (Fig. 3D).
FIGURE 3.

PLD activity is required for in vitro myogenic differentiation. A, [3H]palmitic acid-labeled cells were pretreated for 15 min by 1% 1-butanol and various PLD inhibitors: non isoform-specific fluoro-2-indolyldeschlorohalopemide (FIPI), PLD1-specific I-PLD1, and PLD2-specific I-PLD2. Tritiated phosphatidylbutanol (Pbut) formed was quantified after 10 min of stimulation by 10−7 m AVP. PLD activity is expressed as the percentage of radioactivity in phosphatidylbutanol relative to radioactivity in total phospholipids (*, different from AVP-stimulated cells, p < 0.05). B, L6 cells were cultured in the presence of 10−7 m AVP for 48 h with varying concentrations of PLD inhibitor fluoro-2-indolyldeschlorohalopemide. Cells were lysed, and proteins were subjected to Western blot analysis of myogenin expression. C, L6 cells were cultured in the presence of 10−7 m AVP for 48 h, with varying concentrations of PLD1-specific inhibitor (left panel) or PLD2-specific inhibitor (right panel). Cells were lysed, and proteins were subjected to Western blot analysis of myogenin expression (mean ± S.E. of three determinations; *, significantly different from cells + AVP, p < 0.05). D, shown is immunofluorescence microscopy of nuclear myogenin in non-infected L6 cells (NI) and cells infected with adenovirus coding for GFP, hPLD1 (PLD1), or hPLD2 (PLD2) and cultured in the presence of AVP for 48 h. Total nuclei were visualized by DAPI staining. Differentiation was assessed by the mean percentage of myogenin-positive nuclei, determined in 10 fields. *, significantly different from control cells, p < 0.001. Bar = 40 μm.
In agreement with what was observed by numerous groups with different myogenic cell types, we observed a complete blockade of myogenic differentiation of L6 cells in the presence of rapamycin. However, at variance with what was observed by others in C2C12 cells (4), we observed a total inhibition of myogenic response at the very early steps of myogenin expression (mRNA, protein) and nuclear accumulation, i.e. before the cell fusion step (Fig. 4A). This showed that either mTORC1 or mTORC2 complexes are required for the early myogenic response. To discriminate between the role of each of the two complexes, differentiation was assessed during their respective siRNA-mediated depletion. As shown in Fig. 4B, Raptor silencing (i.e. inactivation of mTORC1) had a positive effect on either myogenin expression or nuclear accumulation. Conversely, Rictor silencing (i.e. inactivation of mTORC2) decreased the early myogenic responses, showing that mTORC2 is required for myogenic differentiation, whereas mTORC1 negatively regulates it. Consistent with this latter conclusion, we observed that overexpression of the mTORC1 effector S6K1 had negative effects on the expression of myogenin and troponin, a sarcomeric protein marker of advanced differentiation (Fig. 4C). Because a negative regulation of mTORC2 by mTORC1/S6K1 cascade through phosphorylation of Rictor on threonine 1135 has been reported (33, 34), we investigated the kinetics of phosphorylation changes of this residue in differentiating L6 myoblasts. AVP stimulation induced a rapid phosphorylation of Thr-1135-Rictor, peaking at 6–30 min (Fig. 4D). This response was enhanced by S6K1 overexpression (Fig. 4E), consistent with the interpretation that mTORC1/S6K1 exerted a negative effect on differentiation by inhibiting mTORC2.
FIGURE 4.

Myogenic differentiation is regulated in an opposite way by mTORC1 and mTORC2. A, cells were cultured for 48 h in the presence of 10−7 m AVP with or without 100 nm rapamycin; myogenin mRNA level was measured by quantitative RT-PCR, myogenin protein content was evaluated by Western blotting analysis, and myogenin nuclear accumulation was evaluated by immunofluorescence microscopy. Bar = 40 μm. B, L6 cells were transfected with control siRNA, Raptor siRNA, or Rictor siRNA for 48 h and then stimulated by AVP for additional 48 h. The percentage of myogenin positive nuclei was measured by immunofluorescence microscopy; shown are the means of three independent experiments, with 10 fields considered in each condition. *, significantly different from control cells, p < 0.02. Bar = 40 μm. The amount of myogenin was evaluated by Western blotting. C, mRNA levels of myogenin and troponin were measured by RT-qPCR, and myogenin protein levels were evaluated by Western blotting in L6 myoblasts overexpressing S6K1 or transfected with the empty vector. *, significantly different from control cells, p < 0.05. D, the levels of phospho-Thr-1135-Rictor were evaluated by Western blotting in L6 cells stimulated for different times by AVP. E, the levels of phospho-Thr-1135-Rictor were evaluated in L6 cells overexpressing or not S6K1. *, significantly different from control cells, p < 0.05.
