

Abstract
2-Methyl-3-buten-2-ol (MBO) is a five-carbon alcohol produced and emitted in large quantities by many species of pine native to western North America. MBO is structurally and biosynthetically related to isoprene and can have an important impact on regional atmospheric chemistry. The gene for MBO synthase was identified from Pinus sabiniana, and the protein encoded was functionally characterized. MBO synthase is a bifunctional enzyme that produces both MBO and isoprene in a ratio of ∼90:1. Divalent cations are required for activity, whereas monovalent cations are not. MBO production is enhanced by K+, whereas isoprene production is inhibited by K+ such that, at physiologically relevant [K+], little or no isoprene emission should be detected from MBO-emitting trees. The Km of MBO synthase for dimethylallyl diphosphate (20 mm) is comparable with that observed for angiosperm isoprene synthases and 3 orders of magnitude higher than that observed for monoterpene and sesquiterpene synthases. Phylogenetic analysis showed that MBO synthase falls into the TPS-d1 group (gymnosperm monoterpene synthases) and is most closely related to linalool synthase from Picea abies. Structural modeling showed that up to three phenylalanine residues restrict the size of the active site and may be responsible for making this a hemiterpene synthase rather than a monoterpene synthase. One of these residues is homologous to a Phe residue found in the active site of isoprene synthases. The remaining two Phe residues do not have homologs in isoprene synthases but occupy the same space as a second Phe residue that closes off the isoprene synthase active site.
Keywords: Enzyme Structure, Evolution, Isoprenoid, Plant, Protein sequence
Introduction
Hemiterpenes are the smallest members of the isoprenoid family. Two are important in biosphere-atmosphere interactions, isoprene (2-methyl-1,3-butadiene) and an isomer of methylbutenol (2-methyl-3-buten-2-ol (MBO)).2 Isoprene is given off by trees such as oak and poplar, and averaged over the globe, the amount of isoprene that enters the atmosphere each year is approximately equal to the input of all other hydrocarbons combined (1). MBO is similar to isoprene and is given off by many species of pine trees in western North America (2–4). Isoprene and MBO are made from dimethylallyl diphosphate (DMADP) (5–7).
Isoprene emission occurs from a wide range of taxa, including members of the moss, fern, gymnosperm, and angiosperm families (4, 8). Two families of isoprene synthase sequences are known from angiosperms (several from Populus species (9–12) and one from kudzu (9)). Both groups of isoprene synthases closely resemble angiosperm monoterpene synthases in sequence and structure (9, 13). MBO emission has been found only in a subset of the pines (gymnosperms) in North America (2, 4).
The plant terpene synthase (TPS) gene family consists of several different subfamilies (14, 15). In angiosperms, monoterpene synthases group together in the TPS-b subfamily and typically have a functional C-terminal α-domain and a vestigial N-terminal β-domain (13, 15). Isoprene synthases from angiosperms (flowering plants) group in the TPS-b subfamily, possess both α- and β-domains, and share the same intron/exon organization (six introns and seven exons) found in the TPS-b subfamily (9, 15). The TPS-b subfamily is found only in the angiosperms and is absent in the gymnosperms.
Gymnosperms contain a diverse assemblage of monoterpene synthases, which represent a distinct lineage of genes that are distantly related to the monoterpene synthase genes (TPS-b) found in angiosperms (14–16). Gymnosperm terpene synthases belong to the TPS-d subfamily (17, 18), which is further subdivided into the d1 (mostly monoterpene synthases), d2 (mostly sesquiterpene synthases), and d3 (mostly diterpene synthases) groups. Structurally, genes in the TPS-d1 family share the α- and β-domains found in the angiosperm TPS-b family but have different intron/exon structures (six introns and seven exons in TPS-b and nine introns and 10 exons in TPS-d1). The sequence and structure of hemiterpene synthases found in gymnosperms and the other major taxonomic groups, which do not have TPS-b genes, are unknown.
There are numerous examples of similar catalytic activities arising in parallel within the angiosperms and gymnosperms from enzymes that share little sequence identity (15). If such a pattern of parallel evolution also occurs with the hemiterpene synthases of gymnosperms, these genes might demonstrate sequences, structures, and mechanisms of forming hemiterpenes that have not been observed in angiosperms.
We set out to find and clone the MBO synthase gene from pines 1) to provide insight into the relationship between hemiterpene synthases in angiosperms and gymnosperms and 2) to investigate the sequence and structural features that distinguish the catalytic activity of MBO synthase from that of monoterpene and isoprene synthases. Understanding the features that distinguish hemiterpene from monoterpene synthases and isoprene from MBO synthases may lead to an understanding of why hemiterpene synthases have evolved in only a small subset of plant taxa.
MATERIALS AND METHODS
RNA Extraction and cDNA Synthesis
Total RNA was extracted from needles of Pinus sabiniana using a procedure adapted from Chang et al. (19) to incorporate 4% polyvinylpyrrolidone 40, 4% polyvinylpolypyrrolidone, and 4% β-mercaptoethanol into cetyltrimethylammonium bromide detergent buffer. Purified RNA was stored at −80 °C until used in cDNA synthesis. First-strand cDNAs were synthesized using Moloney murine leukemia virus reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Primers used to initiate cDNA synthesis are listed in supplemental Table S1.
