

Abstract
Nonstructural protein 5A (NS5A) is essential for hepatitis C virus (HCV) replication and constitutes an attractive target for antiviral drug development. Although structural data for its in-plane membrane anchor and domain D1 are available, the structure of domains 2 (D2) and 3 (D3) remain poorly defined. We report here a comparative molecular characterization of the NS5A-D3 domains of the HCV JFH-1 (genotype 2a) and Con1 (genotype 1b) strains. Combining gel filtration, CD, and NMR spectroscopy analyses, we show that NS5A-D3 is natively unfolded. However, NS5A-D3 domains from both JFH-1 and Con1 strains exhibit a propensity to partially fold into an α-helix. NMR analysis identifies two putative α-helices, for which a molecular model could be obtained. The amphipathic nature of the first helix and its conservation in all genotypes suggest that it might correspond to a molecular recognition element and, as such, promote the interaction with relevant biological partner(s). Because mutations conferring resistance to cyclophilin inhibitors have been mapped into NS5A-D3, we also investigated the functional interaction between NS5A-D3 and cyclophilin A (CypA). CypA indeed interacts with NS5A-D3, and this interaction is completely abolished by cyclosporin A. NMR heteronuclear exchange experiments demonstrate that CypA has in vitro peptidyl-prolyl cis/trans-isomerase activity toward some, but not all, of the peptidyl-prolyl bonds in NS5A-D3. These studies lead to novel insights into the structural features of NS5A-D3 and its relationships with CypA.
Keywords: NMR, Prolyl Isomerase, Protein Domains, Protein Structure, Protein-Protein Interactions, HCV
Introduction
Hepatitis C virus (HCV),3 a small enveloped virus from the Flaviviridae family, is a major cause of chronic liver disease that may lead to steatosis, liver cirrhosis, and hepatocellular carcinoma. Given about 180 million chronically infected individuals worldwide, HCV is an important health challenge (1). Current therapy is based on a combination of pegylated interferon-α and ribavirin but is not fully satisfying because numerous patients are not responding or suffer from serious side effects caused by this treatment. The development of new drugs to treat HCV infections requires a better understanding of the structural and functional features of the viral proteins and their relationships with host cell factors. The HCV genome (∼9.6 kb) encodes for a single polyprotein precursor (∼3,000 aa) that is co- and post-translationally processed by cellular and viral proteases to yield 10 mature proteins (2, 3). They are classified into structural proteins (Core, E1, and E2), which constitute the viral particle, and nonstructural proteins, of which two, p7 and nonstructural protein 2 (NS2), are required for virus assembly. The remainder of the nonstructural proteins (NS3, NS4A, NS4B, NS5A, and NS5B) are involved in HCV RNA replication (reviewed in Refs. 2 and 3).
Cyclophilins are host cell factors that in addition to viral proteins, are equally essential for HCV replication. The human genome encodes up to 16 different cyclophilins (4) that, despite differences in their tissue distribution, subcellular localization, and relative abundances, all share a similar three-dimensional structure and show in vitro peptidyl-prolyl cis/trans-isomerase (PPIase) activity (5). In vivo, they have been assigned numerous cellular functions, including protein folding, immune response, lipid and protein trafficking, or regulation of transcription. Initially, cyclophilins were identified as immune response modulators through their binding to cyclosporin A (CsA) (6) that creates a novel molecular surface leading to inhibition of the calcineurin phosphatase. Recently, the same interaction of CsA with cyclophilins was shown to disrupt an essential role of cyclophilins in the HCV life cycle (7). This replication block is independent of the CsA interaction with calcineurin because the non-imunosuppressive cyclosporin analogues Debio-025 (8–10), NIM811 (11), and SCY635 (12) are equally potent. However, the precise molecular role of the cyclophilins remains to be elucidated. It has been proposed that CypB, CypA, or even Cyp40 is involved (13–17). However, recently, several laboratories have identified CypA as the key player in the HCV replication (17–20).