Because mTORC2 had a positive role in L6 cell differentiation stimulated by AVP, the involvement of the mTORC2 substrate Akt in this response could be expected. The phosphorylation of Akt on Ser-473 observed in these conditions is known to induce, together with PDK1-mediated phosphorylation of Thr-308, a maximal activation of Akt that might participate in the differentiating response of L6 cells in the presence of AVP. To evaluate the role of Akt in this particular setting, the PI3K inhibitor wortmannin was added to the differentiation medium. 100 nm wortmannin is known to be sufficient to block Akt activation but is too low to inhibit mTOR kinase directly (35). At this concentration, wortmannin did not affect myogenic differentiation as evaluated by myogenin nuclear accumulation, although it totally suppressed Ser-473 phosphorylation of Akt (Fig. 5A). Only at a higher concentration (500 nm), at which the compound is no longer selective for PI3K inhibition and affects mTOR, was wortmannin able to inhibit partially differentiation. Similarly, the more stable PI3K inhibitor ZSTK474, which has IC50 values of 16 and 370 nm on PI3Kα and mTOR, respectively (36), had a marginal effect on myogenin expression when used at 50 nm and a limited effect at 100 nm (Fig. 5A). These observations suggested that Akt activation is not required for the cells to differentiate. To further evaluate the Akt role in differentiation, we overexpressed the wild-type Akt1 isoform in L6 cells submitted to differentiating conditions (Fig. 5B). We observed that Akt overexpression scarcely affected myogenin expression and nuclear accumulation. In further support of the lack of major effects of Akt on initiation of differentiation, we also observed that overexpression of either constitutively active or kinase-defective dominant-negative mutants of PI3K did not significantly modify the differentiating response (Fig. 5C).
FIGURE 5.

The IGF/PI3K/Akt pathway is not involved in the first steps of AVP-induced L6 cell differentiation. A, L6 myoblasts were treated with 100 or 500 nm wortmannin (wort) or 20 nm rapamycin (rapa) in the presence of 40 nm IGF-2 or 100 nm insulin (Ins) and stimulated with 10−7 m AVP for 48 h. Immunofluorescence staining of nuclear myogenin was performed. The percentage of myogenin-positive nuclei relative to total number of nuclei evaluated on phase contrast images was calculated on 10 fields (bar = 20 μm). Shown are control Western blots of Ph-Ser-473-Akt, verifying that 100 nm wortmannin blocked Akt activation under our conditions. The effect of the PI3K inhibitor ZSTK474 on myogenin protein expression was evaluated by Western blotting in L6 cells stimulated for 48 h by AVP. *, significantly different from control cells, p < 0.05. B, cells were transfected with Akt or empty vector for 48 h and stimulated with 10−7 m AVP for 48 h, and nuclear myogenin immunostaining was performed and quantified relative to DAPI-stained total nuclei. Bar = 40 μm. As a control of Akt overexpression, Western blotting analysis of Akt protein was performed on cell lysates. C, cells were transfected with empty vector or kinase-dead PI3K (PI3K-KD) or constitutively active PI3K (PI3K-CA) for 48 h and stimulated with 10−7 m AVP for 48 h, and nuclear myogenin immunostaining was performed and quantified relative to DAPI stained total nuclei. Bar = 40 μm. D, L6 myoblasts were treated or not with 100 nm rapamycin (Rapa) in the presence or absence of 10−7 m AVP for 2 or 4 days. Cell lysates were analyzed for IGF-2 content by Western blotting (upper blot, representative result of IGF2 cell content at day 2). Diagrams show the quantitation of IGF2 bands, normalized by tubulin (means ± S.E. of three independent experiments). Alternatively, RNA was extracted, and IGF-2 mRNA content was evaluated by RT-PCR (lower blots). NT, not transfected.
Because it has been reported that rapamycin suppresses the production of IGF-2 by C2C12 cells, thereby preventing an autocrine loop required for differentiation to take place, we examined the ability of IGF-2 and insulin to raise the blockade of L6 myoblast differentiation induced by rapamycin. Neither factor could restore differentiation in the presence of rapamycin (Fig. 5A), although Akt was fully phosphorylated on Ser-473 in these conditions (not shown). Besides, in L6 cells stimulated to differentiate in the presence of AVP, we evaluated the expression of IGF-2 by Western blotting and RT-PCR and observed that rapamycin had no marked influence on IGF-2 protein or mRNA levels (Fig. 5D). The above data were thus consistent with the idea that in our setting the blockade of differentiation induced by rapamycin cannot be ascribed to an inhibition of the IGF/PI3K/Akt pathway but, rather, involves other target(s) of mTOR impinging on the expression of myogenin.