Initial Fragment Isolation
Degenerate primers targeting conserved regions of known conifer monoterpene synthase genes were used in various combinations to prime PCRs with the cDNA template. PCR was performed in 30-μl volumes using 1.25 units of GoTaq DNA polymerase (Promega), 0.25 μl of cDNA, 0.2 mm dNTPs, 0.6 μm each primer, and the manufacturer's buffer at 1× concentration. Amplification was carried out with an initial denaturation at 95 °C for 2 min, followed by 35–45 cycles at 95 °C for 30 s, 50 °C for 30 s, and 72 °C for 3 min and a final extension at 72 °C for 9 min. Reactions producing bands of the expected size (determined by gel electrophoresis) were prepared for direct sequencing using a PCR cleanup kit (Qiagen). Sequencing was performed on an ABI 3730 genetic analyzer at the Michigan State University Research Technology Support Center. The resulting sequence information was analyzed by BLAST (20, 21), and those sequences with significant similarity to terpene synthases were selected for further consideration.
3′- and 5′-Rapid Amplification of cDNA Ends (RACE)
3′- and 5′-RACE was used to obtain the sequence information needed to amplify full-length clones of candidate genes following the procedure described previously (22). Primers used in the RACE procedures are listed in supplemental Table S1. To obtain 5′-ends, a pair of nested gene-specific reverse primers (pon3.1R and pon4.1R) were designed for the target sequence. Synthesis of cDNA was performed using the outer primer (pon3.1R). A poly(A) tail was added to the 3′-end of the single-stranded cDNA using terminal transferase (Invitrogen) and served as an anchor primer for subsequent PCR in which template and primer concentrations were varied over a 10-fold range to find the optimal reaction conditions. Amplification was carried out in two steps with an initial denaturation at 95 °C for 2 min, followed by one cycle of denaturing at 95 °C for 30 s, annealing at 48 °C for 5 min, and extension at 72 °C for 40 min and 35–45 cycles of denaturation at 95 °C for 30 s, annealing at 55–65 °C for 30 s, and extension at 72 °C for 3 min, with a final extension at 72 °C for 9 min. Reactions yielding positive PCR hits were purified and sequenced directly as described above.
Isolation of the Full-length MBO Synthase Gene and Generation of Expression Constructs
Primers designed for the 5′-UTR and 3′-end of the candidate MBO synthase sequence (supplemental Table S1) were used to amplify the full-length gene from cDNA using Phusion high fidelity DNA polymerase (Finnlabs) following the manufacturer's instructions. PCR products were gel-purified using a gel extraction kit (Qiagen), and single overhanging A residues were attached by incubation with dATP and Taq polymerase (New England Biolabs), ligated into pGEM-T (Promega), and transformed into Escherichia coli DH5α for plasmid amplification. Plasmids containing inserts were sequenced to confirm sequence fidelity and the absence of internal stop codons.
The sequence for a codon-optimized gene was designed using the Gene Designer software package and synthesized by DNA2.0, Inc. (Menlo Park, CA), ligated into pJexpress 401, and transformed into E. coli BL21(DE3)-RIL cells (Stratagene). This construct coded for the MBO synthase amino acid sequence without the predicted plastid transport sequence and with a C-terminal His6 tag.
Expression of MBO Synthase in E. coli
Cell cultures of transformed cell lines were grown in LB medium to A600 = 0.8 in 500-ml Erlenmeyer baffle flasks at 37 °C with shaking at 250 rpm. Cultures were induced with 1 mm isopropyl β-d-thiogalactopyranoside and grown for an additional 4 h at 30 °C for protein expression. Cells were harvested by centrifugation (10 min, 4000 × g, 4 °C). To extract proteins, cell pellets were resuspended in lysis buffer (50 mm NaH2PO4, 2 m NaCl, 1 mm DTT, 20 mm β-mercaptoethanol, 0.1% Tween 20, and 40% glycerol), sonicated, and centrifuged to pellet cell fragments (45 min, 5000 × g, 4 °C). This lysate was used in enzyme assays, frozen at −80 °C for storage, or purified on nickel-nitrilotriacetic acid (Ni-NTA)-agarose (Qiagen).
His-tagged proteins were purified on Ni-NTA-agarose columns with all steps performed at 4 °C. Cell lysate was loaded onto a column equilibrated with lysis buffer, and non-His-tagged proteins were eluted with 4 volumes of wash buffer (50 mm NaH2PO4, 2 m NaCl, 1 mm DTT, 20 mm β-mercaptoethanol, 0.1% Tween 20, 40% glycerol, and 20 mm imidazole). His-tagged proteins bound to Ni-NTA-agarose were eluted with 4 volumes of elution buffer (50 mm NaH2PO4, 2 m NaCl, 1 mm DTT, 20 mm β-mercaptoethanol, 0.1% Tween 20, 50% glycerol, and 250 mm imidazole). Fractions (0.5 ml) of the eluate were collected and analyzed by SDS-PAGE with Coomassie Blue staining to identify fractions containing the eluted protein. All steps were performed at 4 °C. Protein concentrations were determined by the Bradford assay (Bio-Rad) using BSA as a standard.
Enzyme Assay and Characterization
Enzyme assays were performed in 20-μl total reaction volumes contained in 2-ml screw cap vials. Reaction mixtures contained 100 mm Hepes (pH 8.0), 20 mm DTT, 40% glycerol, and 5 μl of crude extract or purified enzyme, with concentrations of substrate, cofactors, and pH varied for specific assays. Vials were incubated at 37 °C for 30 min, after which a 2-ml gas sample was withdrawn from the headspace of the vial using a syringe and manually injected onto a stainless steel cryotrapping sample loop immersed in liquid nitrogen and purged with helium. The cryoconcentrated sample was flash-desorbed and injected onto the column of a Shimadzu GC17A chromatograph equipped with a photoionization detector. Separation was performed on a DB1 column (30 m, 0.32 mm inner diameter, 5-μm film; J&W Scientific) held isothermally at 60 °C with a head pressure of 23 p.s.i. Headspace concentrations of volatiles were determined from 4-point standard curves run daily by injecting various amounts of 100 ppm isoprene and MBO from standard tanks obtained from Scott-Marrin (Riverside, CA).