The identification of mutations in NS3, a serine protease, NS5B, the viral RNA-dependent RNA polymerase, and NS5A that all confer in vitro resistance to CsA or non-immunosuppressive CsA analogs, suggested that these proteins are potential cyclophilin binding partners (21–24). Recent studies also suggested that NS2, a cysteine protease, is an additional cyclophilin target (19, 25). NS5A mutations, mostly located in the C-terminal half of the protein, were shown to confer the highest resistance against cyclophilin inhibitors (21, 22). NS5A is a 49-kDa phosphoprotein essential for RNA replication and production of HCV particles (26, 27). Although it has been demonstrated that NS5A has RNA-binding properties (28, 29), its precise function(s) in the HCV life cycle remains to be elucidated. The protein is anchored to the cytoplasmic side of the endoplasmic reticulum membrane via an amphipathic N-terminal α-helix (30). In addition, NS5A is composed of three cytoplasmic domains: NS5A-D1, -D2, and -D3, which are connected by short low complexity sequences. The structure of D1 (residues 27–213) reveals a zinc-binding domain that could adopt at least two kinds of homodimeric structures: a claw-shaped dimer that has been proposed to be properly suited for RNA binding (31) and an alternative structure (32) that may be required for another function of NS5A in viral RNA replication or particle formation. Despite the lack of knowledge of its precise molecular role, D1 appears to be a very promising drug target, as demonstrated by the recent development of the picomolar BMS 790052 inhibitor (33). D2 of NS5A (residues 250–342) is essential for viral RNA replication (27, 34), although a large segment of this domain can be deleted without detectable effects on replication and assembly (26) and has been shown to be natively unfolded (35, 36). However, we showed in a recent study that NS5A-D2 from the JFH-1 strain (genotype 2a) interacts with, and is a substrate for, the PPIase of human cyclophilin A (35). In addition, we could correlate the CypA interaction sites with mutations in D2 that significantly influence viral RNA replication (27) and that confer resistance to cyclophilin inhibitors (14, 22, 37). Finally, D3 (residues 356–447) has been shown, at least in the context of the JFH-1 strain (2a), to be involved in the production and assembly of viral particles (26, 38). Mutations that confer resistance to cyclophilin inhibitors have equally been mapped to NS5A-D3 (19, 21), suggesting that a cyclophilin might (directly) interact with this domain as well.
We recently reported a preliminary molecular characterization of NS5A-D3 of the Con1 (1b) HCV strain and showed that this domain is globally unfolded (39). In the present study, we report a comparative biochemical and molecular characterization of NS5A-D3 from the HCV JFH-1 strain (genotype 2a) and the Con1 (1b) strain. NS5A-D3 from JFH-1 has a 20-residue insertion specific to the genotype 2, raising questions about the structural implications of this insertion. We show that although NS5A-D3 is globally unfolded, the D3 domains from both JFH-1 and Con1 HCV strains have an intrinsic propensity to adopt an α-helical fold. In addition, we investigate the functional relationships between D3 and CypA and show that CypA has in vitro PPIase activity toward some, but not all, of the peptidyl-prolyl bonds in NS5A-D3. These data provide novel insights into the structure of NS5A-D3 and suggest that the interaction with CypA might involve more than one specific NS5A domain.
EXPERIMENTAL PROCEDURES
Sequence Analysis
Sequence analyses were performed using the web site tools of Network Protein Sequence Analysis (NPSA) (40) and of the European HCV Data base available at the Institut de Biologie et Chimie des Protéines. Multiple-sequence alignments and amino acid conservations were carried out with the ClustalW program using default parameters. The repertoire of residues at each amino acid position and their frequencies were computed by using the tool “Extract” in the euHCVdb (41).