Among the few identified substrates of mTORC2, PKCα is especially interesting in the context of myogenic differentiation, as this process is known to require PKC (24). We thus investigated the effects of AVP stimulation of L6 myoblasts on the phosphorylation of serine 657, a PKCα residue targeted by mTORC2. We observed a rapid phosphorylation of Ser-657, peaking at 20–30 min (Fig. 6A), compatible with the involvement of mTORC2. Accordingly, siRNA-mediated Rictor depletion lowered PKCα phosphorylation levels (Fig. 6B). We verified that inhibition of mTORC2 was able to decrease Ser-657-PKCα phosphorylation by using a 24-h rapamycin treatment or PP242 inhibitor, which targets the kinase site of mTOR (37). In both cases, we observed a marked inhibition of AVP-induced PKCα phosphorylation (Fig. 6B). PKCα phosphorylation was also triggered by PA or propranolol cell stimulation (not shown). In view of the negative effects of the mTOR/S6K1 cascade on differentiation (Fig. 4), we evaluated the effects of S6K1 overexpression on phosphorylation of the Ser-657-PKCα position and observed a marked inhibitory effect (Fig. 6C). Finally, to determine whether PKCα was involved in myogenic differentiation, we down-regulated its expression by siRNA-induced depletion. We observed a strong inhibition of differentiation, as evaluated by myogenin protein content, showing that PKCα is required for L6 cells to differentiate (Fig. 6D).
FIGURE 6.

PKCα is phosphorylated on serine 657 in response to AVP stimulation and is required for differentiation. A, the levels of phospho-Ser-657-PKCα were evaluated by Western blotting in L6 cells stimulated for different times by AVP. B, shown are the effects on AVP-induced PKCα phosphorylation of mTORC2 down-regulation induced by Rictor-siRNA (left panel) or 24 h of 100 nm rapamycin (rapa) treatment, or 40 min of treatment by mTOR inhibitor PP242 at 250 nm (right panel). C, phosphorylation of PKCα on Ser-657 was evaluated in cells overexpressing or not S6K1. D, the effects of siRNA-mediated PKCα depletion on myogenin protein expression were evaluated in AVP-stimulated L6 cells by Western blotting. The control of PKCα silencing efficiency is shown. *, significantly different from control cells, p < 0.05. NT, not transfected.
We could thus propose a model in which PLD positively regulates the activity of the mTORC2 complex, which in turn triggers differentiation via an effector that could be PKCα; rapamycin would block differentiation by inducing mTORC2 disassembling and reduction of activity. To validate this model, we overexpressed PLD isoforms in conditions of lowered mTORC2 activity and observed the consequences on myogenic response. When the cells were treated by rapamycin for 48 h to down-regulate mTORC2, the overexpression of PLD1, but not PLD2, enhanced the expression of myogenin (Fig. 7), supporting an important role for PLD1-mediated mTORC2 activation in myogenin expression, a prerequisite for differentiation.
FIGURE 7.
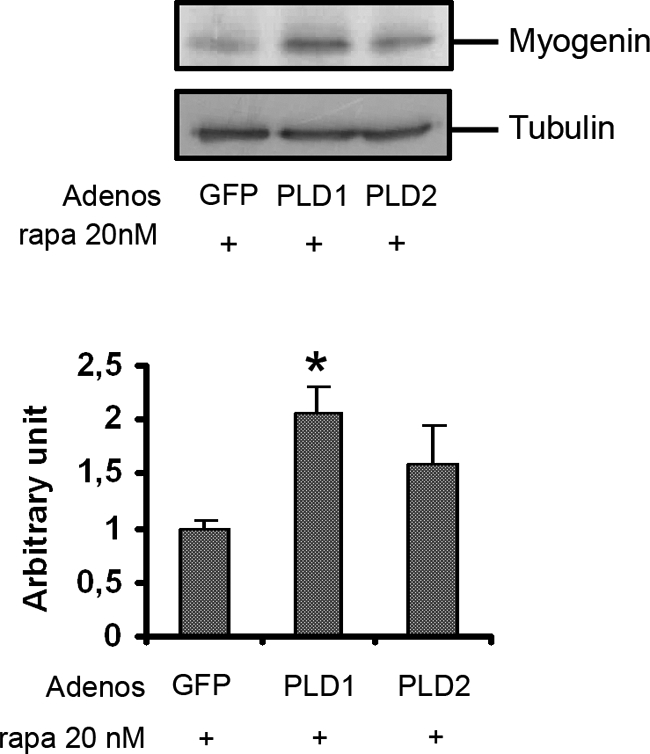
PLD overexpression overcomes rapamycin-induced inhibition of L6 myoblast differentiation. L6 myoblasts were infected with adenoviruses (Adenos) encoding GFP or PLD1 or PLD2 and then treated with 20 nm rapamycin (rapa) in the presence of 10−7 m AVP for 48 h. Cells lysates were analyzed by Western blotting for myogenin expression. Myogenin bands were quantitated and normalized for tubulin amounts. *, significantly different from control cells, p < 0.05.