We corrected for partitioning of MBO into the liquid phase using the Henry's constant of 65 m/atmosphere (23). Using Equation 2 from Ref. 23, it was calculated that 16 times more MBO was in the liquid phase than the gas phase in our system. Because the Henry's constant of isoprene (24) predicts very little partitioning into water, no correction was applied.
Phylogenetic Analysis
Amino acid sequence data for angiosperm and gymnosperm terpene synthases were obtained from GenBankTM (NCBI), combined with the sequence we obtained for MBO synthase, and aligned with the Espresso module of Tcoffee (25) using the crystal structures of Protein Data Bank codes 2J5C, 3N0F, and 5EAU to provide structural information for the alignment process. The resulting alignment was edited by hand to correct minor misalignments and yielded a final alignment containing 1105 sites.
Sequences were analyzed in a Bayesian framework using MrBayes 3.1 (26, 27). Five independent runs each with three heated chains (temperature parameter 0.2) and one cold chain were extended for one million generations with rate variation across sites modeled as an invariant γ-distribution. The Markov chain Monte Carlo algorithm sampled several amino acid substitution matrices and always selected the Jones matrix (28). Trees were sampled every 1000 generations, yielding 1000 trees/run.
Molecular Modeling
Homology models were constructed using the SWISS-MODEL automated mode (29–31) and the crystal structure of bornyl diphosphate synthase (Protein Data Bank code 1N20) as a template. This structure was used in preference to the isoprene synthase structure (13) because 1N20 had greater resolution (2.0 versus 2.8 Å), features helix H3 in the closed conformation, and has both the J-K loop and N-terminal arm fully resolved. Magnesium atoms and substrates were manually docked into the resulting models in positions corresponding to those observed in the 1N20 structure using PyMOL v1.3. Active site residues were identified by searching for atoms within 6 Å of the docked substrate.
RESULTS
Discovery and Functional Confirmation of the MBO Synthase Gene
Using degenerate PCR primers designed to match conserved regions of conifer monoterpene synthase genes, we amplified a series of sequence fragments from cDNA derived from the needles of P. sabiniana. One of these sequences encodes a phenylalanine seven residues upstream of the magnesium-binding site (DDXXD) in a position homologous to that occupied by a Phe residue in angiosperm isoprene synthases (9). Following 3′- and 5′-RACE procedures, a complete sequence (GenBankTM accession number JF719039) coding for a peptide 614 amino acids in length with a 40-amino acid plastid transport sequence predicted by ChloroP was obtained (32). Expression of a codon-optimized sequence in E. coli (with the predicted plastid transport sequence omitted) gave good expression of a soluble 70-kDa protein that could be purified on Ni-NTA-agarose.
Enzyme function was assayed by incubating extracts with DMADP and analyzing the products released into the reaction vial headspace. GC revealed the presence of two peaks with retention times identical to those of authentic isoprene and MBO standards (supplemental Fig. S1). The products were confirmed to be isoprene and MBO by GC/MS (supplemental Fig. S2). Assays of extracts from non-induced E. coli and on boiled purified protein showed only trace levels of isoprene or MBO, whereas those run on crude and purified extracts yielded large amounts of both MBO and isoprene in a ratio of ≈90:1 (Fig. 1).
FIGURE 1.

MBO synthase activity in protein extracts of E. coli containing the recombinant MBO synthase gene. E. coli cells containing recombinant MBO synthase were grown with (induced culture) or without (non-induced) 1 mm isopropyl β-d-thiogalactopyranoside, and soluble protein was extracted as described under “Materials and Methods.” An aliquot of the crude extract derived from the induced culture was boiled for 10 min, and a separate aliquot was purified on Ni-NTA-agarose. Enzyme assays were incubated at 37 °C for 120 min, and product formation was determined by GC. Non-enzymatic conversion of DMADP into isoprene and MBO was measured in paired assays containing no protein and subtracted. Data are means ± S.E. (n = 3). Black bars, data for MBO; gray bars, data for isoprene.
Biochemical Characterization of MBO Synthase
MBO synthase exhibited a broad pH optimum that was nearly flat from pH 6 to 9.5. Incubation at varying pH values had no impact on the relative amounts of isoprene and MBO produced.
Divalent cations were required for enzyme activity (Fig. 2). Isoprene production increased with Mg2+ to saturation at 7.5 mm and remained constant at Mg2+ concentrations up to 100 mm, whereas MBO production increased to a peak at 25 mm Mg2+ and declined at higher concentrations (Fig. 2, upper panel). Enzyme activity peaked at lower concentrations of manganese (2 mm) for both MBO and isoprene production and exhibited inhibition at Mn2+ concentrations greater than 50 mm (Fig. 2, middle panel). Although MBO synthase activity peaked at lower concentrations of Mn2+, MBO synthase activity was 33–35% higher using Mg2+ as a cofactor compared with using Mn2+ (Fig. 2, lower panel).
FIGURE 2.