Expression and Purification of Non-labeled and 15N- and 15N,13C-Labeled NS5A-D3 (JFH-1)
The synthetic sequence coding for domain 3 of NS5A from HCV JFH-1 strain (euHCVdb; GenBankTM accession number AB047639, genotype 2a) was inserted in the pT7.7 plasmid. The recombinant domain 3 (NS5A-D3 (JFH-1)) comprises residues 355–464 of HCV NS5A protein and has extra M- and -LQHHHHHH sequences at N and C termini. Expression of recombinant NS5A-D3 (JFH-1) was done in Escherichia coli BL21(DE3)Star. Bacteria were grown in Luria-Bertani (LB) medium for non-labeled protein or in M9-based semirich medium (M9 medium supplemented with [15N]NH4Cl (1 g/liter), d-[13C6]glucose (2 g/liter) (when 13C labeling was required), Isogro 15N (or 13C,15N) powder growth medium (0.7 g/liter; Sigma-Aldrich), and ampicillin (100 μg/ml). When A600 nm reached ∼0.8, induction was carried out at 37 °C with 0.3 mm isopropyl-β-d-galactopyranoside for 4 h. Harvested cells were lysed using lysozyme and sonication. NS5A-D3 (JFH-1) was first purified on a HisTrap column (1 ml; GE Healthcare). Selected fractions were then pooled, dialyzed again with 20 mm Tris-Cl, pH 7.4, 2 mm EDTA overnight, and loaded on an anion exchange column (ResourceQ (1 ml), GE Healthcare). NS5A-D3 (JFH-1)-containing fractions were selected, using SDS-PAGE analysis, pooled, and dialyzed overnight against 20 mm NaH2PO4/Na2HPO4, pH 6.3, 30 mm NaCl, 0.02% NaN3, 1 mm tris(hydroxypropyl)phosphine. NS5A-D3 (JFH-1) was then concentrated up to 500 μm with a Vivaspin 15 concentrator (cut-off 5 kDa) (Satorius Stedim Biotech). Following filtration at 0.2 μm, NS5A-D3 aliquots were frozen at −80 °C with a few Chelex beads (Sigma-Aldrich).
Expression and Purification of Non-labeled and 15N- and 15N,13C-Labeled NS5A-D3 (Con1)
Expression and purification of domain 3 of NS5A from the HCV Con1 strain (euHCVdb; GenBankTM accession number AJ238799, genotype 1b) were performed as described previously (39). The recombinant domain 3 (NS5A-D3 (Con1)) comprises residues 359–445 of HCV NS5A protein and has extra M- and -LQHHHHHH sequences at the N and C termini.
Circular Dichroism
CD spectra were recorded on a Chirascan dichrograph (Applied Photophysics, Surrey, UK) calibrated with (1S)-(+)-10-camphorsulfonic acid. Measurements were carried out at room temperature in a 0.1-cm path length quartz cuvette, with protein concentrations ranging from 8 to 10 μm. Spectra were recorded in the 185–260-nm wavelength range with a 0.5-nm increment and a 2-s integration time. Spectra were processed, base line-corrected, and smoothed using Chirascan software. Spectral units were expressed as the molar ellipticity per residue by using protein concentrations determined from measurements of UV light absorbance of tyrosine and tryptophan at 280 nm (molar extinction coefficients of 1,536 and 5,600 m−1·cm−1, respectively). The α-helix content was estimated using the method of Chen et al. (42), assuming that the residue molar ellipticity at 222 nm is exclusively due to α-helix.
NMR Data Collection and Assignments
All of the NMR spectra used for the backbone assignment were acquired on a Bruker Avance 800-MHz spectrometer with a standard triple resonance probe (Bruker Biospin, Karlsruhe, Germany). The proton chemical shifts were referenced using the methyl signal of sodium 3-trimethyl-sill-[2,2′,3,3′-d4]propionate at 0 ppm. Spectra were processed with the Bruker TopSpin software package 1.3 and analyzed using the product plane approach developed in our laboratory (43). Backbone assignments were achieved using two-dimensional 1H,15N HSQC and triple resonance three-dimensional 1H,15N,13C HNCO, HN(CA)CO, HNCACB, HN(CO)CACB, and HN(CA)NNH spectra.
For assignment of NS5A-D3 (JFH-1) resonances without and in the presence of 50% TFE, NMR experiments were, respectively, acquired with 500 μm 15N,13C-labeled NS5A-D3 (JFH-1) samples in 20 mm NaH2PO4/Na2PO4, pH 6.3, 30 mm NaCl, 1 mm tris(hydroxypropyl)phosphine, 0.02% NaN3 with or without 50% TFE-d3 at 298 K (Biological Magnetic Resonance Data Bank accession numbers 16798 and 16799).
Assignments of NS5A-D3 (Con1) in aqueous buffer were described in a previous study from our laboratory (39) (Biological Magnetic Resonance Data Bank accession number 16166). Assignment of NS5A-D3 (Con1) resonances in the presence of 50% TFE were acquired on a 440 μm 15N,13C-labeled NS5A-D3 (Con1) sample in 20 mm NaH2PO4/Na2PO4 pH 6.3, 30 mm NaCl, 1 mm tris(hydroxypropyl)phosphine, 0.02% NaN3, 50% TFE-d3 at 298K (Biological Magnetic Resonance Data Bank accession number 16800).