DISCUSSION
In this work we observed in L6 myoblasts that induction of differentiation by AVP was accompanied by stimulation of both mTORC1 and mTORC2 complexes. mTORC1 activation was evidenced by phosphorylation of S6K1 at threonine 389/412, whereas mTORC2 activation resulted in the phosphorylation of Akt on serine 473. PLD activity was involved in the activation of both mTOR complexes, as shown by PLD overexpression and siRNA silencing experiments, and by the ability of exogenous PA and propranolol, a compound that induces endogenous PA accumulation, to reproduce the effects of the PLD agonist AVP. AVP is present in human fetal muscle (38), and AVP receptors are expressed in human skeletal muscle (39) and myoblasts (23). Moreover, circulating AVP levels are increased during physical exercise (40), and AVP injection improves in vivo muscle regeneration (41). Based on these observations, AVP is likely to play a physiological role in skeletal muscle development and homeostasis.
The involvement of PLD in the activation of both mTOR complexes has been reported to occur in cancer cell lines by Foster and co-workers (9). These authors have shown, by expressing dominant negative PLD mutants, that PA, which can be produced by either of the PLD isoforms, is required for the assembly of both mTORC1 and mTORC2, in agreement with previous studies showing that PA binds to the mTOR protein, common to the two complexes, at the FRB site (9, 10, 42). In addition, Foster's group (9) proposed that PA also participates in mTORC1 activation by preventing the binding of the PRAS40 inhibitory protein. PA has also been shown to influence the oligomerization of mTORC1 and mTORC2 complexes, with potential consequences on their activities (43). Another proposed mechanism of mTOR activation by PA involves the displacement of the inhibitory protein FKBP38 by competition for the same binding site (5). However, the role of FKBP38 in mTOR regulation has been recently questioned (44). Thus, although a tridimensional model of PA/mTOR interaction has been established (10), the mechanism of mTOR activation remains unclear. In particular, there is no evidence of a trans-conformation of the FRB domain under PA binding (10). Based on the report that localization of mTORC2 complex into the raft fraction of endothelial cell plasma membrane is required for its activity, an intriguing possibility is that PA drives the targeting of mTORC2 to lipid rafts (45). Interestingly, a link between PLD and mTOR activation has been recently described in muscle tissue. Mechanical stimulation of murine muscle activates mTOR signaling independently of the PI3K/Akt pathway by a mechanism requiring PLD and PA (46).
Concerning the involvement of PLD in the regulation of the mTORC1 substrate S6K1 in differentiating L6 cells, additional levels of complexity are emerging. S6K1 kinase exists under two forms that are produced by translation of a single mRNA with usage of two different translational start sites (47). It is noticeable that the kinetics of activation of the two S6K1 isoforms were different, p85 reaching a maximal phosphorylation at 10 min versus 40 min for p70 (Fig. 1A), suggesting that different mechanisms could be involved. Indeed, whereas PLD activates the p70-SK1 isoform entirely through mTORC1 activation, as shown by complete suppression of AVP- or propranolol-induced p70 phosphorylation by rapamycin treatment, the p85 isoform phosphorylation is incompletely sensitive to rapamycin inhibition and is thus in part independent of mTORC1. A direct activation of S6K1 by PA, bypassing mTORC1, has been proposed by others, although the possibility of a differential sensitivity of the two isoforms was not considered (48). The present results suggest that p85 can be the S6K1 isoform submitted to direct activation by PLD-produced PA. The two S6K1 isoforms thus appear to be differentially regulated in skeletal myoblasts, as described previously in cardiomyocytes (49, 50). In line with a differential regulation of the two forms, their subcellular localization has been reported to be different, the p70 isoform being predominantly cytosolic, whereas the p85 isoform carrying a nuclear localization signal sequence might be mainly present in the nucleus, where it ensures a specific role in the control of mitogenesis (51–53).
As widely described, myogenic differentiation is dependent on mTOR, based on its total suppression by the mTOR inhibitor rapamycin (18, 19) and rescue by overexpression of rapamycin-resistant mTOR mutant (20, 21). However, the link between mTOR and the myogenic response is still a matter of controversy. mTOR might be required to ensure the activation of p38-MAP kinase (19) of cyclin-dependent kinase-5 (54). Besides, Chen and Erbay (20) reported that initial C2C12 myogenic cell differentiation is controlled by mTOR in a kinase-independent way through regulation of IGF-2 expression, rapamycin blocking the differentiation process by preventing the autocrine stimulatory effect of IGF-2. The kinase-independent myogenic function of mTOR has been disputed (21). In contrast, a late-stage fusion step might be regulated in a kinase-dependent way by mTOR through a yet-to-be identified secreted factor (55). Moreover, the initial observations of mTOR implication in rapamycin effect on myogenesis implied that signaling through mTORC1 was critical in the process, as rapamycin was considered to selectively target this complex. The later recognition that persistent inhibition by rapamycin can affect the assembly of mTORC2 complex (3) raised the question of whether rapamycin exerted its inhibitory effect on differentiation by modulating mTORC2 (4).