Divalent cation dependence of MBO synthase activity. MBO synthase was extracted in sodium phosphate buffer free of divalent cations. Divalent cation-free enzyme was then assayed in a reaction mixture containing final concentrations of 100 mm Hepes (pH 8.0), 100 mm KCl, 11.2 mm DMADP, and 20 mm DTT. Other details are as described in the legend to Fig. 1. Data were normalized to the amount of MBO produced at 20 mm MgCl2 for each trial. ●, isoprene; ○, MBO. Black bars, data for MBO; gray bars, data for isoprene.
MBO synthase did not require a monovalent cation for activity; however, some monovalent cations could alter both enzyme activity and the MBO/isoprene product ratio (Fig. 3, upper panel). Most monovalent cations (Li+, Na+, K+, Rb+, and Cs+ as chloride salts) inhibited isoprene production. Li+ and Cs+ strongly inhibited MBO production, whereas NH4+ and Na+ inhibited MBO production slightly. In contrast, K+ and Rb+ strongly enhanced MBO production. MBO production was 3.1 times higher when incubated with K+ (100 mm) and 2.4 times higher when incubated with Rb+ (100 mm) than in the absence of a monovalent cation. K+ was the preferred cation for MBO production, with MBO synthase exhibiting 22% more activity with K+ than with Rb+.
FIGURE 3.

Monovalent cation dependence of MBO synthase activity. MBO synthase was extracted in sodium phosphate buffer free of divalent and monovalent cations. Cation-free enzyme was assayed in a reaction mixture as described in the legend to Fig. 1 except for the monovalent cation concentrations. MBO and isoprene emission data were normalized to the amount of MBO produced at 50 mm KCl for each trial. Data are means ± S.E. (n = 4). ●, isoprene; ○, MBO. Black bars, data for MBO; gray bars, data for isoprene.
MBO production by MBO synthase exhibited a type II response (33) to K+, with saturation occurring above 100 mm (Fig. 3, lower panel). However, at higher K+ concentrations, isoprene production exhibited an exponential decline, which mirrored the shape of the rise in MBO production (Fig. 3, lower panel). However, because of the nearly 100-fold different scales of MBO and isoprene production, the mirrored curves do not represent a 1:1 trade-off in response to increasing K+.
MBO synthase activity increased with DMADP concentration to a maximum of ∼16 mm for both MBO and isoprene production, but higher concentrations of substrate inhibited enzyme activity (Fig. 4). The ratio of MBO to isoprene produced remained constant over the range of DMADP concentrations examined. Incubating MBO synthase with either geranyl diphosphate (GDP) or isopentenyl diphosphate did not result in the detectable formation of monoterpene or hemiterpene products.
FIGURE 4.

Substrate dependence of MBO synthase activity. Crude extract of MBO synthase was assayed with final concentrations of 100 mm Hepes (pH 8.0), 20 mm MgCl2, 100 mm KCl, and 20 mm DTT. Other details are as described in the legend to Fig. 1. Data are means ± S.E. (n = 3), and the lines were fitted using the Michaelis-Menten equation with allowance for a Hill coefficient and substrate inhibition. ●, isoprene; ○, MBO.
A modified Michaelis-Menten equation that allows for cooperativity and substrate inhibition was fitted to the MBO and isoprene production data in Fig. 4 (Equation 1),
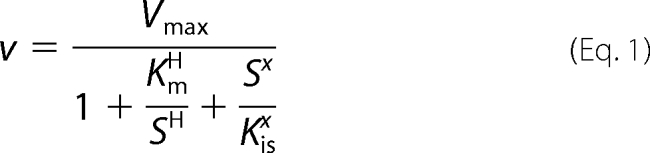 |
where H is the Hill coefficient describing cooperativity, Kis is the binding affinity of the substrate for the enzyme-substrate complex, and x is the number of substrate molecules that could bind to and inactivate the enzyme-substrate complex (see supplemental Fig. S3 for the complete kinetic model) (34, 35). Fitting by finding the minimum of the sum of squared errors yielded a Km of 20.0 mm DMADP and a Vmax of 212 pmol mg−1 s−1 for MBO production and a Km of 18.2 mm DMADP and a Vmax of 5.6 pmol mg−1 s−1 for isoprene production. The Hill coefficients were 1.2 and 1.4 for MBO and isoprene, respectively, indicating little cooperativity for MBO synthase. A simpler fitting procedure fitting only the data showing no obvious substrate inhibition but allowing for cooperativity gave a Km of 10.3 mm DMADP, a Vmax of 116 pmol mg−1 s−1, and a Hill coefficient of 1.2 for MBO production. The enzyme kinetic parameters for MBO synthase are compared with those of isoprene synthases and other terpene synthases in Table 1. With the exception of a larger Hill coefficient in the kudzu isoprene synthase, hemiterpene synthases behave similarly, with kcat values comparable with those reported for both monoterpene and sesquiterpene synthases.
TABLE 1.