Structural NMR Data Collection in 50% TFE
Additional three-dimensional 1H,15N,1H NOESY-HSQC and 1H,15N,1H HNHA spectra were acquired on the NS5A-D3 samples in the presence of 50% TFE-d3, using a Bruker Avance 600-MHz spectrometer equipped with a cryogenic triple resonance probe head (Bruker). These experiments were used to measure distance-based (NOE) and backbone dihedral angle-based (3JHN-Hα (Hz) coupling constants) experimental restraints, respectively, on NS5A-D3 samples. Due to partial overlap in the proton dimension maximum, peak intensities rather than peak integrals were measured for NOE cross-peaks. The 3JHN-Hα coupling constants (Hz) are related to the backbone dihedral angle Φ via the Karplus equation, 3JHN-Hα = A·cos2(Φ-60) + B·cos(Φ-60) + C (where A = 6.51, B = −1.76, and C = 1.60) (44). Experimental 3JHN-Hα constants were extracted from the HNHA spectra using Equation 1,
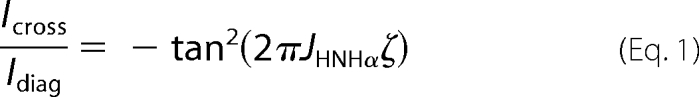 |
where Icross and Idiag represent the intensity of the cross-peak (HNHα) and of the corresponding diagonal peak (HNHN), respectively (45). The 2ζ parameter corresponds to the total evolution time for the 3JHN-Hα coupling and was set to 26.1 ms. The PyMOL software (available on the World Wide Web) was used for molecular graphics.
Structural NMR Structure Model Calculation of NS5A-D3 in 50% TFE
Three-dimensional structure calculations of NS5A-D3 (JFH-1 and Con1) in 50% TFE were done using the CNS software (46). 13C chemical shift values, 3JHN-Hα coupling constant values, and a list of NOE cross-peaks (see supplemental Table 2) were used as experimental restraints using the CNS dynamical annealing protocol; about 500 structures were calculated for NS5A-D3 (JFH-1) and for NS5A-D3 (Con1). In each case, on the initial set of accepted structures that fulfill the experimental restraints, an additional round of selection was made with acceptance criteria being no distance restraint violations >0.5 Å and no 3JHN-Hα coupling constant violations of >1 Hz for the values that were <5 Hz. This stricter selection step led to 111 accepted structures for NS5A-D3 (JFH-1) and 65 accepted structures for NS5A-D3 (Con1). Finally, from these accepted ensembles, only the 20 lowest energy structures were retained for NS5A-D3 (JFH-1) and for NS5A-D3 (Con1) in 50% TFE (see statistics in supplemental Table 2).
Interaction between NS5A-D3 and Cyclophilin A
Recombinant expression and purification of human cyclophilin A were performed as described previously. Unlabeled CypA was mixed with [15N]NS5A-D3, from the JFH-1 or Con1 strain, at equimolar concentration (100 μm each), and 1H,15N HSQC spectra were acquired. Interaction was assessed by comparing the 1H,15N HSQC spectra of NS5A-D3 recorded without and in the presence of CypA and in the latter case without or with cyclosporin A.
PPIase Activity of Cyclophilin A on NS5A-D3 Measured by NMR
The PPIase activity of CypA toward NS5A-D3 from the JFH-1 and Con1 strains was assessed using heteronuclear 1H,15N z-exchange spectra (47, 48). 15N z-exchange spectra, with 25-, 50-, 100-, 200-, 300-, and 400-ms mixing times, were recorded in triplicate for both NS5A-D3 using a 900-MHz spectrometer equipped with a cryoprobe. Experiments were recorded on a sample of 220 μm [15N]NS5A-D3 and 23 μm CypA and on a sample of 300 μm [15N]NS5A-D3 and 30 μm CypA. The exchange rates (kexch), as a function of mixing time (MT), were determined as described previously (35).