We had previously shown that PLD activity and PA production are required for myogenic differentiation. Here, we confirm this finding by showing that adenovirus-mediated PLD1 overexpression enhances differentiation, whereas a PLD1-specific synthetic inhibitor or an isoform-aspecific PLD inhibitor both potently prevent differentiation. We, therefore, examined the hypothesis that modulation of mTOR complexes is responsible for the positive effects of PLD on differentiation. We observed by using selective siRNA silencing that only mTORC2 complex is positively involved in differentiation, in agreement with what was described by others in C2C12 cells (4) and human primary myoblasts (56), and that, in the opposite, mTORC1 complex has a negative effect on differentiation. This latter observation is consistent with the reported negative regulation exerted by mTORC1 on mTORC2 through S6K1-mediated Thr-1135 phosphorylation of Rictor (33, 34) and with our observation of a negative effect of S6K1 overexpression on differentiation. In further support to this model, we verified that AVP stimulation of L6 cells induced a rapid phosphorylation of Rictor on Thr-1135, the residue targeted by S6K1, and that this effect was reproduced by S6K1 overexpression.
The fact that PLD can activate both mTOR complexes, which act in an opposite way on differentiation, raises the question of how PLD action can result in a positive response. Because mTORC2 is known to activate mTORC1 via the Akt-mediated phosphorylation and inactivation of two negative regulators, the TSC complex and the PRAS40 protein (35, 57), mTORC1 activation might be viewed as a negative feedback loop limiting the extent of mTORC2-stimulated differentiation. Thus, activation of mTORC2 appears to be a possible pathway through which PLD activity participates together with other signals in myogenic differentiation. The ability of PLD overexpression to raise in part the inhibition of myogenin expression induced by prolonged rapamycin treatment, i.e. by mTORC2 disassembling, is consistent with this proposal. It is not clear which effector(s) downstream mTORC2 is involved in differentiation. In C2C12 cells, Akt seems to be the candidate, because ectopic expression of constitutively active Akt rescues differentiation of cells depleted in Rictor and, although with much delay and incompletely, of cells treated by rapamycin (4). In AVP-stimulated L6 cells we did not observe such a major role for Akt, as shown by the limited effects of PI3K inhibitors on differentiation, by an absence of correlation between Akt-Ser-473 phosphorylation status and the extent of myogenic response in the presence of wortmannin or rapamycin plus insulin, and by the modest effects of Akt or PI3K overexpression on early myogenesis steps. In line with these observations, it has been reported that overexpression of a dominant-negative Akt mutant in primary mouse myoblasts had no noticeable effect on the levels and timing of expression of differentiation markers and cell fusion (58). Moreover, in view of the mild inhibition of Akt function in L6 myotubes under mTORC2 blockade by pharmacological inhibitors directed at the kinase site (37), it seems unlikely that the drastic effect of rapamycin on myogenesis is mediated by Akt inhibition resulting from mTORC2 disassembly. Thus, there seem to exist a number of differences between L6 and C2C12 myogenic cell models regarding the signaling set in motion at the onset of differentiation. In particular, the role of Akt seems to be much less critical in initiation of L6 differentiation, and the autocrine activation of differentiation by IGF-2 is not the target of rapamycin in L6, in agreement with the previously reported suppression of L6A1 myoblast differentiation by rapamycin in the presence of IGF-1 (18).
As for the mTORC2 effector(s) involved in L6 cell differentiation, other proteins identified as targets of mTORC2 (57) could be considered, especially PKCα, because of the importance of PKCs in myogenic response (24). We observed a rapid phosphorylation of the HM motif of PKCα, a mTORC2 target (59), in response to AVP stimulation. This response was attenuated by S6K1 overexpression and by general mTOR inhibitors, as expected for a mTORC2-mediated event. Besides, we established that PKCα is required for myogenesis by showing that its depletion strongly inhibits myogenin expression. Because previous studies have demonstrated that mTORC2-dependent phosphorylation of PKCα is essential for the stability and activity of the protein (60), we can propose that mTORC2 is involved in myogenic differentiation through its effects on PKCα. It is interesting to note that PKCα is an activator of PLD1 (61) and could thereby participate in a positive feedback loop. In support to this assumption, we had observed a significant decrease in PLD response to AVP after PKC down-regulation induced by a 24-h 10−7 m 12-O-tetradecanoylphorbol-13-acetate treatment (25). Another possible differentiation promoting pathway downstream mTORC2 might involve actin cytoskeleton rearrangements. mTORC2 has indeed been reported to regulate actin polymerization (59, 62), and we have reported that PLD is involved in the formation of stress fibers in differentiating L6 myoblasts (25).