Comparison of selected terpene synthase enzyme kinetic parameters
| Enzyme | Product | kcat | Km | Hill coefficient | Ref. |
|---|---|---|---|---|---|
| s−1 | |||||
| Malus domestica farnesene synthase | Farnesene | 0.0553 | 10–25 μma | 37, 47 | |
| M. domestica farnesene synthase | Monoterpenes | 0.00028 | 50 μma | 37 | |
| Mentha spicata limonene synthase | Limonene | 0.024 | 6.7 μm | 41 | |
| M. spicata 1,8-cineole synthase | 1,8-Cineole | 0.053 | 65 μm | 48 | |
| P. alba isoprene synthase | Isoprene | 0.03 | 8.7 mm | 11 | |
| Populus x canescens isoprene synthase | Isoprene | 0.26 | 2.45 mm | 49 | |
| P. montana isoprene synthase | Isoprene | 0.088 | 7.7 mm | 4.1 | 9 |
| P. sabiniana MBO synthase | MBO | 0.015 | 20.0 mm | 1.2 | This study |
| P. sabiniana MBO synthase | Isoprene | 0.0004 | 18.2 mm | 1.4 | This study |
a This is the concentration used in the assay when the Km was not reported.
MBO Synthase and Isoprene Synthase Evolved Independently
A gene tree was constructed to examine the placement of MBO synthase within the broader terpene synthase gene family (Fig. 5). The gene most closely related to MBO synthase is a linalool synthase from Picea abies, with which MBO synthase shares 82% amino acid identity. Also closely related to MBO synthase are farnesene synthases from P. abies and Pinus taeda. Together, these enzymes form a strongly supported clade (posterior probability of 96%) of related enzymes producing an acyclic hemiterpene alcohol (MBO), an acyclic monoterpene alcohol (linalool), and an acyclic sesquiterpene (E-α-farnesene) nested within what is otherwise a clade dominated by enzymes producing cyclic monoterpenes.
FIGURE 5.

Relationships of hemiterpene synthases to angiosperm and gymnosperm terpene synthases. Amino acid sequences were analyzed in a Bayesian framework using MrBayes with five independent runs and four chains for 106 generations. Terminal positions in the tree list the species, enzyme product(s), and class of terpene produced by each enzyme (C5 = hemiterpene, C10 = monoterpene, C15 = sesquiterpene, and C20 = diterpene). Numbers at nodes represent posterior probabilities of splits. ent-Kaurene synthase from P. patens was included as an outgroup, and the tree was rooted midway between the ent-kaurene synthase of primary metabolism and terpene synthases of secondary metabolism. Methylbutenol synthase and isoprene synthase genes are highlighted by gray boxes.
Comparison of the placement of MBO synthase with the placement of the isoprene synthases found in angiosperms shows that these two hemiterpene-forming enzymes are distantly related. MBO synthase shares only 33–34% amino acid identity with the Populus alba and Pueraria montana (kudzu) isoprene synthases. MBO synthase nests well within the TPS-d1 clade of gymnosperm monoterpenes synthases, whereas isoprene synthases from P. alba and P. montana nest well within the TPS-b clade of angiosperm monoterpene synthases. This indicates that MBO synthases in pines and isoprene synthases in angiosperms likely evolved independently from monoterpene synthase genes present in their respective groups.
Methylbutenol Synthase Evolved by Active Site Reduction
MBO synthase was modeled on the crystal structure of bornyl diphosphate synthase (BPPS; Protein Data Bank code 1N20) using residues 50–614 of the MBO synthase gene, which correspond to the complete amino acid sequence following the RRX8W motif. The BPPS template chosen featured a closed active site conformation with Mg2+ and a GDP substrate analog bound in the active site. MBO synthase and BPPS share 33% sequence identity. Residue numbers described hereafter correspond to the consecutive numbering of the full-length amino acid sequence of a given gene beginning with the start codon.
The homology model revealed that Phe-358 of MBO synthase is important in reducing the size of the active site of MBO synthase relative to that of monoterpene synthases. Phe-358 extends into the active site from helix D in a location identical to that observed for isoprene synthase (9, 13). Comparison of the active site surface of MBO synthase and the placement of GDP based on its position in BPPS shows that in MBO synthase, GDP extends outside the active site volume into space occupied by Phe-358 (Fig. 6). In contrast, DMADP, the substrate for MBO synthase, fits well within the boundary of the modeled MBO synthase active site.
FIGURE 6.

Comparison of MBO synthase and linalool synthase active site volumes. The active site volumes of MBO synthase (blue mesh) and linalool synthase (red mesh) are modeled on BPPS (Protein Data Bank code 1N20). The green spheres represent magnesium atoms, and the stick figure structure shows the placement of the DMADP carbon skeleton (green) and the GDP carbon skeleton (green and purple) in the active site. The orange and red portion indicates the diphosphate moiety. Phe-358 of MBO synthase is shown in blue to highlight the role it plays in reducing active site volume relative to that of linalool synthase.
To better understand the differences between MBO synthase and the related linalool synthase, we compared the residues making up the active sites. MBO synthase differs from linalool synthase by only three residues in the active site (F358A, A361G, and T513S). In each case, the amino acid found in MBO synthase is larger than that found in linalool synthase; however, Phe-358 is the only amino acid difference that significantly alters active site shape (reducing its size). Replacing Phe-358 in the homology model with alanine (found in linalool synthase) increased the size of the active site such that GDP will fit easily (Fig. 6). This suggests that MBO synthase might have evolved from a linalool synthase by a single amino acid change.
Methylbutenol Synthase and Isoprene Synthase Achieve Active Site Reduction Using Different Amino Acid Residues
In isoprene synthase, a second Phe residue (Phe-485) extending from helix H toward Phe-338 on helix D appears to be involved in reducing the active site volume. Together, Phe-338 and Phe-485 in isoprene synthase (ISPS) form a wall at the back and sides of the active site. The position occupied by Phe-338 in P. alba ISPS is also occupied by a Phe residue in MBO synthase (Phe-358); however, the residue that is homologous to Phe-485 in P. alba ISPS is Leu-505 in MBO synthase. Leu-505 is smaller than Phe and therefore less effective in reducing the active site volume on the side adjacent to helix H.