RESULTS
Sequence Analysis of NS5A-D3 (JFH-1)
Sequence alignment of the NS5A-D3 domains shows that the sequence of JFH-1 (genotype 2a) differs by a small 3-amino acid residue insertion (positions 378–380) and a long 20-residue insertion (positions 431–450) when compared with the Con1 strain (genotype 1b) (Fig. 1 and supplemental Fig. 1). The latter is located near the C terminus of the protein and is only observed in genotype 2 sequences (see supplemental Fig. 1C). Moreover, it is located within a region of high conservation in all genotypes (see supplemental Fig. 1C), arguing for an important function for the viral replication cycle. Amino acid sequence comparison also highlights additional intergenotypic conservation in the N-terminal region of NS5A-D3 (positions 363–380 in genotype 1b, supplemental Fig. 1C). Except for the conserved C-terminal residues of NS5A-D3, which are already part of the NS5A-NS5B cleavage site, the remaining sequence is poorly conserved between genotypes. However, the amino acid repertoire derived from the alignment of 363 and 21 sequences of genotype 1b and 2a, respectively, revealed the strict conservation of residues at some positions and the conservation of the physicochemical character of residues at most other positions (see supplemental Fig. 1, A and B). Together, these data indicate that the overall structure of NS5A-D3 is conserved across the different HCV genotypes and that the 20-residue insertion in genotype 2 probably corresponds to an extra loop.
FIGURE 1.

Sequence comparison of NS5A-D3 from JFH-1 and Con1 HCV strains and interaction with cyclophilin A. The NS5A-D3 sequence from the HCV JFH-1 strain (aa 355–464; genotype 2a; GenBankTM accession number AB047639) is aligned with the NS5A-D3 sequence from the HCV Con1 strain (aa 359–445; genotype 1b; AC number, AJ238799). Amino acids are numbered with respect to the respective NS5A proteins. The hyphens indicate gaps compared with the sequence alignment. Similarities between NS5A sequences are depicted according to ClustalW convention (asterisk, invariant; colon, highly similar; dot, similar). Proline residues (which are not visible in the 1H-15N HSQC NMR experiment; see “Experimental Procedures”) are highlighted in gray, and those conserved in both strains are in boldface type. The perturbations of chemical shifts observed for each residue in the 1H,15N HSQC spectrum of [15N]NS5A-D3 (100 μm) in the presence of cyclophilin A (100 μm) are indicated as follows: residue in blue, chemical shift perturbation and/or decreased intensity; residue in red, broadening of the resonance beyond detection; residue underlined, residue with its minor form perturbed (chemical shift perturbation and/or decreased intensity).
Analysis of the amino acid composition of NS5A-D3 indicates that this domain is composed of numerous residues generally observed in flexible structures. As exemplified with NS5A-D3 of JFH-1, a mere 2 (Ser and Gly) or 4 (Ser, Gly, Pro, and Glu) amino acids represent 34 or 56% of the complete sequence, respectively. The domain is highly acidic with a calculated isoelectric point (pI) of 3.5 (Asp + Glu = 15 versus Arg + Lys = 2).
Biochemical Characterization of HCV NS5A-D3 (JFH-1)
NS5A-D3 from the JFH-1 strain (residues 355–464) was overexpressed in E. coli and purified to homogeneity by using a two-step purification procedure (Fig. 2A). Although the molecular mass of NS5A-D3 (JFH-1) was 11,837 Da, as determined by mass spectroscopy, the purified protein migrates as a ∼27-kDa protein by SDS-PAGE (Fig. 2A). This discrepancy, already observed previously for NS5A-D3 of Con1 (39) (Fig. 2A), can be explained by the high content of acidic amino acids and proline residues (Fig. 1) (49). When analyzed by gel filtration chromatography, the elution time of NS5A-D3 (JFH-1) corresponds to a ∼43-kDa globular protein (Fig. 2B). This behavior is frequently observed for proteins that lack a globular organization (50, 51) and is similar to that observed for the unfolded NS5A-D3 of the Con1 strain (Fig. 2B) (39). Although NS5A-D3 from JFH-1 exhibits 23 additional residues as compared with that of the Con1 strain, its elution volume was slightly higher using the same experimental conditions. This unexpected behavior suggests a slightly higher level of structuring in NS5A-D3 of JFH-1.
FIGURE 2.