Because PLD activity in mammalian cells is ensured by two different isoforms, PLD1 and PLD2, which exhibit different regulations and subcellular locations, the question of the respective role of the two isoforms in mTOR regulation and myogenic differentiation can be raised. The issue of whether one of PLD isoforms is more related to mTOR regulation has been extensively discussed (5, 6). It appears that, depending on the particular system considered, either PLD isoform can ensure mTOR activation. In the present work we observed that in L6 model, mTOR activation and myogenic differentiation were both more sensitive to PLD1 than to PLD2 expression changes or inhibition.
On the whole it appears that PLD and, more specifically the PLD1 isoform, is involved in an intricate regulation of the mTOR system in differentiating L6 myogenic cells (Fig. 8). This regulation participates in the control of myogenic response both positively, through mTORC2 complex, and negatively, through mTORC1 complex. In addition, PLD1 might directly impinge on the longer p85 isoform of S6K1, possibly through PA binding, suggesting that this isoform might have a specific role in differentiating myoblasts, which deserves further investigations. In view of the role played by the PLD/mTOR pathway in major muscle functions, including myogenic differentiation, PLD may constitute a critical factor for muscle tissue maintenance and regeneration and be considered as a potential therapeutic target in disorders affecting this tissue.
FIGURE 8.

Proposed model for the implication of PLD-mediated modulation of the mTOR system in myogenic differentiation of L6 cells. 1, PA produced by the action of PLD binds to and activates mTORC1. In turn, this complex activates S6K1, in particular the p70 isoform. 2, PA may also act directly upon the p85 S6K1 isoform. 3, PA also activates mTORC2. 4, this complex then ensures the phosphorylation of Akt on Ser-473 and through the activation of another effector, possibly PKCα, promotes myogenin expression and early myogenic differentiation. Akt activation might, rather, influence late steps of myotube maturation. 5, mTORC1 exerts a negative effect on mTORC2, mediated by S6K1, possibly acting by Rictor phosphorylation on Thr-1135.
This work has been supported by a grant from the Association Française contre les Myopathies (MNM2 2007) and a fellowship from the Association pour la Recherche sur le Cancer (to R. J.).
- mTOR
- mammalian target of rapamycin
- S6K1
- S6 kinase 1
- AVP
- arginine-vasopressin
- IGF
- insulin-like growth factor
- PA
- phosphatidic acid
- PLD
- phospholipase D.
REFERENCES
- 1. Laplante M., Sabatini D. M. (2009) J. Cell Sci. 122, 3589–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosner M., Hengstschläger M. (2008) Hum. Mol. Genet. 17, 2934–2948 [DOI] [PubMed] [Google Scholar]
- 3. Sarbassov D. D., Ali S. M., Sengupta S., Sheen J. H., Hsu P. P., Bagley A. F., Markhard A. L., Sabatini D. M. (2006) Mol. Cell 22, 159–168 [DOI] [PubMed] [Google Scholar]
- 4. Shu L., Houghton P. J. (2009) Mol. Cell. Biol. 29, 4691–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun Y., Chen J. (2008) Cell Cycle 7, 3118–3123 [DOI] [PubMed] [Google Scholar]
- 6. Foster D. A. (2009) Biochim. Biophys. Acta 1791, 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raghu P., Manifava M., Coadwell J., Ktistakis N. T. (2009) Biochim. Biophys. Acta 1791, 889–897 [DOI] [PubMed] [Google Scholar]
- 8. Fang Y., Vilella-Bach M., Bachmann R., Flanigan A., Chen J. (2001) Science 294, 1942–1945 [DOI] [PubMed] [Google Scholar]
- 9. Toschi A., Lee E., Xu L., Garcia A., Gadir N., Foster D. A. (2009) Mol. Cell. Biol. 29, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veverka V., Crabbe T., Bird I., Lennie G., Muskett F. W., Taylor R. J., Carr M. D. (2008) Oncogene 27, 585–595 [DOI] [PubMed] [Google Scholar]