In MBO synthase, Phe-466 and Phe-583 extend into the active site from different helices and fill the same space as Phe-485 in ISPS (Fig. 7A). Phe-466 extends upward from the helix G1/G2 hinge forming the side wall of the active site adjacent to helix H, and Phe-583 extends into the active site from helix J. Spatially, Phe-485 in ISPS fits in between Phe-466 and Phe-583 in MBO synthase (Fig. 7B). Comparison of the active sites in ISPS and MBO synthase shows that they are nearly identical in shape and size, despite using different Phe residues on the helix H side to define active site boundary. This illustrates that MBO synthase and isoprene synthase obtain similar active site conformations but do so using different amino acid residues.
FIGURE 7.

Impact of helix H side residues on MBO synthase and isoprene synthase active sites. A, active site volume of native MBO synthase (blue mesh) or MBO synthase with F466S and F583L mutations (red mesh) modeled on BPPS (Protein Data Bank code 1N20). Green spheres represent magnesium atoms. The purple stick figure represents the placement of GDP based on its position in BPPS. The green stick figure shows GDP rotated to fit into the larger volume created by the F466S and F583L mutations. The cyan residue shows the position of Phe-485 in isoprene synthase. B, active site volume of native isoprene synthase (blue mesh) or isoprene synthase with an F485L mutation (red mesh) modeled on BPPS (Protein Data Bank code 1N20). Green spheres represent magnesium atoms. The purple stick figure represents the placement of GDP based on its position in BPPS. The green stick figure shows GDP rotated to fit into the larger volume created by the F485L mutation. The cyan residues show the positions of Phe-466 and Phe-583 in MBO synthase.
To determine the importance of Phe-485 in ISPS and Phe-466 and Phe-583 in MBO synthase for restricting the active site volume and preventing the binding of GDP, we performed in silico mutation of the MBO synthase and ISPS genes, replacing the Phe residues with smaller amino acids commonly found at these sites in monoterpene synthases. To identify biologically relevant alternative residues at Phe-485, Phe-466, and Phe-583, we examined a sequence alignment containing numerous angiosperm and gymnosperm monoterpene synthase genes. In angiosperms, the position occupied by Phe-485 in ISPS is most commonly occupied by Leu in monoterpene synthases. Phe-466 in MBO synthase is most commonly replaced with Ser in gymnosperm monoterpene synthases, and Phe-583 is most commonly replaced with Leu. Replacing Phe residues with the smaller amino acids extended the size of the active site alongside helix H in both the MBO synthase (Fig. 7A) and isoprene synthase (Fig. 7B) homology models. This enlargement was such that GDP could refold into the extra space alongside helix H even if the helix D residues Phe-338 and Phe-358 constrained its placement along the helix D side of the active site. However, when the native helix H side Phe residues found in ISPS (Phe-485) and MBO synthase (Phe-466 and Phe-583) were present, the active site dimensions along helix H were small enough such that even after refolding, GDP would be forced to occupy space outside the active site surface. This suggests that both helix D and helix H Phe residues found in MBO synthase and isoprene synthase are important for preventing the activity of hemiterpene synthases with GDP.
DISCUSSION
Biochemical characterization of the recombinant MBO synthase shows that the enzyme behaves in a fashion broadly similar to that seen in the native plant enzyme. However, a few key differences were observed. MBO synthase was completely dependent on the presence of divalent cations and had more affinity for Mn2+ than for Mg2+, as has been reported for MBO synthase by Fisher et al. (5). We observed higher activity with Mg2+ and inhibition by Mn2+ at high concentrations. This latter effect has also been seen for some monoterpene synthases (36). MBO synthase exhibited a type II response to potassium, as is commonly observed in gymnosperm terpene synthases. The enhancement of activity by K+ has been traced to a specific amino acid in the H-α1 loop (37). Enzymes that have a positively charged amino acid at this site do not require K+, whereas those with neutral amino acids do. Gymnosperm terpene synthases generally have a Ser residue in this position, and MBO synthase follows this trend. In addition to the MBO synthase activity, we also observed a small isoprene synthase activity that was strongest in the absence of K+ and inhibited at high K+ concentrations. Potassium levels in plant cells may be in the range of 40–80 mm (38, 39), so the in vivo activity of MBO synthase would produce >99% MBO, explaining why isoprene emission has not been observed from MBO-emitting pines.
Silver and Fall (7) suggested that MBO synthesis and isoprene synthesis could have similar mechanisms, but all isoprene synthases reported to date have made only isoprene. The dual MBO synthase and isoprene synthase activities of MBO synthase could arise by formation of a carbocation that is quenched either by water to form MBO or by abstraction of any of the six methyl hydrogens to form isoprene (Fig. 8). A similar bifunctionality was reported for kaurene synthase from Physcomitrella patens (40). In that case, a specific amino acid that allowed water to quench the carbocation (Ser-710) was identified. MBO synthase has a serine in the analogous position (Ser-440), whereas isoprene synthases have phenylalanine in this position. The smaller amino acid in MBO synthase could allow water to stay in the active site when it closes, allowing synthesis of MBO in preference to isoprene. However, the linalool synthase of Arabidopsis has a tyrosine in the analogous position (Tyr-406), and both R- and S-stereoisomers of linalool are known, indicating flexibility in the source of water for cation quenching.