Purification and gel filtration analyses of HCV NS5A-D3. A, purified recombinant proteins from JFH-1 (11.8 kDa) and Con1 (9.9 kDa) HCV strains were analyzed by 15% SDS-PAGE and stained with Coomassie Blue. B, the gel filtration analyses of NS5A-D3 (Con1) (black curve) and NS5A-D3 (JFH-1) (gray curve) were performed on a Superdex S200 column equilibrated in 50 mm sodium phosphate, pH 7.4, 1 mm TCEP, with a flow rate of 0.5 ml/min. Elution volumes of globular protein standards are indicated by black arrows with the following corresponding molecular masses. 1, thyroglobulin (669,000 Da); 2, ferritin (440,000 Da); 3, aldolase (158,000 Da); 4, conalbumin (75,000 Da); 5, ovalbumin (43,000 Da); 6, chymotrypsin (25,000 Da); 7, ribonuclease (13,700 Da); 8, vitamin B12 (1355 Da).
NMR Analysis of NS5A-D3 (JFH-1)
NMR spectroscopy was used to gain structural and functional information of NS5A-D3 at an atomic level. At 800 MHz, the 1H,15N HSQC of NS5A-D3 (JFH-1) proved sufficiently well resolved despite the limited 0.85-ppm dispersion of the amide proton resonances (Fig. 3A). Both the narrow amide proton dispersion and the clustering of glycine and serine/threonine resonances confirm the non-structured nature of NS5A-D3 (JFH-1). Using standard triple resonance experiments on a doubly labeled [15N,13C]NS5A-D3 (JFH-1) sample and using our product plane-based assignment method (43), all of the backbone proton amide resonances were assigned with the exception of the first methionine and the proline residues (Fig. 3A). Several less intense peaks were assigned to residues that are close to one of the 14 prolines in NS5A-D3 (JFH-1) and for which their amide proton resonance is sensitive to the cis/trans equilibrium of this proline. Up to 25 of these less intense resonances were assigned (see supplemental Table 1). In addition to proton amide, 13Cα, 13Cβ, and 13CO resonances could also be assigned for almost all NS5A-D3 (JFH-1) residues. The unassigned 13C resonances correspond to Pro379, Pro417, Pro437, and Pro438 that are located in (Pro)2 or (Pro)3 motifs. These 13C chemical shifts were used to assess the secondary structure content in NS5A-D3 (JFH-1), at a per residue level. The chemical shift index (52) values for 13Cα are essentially negative, whereas the ones for 13Cβ are mostly positive (Fig. 3B). These values would indicate an extended structure for NS5A-D3 (JFH-1). Regarding the 13CO, chemical shift index (CSI) values are distributed between positive and negative values. The resulting zero CSI consensus values thereby confirm the absence of stable secondary structure elements. Nevertheless, for residues Glu366-Ala367-Leu368, the CSI consensus values are −1 (Fig. 3B) and might correspond to a single α-helical turn. In conclusion, in aqueous buffer, NS5A-D3 (JFH-1) is mostly unfolded but has a short and stable α-helical secondary structure element centered around the N-terminal Ala367.
FIGURE 3.

Molecular NMR characterization of NS5A-D3 (JFH-1). A, assigned 1H,15N HSQC spectrum of domain 3 of NS5A (JFH1). The small inset shows the spectrum region corresponding to the unique tryptophan side chain. B, CSI analysis of NS5A-D3 (JFH-1). NS5A-D3 (JFH-1) 13Cα, 13Cβ, and 13CO chemical shifts were analyzed using the CSI software (52). The resulting CSI consensus is shown, at the bottom, along the NS5A-D3 (JFH-1) sequence.