- 11. Bodine S. C., Stitt T. N., Gonzalez M., Kline W. O., Stover G. L., Bauerlein R., Zlotchenko E., Scrimgeour A., Lawrence J. C., Glass D. J., Yancopoulos G. D. (2001) Nat. Cell Biol. 3, 1014–1019 [DOI] [PubMed] [Google Scholar]
- 12. Hornberger T. A., Chu W. K., Mak Y. W., Hsiung J. W., Huang S. A., Chien S. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 4741–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kubica N., Bolster D. R., Farrell P. A., Kimball S. R., Jefferson L. S. (2005) J. Biol. Chem. 280, 7570–7580 [DOI] [PubMed] [Google Scholar]
- 14. Park I. H., Erbay E., Nuzzi P., Chen J. (2005) Exp. Cell Res. 309, 211–219 [DOI] [PubMed] [Google Scholar]
- 15. Rommel C., Bodine S. C., Clarke B. A., Rossman R., Nunez L., Stitt T. N., Yancopoulos G. D., Glass D. J. (2001) Nat. Cell Biol. 3, 1009–1013 [DOI] [PubMed] [Google Scholar]
- 16. Ohanna M., Sobering A. K., Lapointe T., Lorenzo L., Praud C., Petroulakis E., Sonenberg N., Kelly P. A., Sotiropoulos A., Pende M. (2005) Nat. Cell Biol. 7, 286–294 [DOI] [PubMed] [Google Scholar]
- 17. Ge Y., Wu A. L., Warnes C., Liu J., Zhang C., Kawasome H., Terada N., Boppart M. D., Schoenherr C. J., Chen J. (2009) Am. J. Physiol. Cell Physiol. 297, C1434–C1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coolican S. A., Samuel D. S., Ewton D. Z., McWade F. J., Florini J. R. (1997) J. Biol. Chem. 272, 6653–6662 [DOI] [PubMed] [Google Scholar]
- 19. Cuenda A., Cohen P. (1999) J. Biol. Chem. 274, 4341–4346 [DOI] [PubMed] [Google Scholar]
- 20. Erbay E., Chen J. (2001) J. Biol. Chem. 276, 36079–36082 [DOI] [PubMed] [Google Scholar]
- 21. Shu L., Zhang X., Houghton P. J. (2002) J. Biol. Chem. 277, 16726–16732 [DOI] [PubMed] [Google Scholar]
- 22. Minotti S., Scicchitano B. M., Nervi C., Scarpa S., Lucarelli M., Molinaro M., Adamo S. (1998) Cell Growth Differ. 9, 155–163 [PubMed] [Google Scholar]
- 23. Breton C., Haenggeli C., Barberis C., Heitz F., Bader C. R., Bernheim L., Tribollet E. (2002) J. Clin. Endocrinol. Metab. 87, 1415–1418 [DOI] [PubMed] [Google Scholar]
- 24. Komati H., Minasi A., Naro F., Lagarde M., Prigent A. F., Adamo S., Némoz G. (2004) FEBS Lett. 577, 409–414 [DOI] [PubMed] [Google Scholar]
- 25. Komati H., Naro F., Mebarek S., De Arcangelis V., Adamo S., Lagarde M., Prigent A. F., Némoz G. (2005) Mol. Biol. Cell 16, 1232–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mebarek S., Komati H., Naro F., Zeiller C., Alvisi M., Lagarde M., Prigent A. F., Némoz G. (2007) J. Cell Sci. 120, 407–416 [DOI] [PubMed] [Google Scholar]
- 27. Yoon M. S., Chen J. (2008) J. Cell Sci. 121, 282–289 [DOI] [PubMed] [Google Scholar]
- 28. Monovich L., Mugrage B., Quadros E., Toscano K., Tommasi R., LaVoie S., Liu E., Du Z., LaSala D., Boyar W., Steed P. (2007) Bioorg. Med. Chem. Lett. 17, 2310–2311 [DOI] [PubMed] [Google Scholar]
- 29. Su W., Yeku O., Olepu S., Genna A., Park J. S., Ren H., Du G., Gelb M. H., Morris A. J., Frohman M. A. (2009) Mol. Pharmacol. 75, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scott S. A., Selvy P. E., Buck J. R., Cho H. P., Criswell T. L., Thomas A. L., Armstrong M. D., Arteaga C. L., Lindsley C. W., Brown H. A. (2009) Nat. Chem. Biol. 5, 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bondeva T., Pirola L., Bulgarelli-Leva G., Rubio I., Wetzker R., Wymann M. P. (1998) Science 282, 293–296 [DOI] [PubMed] [Google Scholar]
- 32. Zeiller C., Mebarek S., Jaafar R., Pirola L., Lagarde M., Prigent A. F., Némoz G. (2009) Biochim Biophys Acta 1793, 1236–1249 [DOI] [PubMed] [Google Scholar]
- 33. Julien L. A., Carriere A., Moreau J., Roux P. P. (2010) Mol. Cell. Biol. 30, 908–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dibble C. C., Asara J. M., Manning B. D. (2009) Mol. Cell. Biol. 29, 5657–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang L., Harris T. E., Roth R. A., Lawrence J. C., Jr. (2007) J. Biol. Chem. 282, 20036–20044 [DOI] [PubMed] [Google Scholar]