FIGURE 8.

Reaction mechanism for MBO and isoprene synthesis and corresponding acyclic monoterpene and sesquiterpene synthesis reactions. The reaction is initiated by loss of the diphosphate, resulting in a carbocation, which is quenched by proton abstraction to make isoprene or addition of water to make the tertiary alcohol MBO. Any of six protons can be abstracted to make isoprene. Various acyclic monoterpenes and sesquiterpenes can be formed depending on which proton is abstracted or which stereoisomer of the alcohol is made.
The mechanism of hemiterpene formation corresponds to the mechanisms of acyclic terpene synthases. In all cases, the carbocation stays at carbon 3 (Fig. 8) instead of migrating down the chain to facilitate ring formation. Only two hemiterpenes can be formed, isoprene by proton abstraction or MBO by hydroxyl addition. For acyclic monoterpenes and sesquiterpenes, the specific proton abstracted or the stereochemistry of the hydroxyl addition yields different products. Most of the possible products have been reported, indicating that these synthases are likely to be opportunistic in the base used to quench the carbocation (Fig. 8). The mechanism of hemiterpene synthases and other acyclic terpene synthases also would not require the rotation around the 2–3 bond that cyclized terpene synthases require and that requires the canonical double arginine at the N terminus (41). Acyclic monoterpene synthases without the canonical double arginine are known (42). It may be possible to find/engineer hemiterpene synthases without the double arginine.
The Km for DMADP exhibited by MBO synthase is comparable with that of isoprene synthases but much larger than the substrate Km observed for monoterpene synthases (millimolar versus micromolar). Some hemiterpene synthases exhibit cooperativity (9), but MBO synthase shows very little. The Km values of both the MBO synthase and isoprene synthase activities are the same, indicating that the enzyme is likely limited by the rate of formation of the carbocation and that, once it is formed, its fate as either MBO or isoprene is independent of the initial substrate concentration.
Identification of the MBO synthase gene supports the idea that hemiterpene synthases evolved from monoterpene synthases by active site reduction brought about by replacement of key amino acid residues with phenylalanines. However, MBO synthase also illustrates that such active site reduction can occur in more than one fashion. In both MBO synthase and isoprene synthase, a Phe residue extending from helix D closes off the back of the active site, yet MBO synthase and isoprene synthase use different amino acid residues to close off the helix H side of the active site volume. Isoprene synthases use Phe-485, whereas MBO synthase uses Phe-466 and Phe-583. Modeling shows that these residues function in the same way to reduce the size of the active site, showing flexibility in how the active site shape needed for hemiterpene synthesis is achieved. The large number of Phe residues in the active site may also be important because of the large presence of π-electrons in these aromatic groups. These could help stabilize the carbocation intermediate by cation-π interactions (43–46), allowing time for catalysis.
Comparison of the placement of MBO synthase and isoprene synthases within the terpene synthase gene family clearly demonstrates that hemiterpene synthesis evolved independently in angiosperms and gymnosperms. MBO synthase clusters with gymnosperm monoterpene synthases, isoprene synthases cluster with angiosperm monoterpene synthases, and these gene families diverged between 250 and 290 million years ago (17). Molecular modeling has shown that linalool synthase differs from MBO synthase by three amino acids, of which one appears to be sufficient to eliminate activity with GDP. Failure to observe linalool production when assayed with GDP shows that MBO synthase is not a linalool synthase with a broader substrate acceptance. Given that linalool synthases are found in many taxa and that the shift from linalool synthase activity to MBO synthase activity appears to require a relatively simple change, it remains a puzzle why MBO emission has not been found outside of Pinus.
Supplementary Material
This work was supported by National Science Foundation Grant IOS-0950574 and a gift from ZuvaChem, Inc. A patent has been filed covering the MBO gene sequence.
The nucleotide sequence(s) reported in this paper has been submitted to the Gen-BankTM/EBI Data Bank with accession number(s) JF19039.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3, Table S1, and additional references.
- MBO
- 2-methyl-3-buten-2-ol
- DMADP
- dimethylallyl diphosphate
- TPS
- terpene synthase
- RACE
- rapid amplification of cDNA ends
- Ni-NTA
- nickel-nitrilotriacetic acid
- GDP
- geranyl diphosphate
- BPPS
- bornyl diphosphate synthase
- ISPS
- isoprene synthase.