NS5A-D3 Has α-Helical Propensity
We showed previously that NS5A-D3 (Con1) is unfolded in an aqueous environment, although 13Cα and 13CO values in the 369–379 region indicated some helical tendency (39). This region corresponds to the same location where we observe the short α-helix in NS5A-D3 (JFH-1) (Figs. 1 and 3). Disorder predictors (including the VL-XT PONDR® algorithm (53, 54) and the metaPrDOS meta server (55)) indeed predicted a high degree of disorder in both NS5A-D3 sequences of the JFH-1 and the Con1 HCV strains, with, however, disorder scores close to the order/disorder limit value (0.5) for the N-terminal region (∼25 first residues) and the extreme C termini (Fig. 4). In these locations, the VL-XT PONDR® algorithm gave scores that would correspond to slightly ordered regions, whereas the metaPrDOS predictor gave scores above the disorder threshold (score >0.5), although the values marked a significant decrease. The metaPrDOS score is below the 0.5 threshold only for residues 365–369 of NS5A-D3 from JFH-1. This prediction exactly matches with the short α-helical elements that we identified on the basis of the NMR CSI scores (Fig. 3). We next reanalyzed our experimental 13Cα and 13Cβ chemical shifts of both NS5A-D3 domains in aqueous conditions with the SSP (Secondary Structure Propensities) program (56), developed to probe the propensities of unfolded proteins to form secondary structures from experimental NMR data. SSP analyses showed that both NS5A-D3 (JFH-1) and NS5A-D3 (Con1) have significant positive SSP scores (corresponding to an α-helix) in their N- and C-terminal regions and detected only significantly negative scores for residues 439–444 from JFH-1 located in the genotype 2-specific insertion (Fig. 4). In the N-terminal region of NS5A-D3, the SSP score is higher for JFH-1 than for Con1, with a maximal value of 0.398 for Ala367 in JFH-1 compared with 0.233 for Ala371 in Con1. Altogether, these results show that, despite being globally unfolded in aqueous solution, both NS5A-D3 (JFH-1) and NS5A-D3 (Con1) have intrinsic α-helical propensity in their N-terminal (residues 356–375 for JFH-1 and 360–379 for Con1) and C-terminal (residues 460–464 for JFH-1 and 442–445 for Con1) regions (Fig. 4).
FIGURE 4.

Disorder predictions and helical propensity of NS5A-D3. In the graphics the sequences of NS5A-D3 (JFH-1, 2a) (top) and NS5A-D3 (Con1, 1b) (bottom) are kept aligned as shown in Fig. 1. Disorder predictions were performed on the sequences of NS5A-D3 using the PONDR software (solid curve) and the MetaPrDOS meta server (dashed curve). A disorder score (left axes) above the 0.5 threshold indicates a disorder state, whereas a lower score indicates an order state. Results of the SSP analyses of the Cα and Cβ NMR chemical shifts of NS5A-D3 residues are indicated as gray bars. The positive SSP scores (right axes) correspond to helical propensity, whereas negative scores indicate extended regions.
Folding Propensity of NS5A-D3
The CD spectrum of NS5A-D3 is typical for a poorly folded protein with a large negative peak centered at 198 nm. Only the small but reproducible shoulder at 222 nm points toward a possible helical contribution (Fig. 5, A and D). To further probe the conformational preferences of NS5A-D3, we used TFE as a co-solvent known to stabilize transient secondary structures, especially α-helices (57). Once dissolved in a TFE/water (50:50) mixture, the far-UV CD difference spectra of both NS5A-D3 of JFH-1 and of Con1 exhibited the distinct minima at 208 and 222 nm and the maximum at 192 nm, both characteristic for α-helical folding (Fig. 5, A and D). Titration experiments with increasing concentrations of TFE, ranging from 0 to 75%, showed that maximal CD helical signals were obtained around 45% TFE for NS5A-D3 (JFH-1) (Fig. 5G). This is consistent with the suggestion of Jasanoff and Fersht (58) that, for a peptide with helical propensity, helicity is generally at a maximum by 20–30% TFE, and folding is complete by 50%. Above this concentration, TFE could induce irrelevant folding, as is likely the case for NS5A-D3 (Con1). Using the CD signal at 222 nm, the α-helical content in 50% TFE was estimated to be ∼19% for NS5A-D3 (JFH-1) and ∼27% for NS5A-D3 (Con1). Taking into account the respective lengths of the two proteins (119 and 96 residues, including the affinity tag), this means that ∼23 and ∼26 residues adopt an α-helical conformation in NS5A-D3 from JFH-1 and Con1, respectively. Similar α-helical folding was observed by CD in the presence of detergents (SDS, n-dodecyl-β-d-maltoside (DM), or dodecyl phosphocholine (DPC)) mimicking a hydrophobic environment (Fig. 5, B, C, E, and F).
FIGURE 5.