- 36. Kong D., Dan S., Yamazaki K., Yamori T. (2010) Eur. J. Cancer 46, 1111–1121 [DOI] [PubMed] [Google Scholar]
- 37. Feldman M. E., Apsel B., Uotila A., Loewith R., Knight Z. A., Ruggero D., Shokat K. M. (2009) PLoS Biol. 7, e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smith A., Stephen R. I., Arkley M. M., McIntosh N. (1992) Early Hum. Dev. 28, 215–222 [DOI] [PubMed] [Google Scholar]
- 39. Thibonnier M., Graves M. K., Wagner M. S., Auzan C., Clauser E., Willard H. F. (1996) Genomics 31, 327–334 [DOI] [PubMed] [Google Scholar]
- 40. Convertino V. A., Keil L. C., Greenleaf J. E. (1983) J. Appl. Physiol. 54, 508–514 [DOI] [PubMed] [Google Scholar]
- 41. Moresi V., Garcia-Alvarez G., Pristerà A., Rizzuto E., Albertini M. C., Rocchi M., Marazzi G., Sassoon D., Adamo S., Coletti D. (2009) PloS one 4, e5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fang Y., Park I. H., Wu A. L., Du G., Huang P., Frohman M. A., Walker S. J., Brown H. A., Chen J. (2003) Curr. Biol. 13, 2037–2044 [DOI] [PubMed] [Google Scholar]
- 43. Takahara T., Hara K., Yonezawa K., Sorimachi H., Maeda T. (2006) J. Biol. Chem. 281, 28605–28614 [DOI] [PubMed] [Google Scholar]
- 44. Uhlenbrock K., Weiwad M., Wetzker R., Fischer G., Wittinghofer A., Rubio I. (2009) FEBS Lett. 583, 965–970 [DOI] [PubMed] [Google Scholar]
- 45. Partovian C., Ju R., Zhuang Z. W., Martin K. A., Simons M. (2008) Mol. Cell 32, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. O'Neil T. K., Duffy L. R., Frey J. W., Hornberger T. A. (2009) J. Physiol. 587, 3691–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grove J. R., Banerjee P., Balasubramanyam A., Coffer P. J., Price D. J., Avruch J., Woodgett J. R. (1991) Mol. Cell. Biol. 11, 5541–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lehman N., Ledford B., Di Fulvio M., Frondorf K., McPhail L. C., Gomez-Cambronero J. (2007) FASEB J. 21, 1075–1087 [DOI] [PubMed] [Google Scholar]
- 49. Laser M., Kasi V. S., Hamawaki M., Cooper G., 4th, Kerr C. M., Kuppuswamy D. (1998) J. Biol. Chem. 273, 24610–24619 [DOI] [PubMed] [Google Scholar]
- 50. Kenessey A., Ojamaa K. (2006) J. Biol. Chem. 281, 20666–20672 [DOI] [PubMed] [Google Scholar]
- 51. Kim S. J., Kahn C. R. (1997) Biochem. Biophys. Res. Commun. 234, 681–685 [DOI] [PubMed] [Google Scholar]
- 52. Coffer P. J., Woodgett J. R. (1994) Biochem. Biophys. Res. Commun. 198, 780–786 [DOI] [PubMed] [Google Scholar]
- 53. Reinhard C., Fernandez A., Lamb N. J., Thomas G. (1994) EMBO J. 13, 1557–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sarker K. P., Lee K. Y. (2004) Oncogene 23, 6064–6070 [DOI] [PubMed] [Google Scholar]
- 55. Park I. H., Chen J. (2005) J. Biol. Chem. 280, 32009–32017 [DOI] [PubMed] [Google Scholar]
- 56. Trendelenburg A. U., Meyer A., Rohner D., Boyle J., Hatakeyama S., Glass D. J. (2009) Am. J. Physiol. Cell Physiol. 296, C1258–C1270 [DOI] [PubMed] [Google Scholar]
- 57. Huang J., Wu S., Wu C. L., Manning B. D. (2009) Cancer Res. 69, 6107–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bois P. R., Grosveld G. C. (2003) EMBO J. 22, 1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sarbassov D. D., Ali S. M., Kim D. H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2004) Curr. Biol. 14, 1296–1302 [DOI] [PubMed] [Google Scholar]
- 60. Ikenoue T., Inoki K., Yang Q., Zhou X., Guan K. L. (2008) EMBO J. 27, 1919–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hu T., Exton J. H. (2003) J. Biol. Chem. 278, 2348–2355 [DOI] [PubMed] [Google Scholar]
- 62. Jacinto E., Loewith R., Schmidt A., Lin S., Rüegg M. A., Hall A., Hall M. N. (2004) Nat. Cell Biol. 6, 1122–1128 [DOI] [PubMed] [Google Scholar]