REFERENCES
- 1. Guenther A., Karl T., Harley P., Wiedinmyer C., Palmer P. I., Geron C. (2006) Atmos. Chem. Phys. 6, 3181–3210 [Google Scholar]
- 2. Harley P., Fridd-stroud V., Greenberg J., Guenther A., Vasconcellos P. (1998) J. Geophys. Res. 103, 25479–25486 [Google Scholar]
- 3. Schade G. W., Goldstein A. H., Gray D. W., Lerdau M. T. (2000) Atmos. Environ. 34, 3535–3544 [Google Scholar]
- 4. Lerdau M., Gray D. (2003) New Phytol. 157, 199–211 [DOI] [PubMed] [Google Scholar]
- 5. Fisher A. J., Baker B. M., Greenberg J. P., Fall R. (2000) Arch. Biochem. Biophys. 383, 128–134 [DOI] [PubMed] [Google Scholar]
- 6. Silver G. M., Fall R. (1991) Plant Physiol. 97, 1588–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Silver G. M., Fall R. (1995) J. Biol. Chem. 270, 13010–13016 [DOI] [PubMed] [Google Scholar]
- 8. Hanson D. T., Swanson S., Graham L. E., Sharkey T. D. (1999) Am. J. Bot. 86, 634–639 [PubMed] [Google Scholar]
- 9. Sharkey T. D., Yeh S., Wiberley A. E., Falbel T. G., Gong D., Fernandez D. E. (2005) Plant Physiol. 137, 700–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller B., Oschinski C., Zimmer W. (2001) Planta 213, 483–487 [DOI] [PubMed] [Google Scholar]
- 11. Sasaki K., Ohara K., Yazaki K. (2005) FEBS Lett. 579, 2514–2518 [DOI] [PubMed] [Google Scholar]
- 12. Vickers C. E., Possell M., Nicholas Hewitt C., Mullineaux P. M. (2010) Plant Mol. Biol. 73, 547–558 [DOI] [PubMed] [Google Scholar]
- 13. Köksal M., Zimmer I., Schnitzler J. P., Christianson D. W. (2010) J. Mol. Biol. 402, 363–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bohlmann J., Meyer-Gauen G., Croteau R. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 4126–4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen F., Tholl D., Bohlmann J., Pichersky E. (2011) Plant J. 66, 212–229 [DOI] [PubMed] [Google Scholar]
- 16. Trapp S. C., Croteau R. B. (2001) Genetics 158, 811–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin D. M., Fäldt J., Bohlmann J. (2004) Plant Physiol. 135, 1908–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keeling C. I., Weisshaar S., Ralph S. G., Jancsik S., Hamberger B., Dullat H. K., Bohlmann J. (2011) BMC Plant Biol. 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang S., Puryear J., Cairney J. (1993) Plant Mol. Biol. Reporter 11, 113–116 [Google Scholar]
- 20. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 21. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frohman M. A. (1993) Methods Enzymol. 218, 340–356 [DOI] [PubMed] [Google Scholar]
- 23. Iraci L. T., Baker B. M., Tyndall G. S., Orlando J. J. (1999) J. Atmos. Chem. 33, 321–330 [Google Scholar]
- 24. Copolovici L. O., Niinemets U. (2005) Chemosphere 61, 1390–1400 [DOI] [PubMed] [Google Scholar]
- 25. Notredame C., Higgins D. G., Heringa J. (2000) J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 26. Huelsenbeck J. P., Ronquist F. (2001) Bioinformatics 17, 754–755 [DOI] [PubMed] [Google Scholar]
- 27. Ronquist F., Huelsenbeck J. P. (2003) Bioinformatics 19, 1572–1574 [DOI] [PubMed] [Google Scholar]
- 28. Jones D. T., Taylor W. R., Thornton J. M. (1992) Comput. Appl. Sci. 8, 275–282 [DOI] [PubMed] [Google Scholar]
- 29. Guex N., Peitsch M. C. (1997) Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 30. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 32. Emanuelsson O., Nielsen H., von Heijne G. (1999) Protein Sci. 8, 978–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Page M. J., Di Cera E. (2006) Physiol. Rev. 86, 1049–1092 [DOI] [PubMed] [Google Scholar]
- 34. Neet K. E. (1995) Methods Enzymol. 249, 519–567 [DOI] [PubMed] [Google Scholar]
- 35. LiCata V. J., Allewell N. M. (1997) Biophys. Chem. 64, 225–234 [DOI] [PubMed] [Google Scholar]
- 36. Rajaonarivony J. I., Gershenzon J., Croteau R. (1992) Arch. Biochem. Biophys. 296, 49–57 [DOI] [PubMed] [Google Scholar]
- 37. Green S., Squire C. J., Nieuwenhuizen N. J., Baker E. N., Laing W. (2009) J. Biol. Chem. 284, 8661–8669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walker D. J., Leigh R. A., Miller A. J. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 10510–10514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gobert A., Isayenkov S., Voelker C., Czempinski K., Maathuis F. J. M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 10726–10731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kawaide H., Hayashi K., Kawanabe R., Sakigi Y., Matsuo A., Natsume M., Nozaki H. (2011) FEBS J. 278, 123–133 [DOI] [PubMed] [Google Scholar]
- 41. Williams D. C., McGarvey D. J., Katahira E. J., Croteau R. (1998) Biochemistry 37, 12213–12220 [DOI] [PubMed] [Google Scholar]
- 42. Dudareva N., Martin D., Kish C. M., Kolosova N., Gorenstein N., Fäldt J., Miller B., Bohlmann J. (2003) Plant Cell 15, 1227–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dougherty D. A. (1996) Science 271, 163–168 [DOI] [PubMed] [Google Scholar]
- 44. Gallivan J. P., Dougherty D. A. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 9459–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lesburg C. A., Zhai G., Cane D. E., Christianson D. W. (1997) Science 277, 1820–1824 [DOI] [PubMed] [Google Scholar]
- 46. Christianson D. W. (2006) Chem. Rev. 106, 3412–3442 [DOI] [PubMed] [Google Scholar]
- 47. Pechous S. W., Whitaker B. D. (2004) Planta 219, 84–94 [DOI] [PubMed] [Google Scholar]
- 48. Kampranis S. C., Ioannidis D., Purvis A., Mahrez W., Ninga E., Katerelos N. A., Anssour S., Dunwell J. M., Degenhardt J., Makris A. M., Goodenough P. W., Johnson C. B. (2007) Plant Cell 19, 1994–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schnitzler J. P., Zimmer I., Bachl A., Arend M., Fromm J., Fischbach R. J. (2005) Planta 222, 777–786 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.