Far UV circular dichroism analyses of NS5A-D3. CD spectra of NS5A-D3 from JFH-1 (A–C) and from Con1 (D–F) were recorded in 10 mm sodium phosphate buffer, pH 7.4 (H2O solution, solid line), complemented with either 50% TFE (large dashed line) (A and D) or with the following detergents: 100 mm SDS (small dashed line), 100 mm n-dodecyl-β-d-maltoside (DM, medium dashed line), or 100 mm dodecyl phosphocholine (DPC; dotted lines, B and E). The difference spectra (dotted lines in A and D) were obtained by subtracting the spectrum recorded in the presence of 50% TFE from that recorded in H2O. In the presence of the various detergents, the corresponding difference spectra are reported in C and D. In all cases, the difference spectra were typical of α-helix folding with two minima at 208 and 222 nm and a maximum at 192 nm. G, the α-helix contents in NS5A-D3 JFH-1 (black circles) and in NS5A-D3 Con1 (white circles) are plotted as a function of percent TFE.
To gain further structural insights at the atomic level, we next investigated the α-helical conformation of NS5A-D3 by NMR spectroscopy in 50% TFE. Under these experimental conditions, the 1H,15N HSCQ spectrum of NS5A-D3 became better resolved with the amide proton resonances for NS5A-D3 (JFH-1) comprised between 7.6 and 9.15 ppm and those of NS5A-D3 (Con1) between 7.65 and 8.95 ppm (Fig. 6 and supplemental Fig. 2). Chemical shift values were very different upon the addition of 50% TFE (supplemental Fig. 2), forcing us to reassign the proton amide resonances of NS5A-D3 in these novel conditions (Fig. 6). Nearly complete assignments of 13Cα, 13Cβ, and 13CO resonances were achieved for both NS5A-D3 domains and showed significant deviations of 13C chemical shifts (Δ13C) from random coil values in two regions of both NS5A-D3 (JFH-1) and NS5A-D3 (Con1) (Fig. 7A). In NS5A-D3 (JFH-1), two stretches of consecutive positive Δ13Cα and Δ13CO values (Δ13C ≥ 0.7ppm), corresponding to residues Glu361–Phe376 and Thr462–Val464, are typical for α-helical secondary structure. Similar observations were made for NS5A-D3 (Con1) in regions Ser364–Gly381 and Ser441–Val445. Negative Δ13Cβ values, which indicate α-helices, are observed for some but not all residues in these regions. In addition, we measured the 3JHN-Hα coupling constants in both NS5A-D3 domains in 50% TFE using a three-dimensional HNHA experiment (45). Because the 3JHN-Hα is related to the backbone dihedral angle Φ via the Karplus equation (44), this parameter is also a good indicator of the secondary structure content. 3JHN-Hα values are close to random coil values (3JHN-Hα ≈ 6.5Hz) over most of the NS5A-D3 sequences, but significantly lower values corresponding to dihedral angles Φ found in α-helices were measured in the N-terminal regions (Fig. 7A). The coupling constant values are comprised between 3.13 and 5.15 Hz for residues Glu361–Lys374 in NS5A-D3 (JFH-1) and between 3.3 and 4.98 Hz for residues Glu365–Lys378 in NS5A-D3 (Con1). Concerning the C-terminal region, low 3JHN-Hα values were found for residues Glu442–Val445 in NS5A-D3 (Con1) but only Thr463 in NS5A-D3 (JFH-1). Analysis of the heteronuclear three-dimensional NOESY-HSQC spectra recorded in 50% TFE gave a further overview of the NOE connectivities in NS5A-D3 (JFH-1 and Con1). Typical short range (i, i + 1) and medium range (i, i + 2), (i, i + 3), and (i, i + 4) NOE connectivities confirmed the presence of a α-helix in the Ser360–Gly377 region of the JFH-1 domain and in the Ser364–Gly381 region of Con1 (Fig. 7B), although no long range NOEs could be identified. Similar characteristic NOE patterns were observed for a shorter region (i.e. 3–4 residues) at the C terminus of each NS5A-D3. Elsewhere in the sequence, only a few sequential NOE cross-peaks were detected, in agreement with an unstructured state.
FIGURE 6.

NMR analysis of NS5A-D3 in 50% TFE. Shown are assigned 1H,15N HSQC spectra of NS5A-D3 from the JFH-1 (A) and the Con1 (B) HCV strains that have been recorded in the presence of 50% TFE using an 800-MHz NMR spectrometer.
FIGURE 7.

Structural NMR parameters of NS5A-D3 in 50% TFE. A, the 13C chemical shift deviations from random coil values (Δ13C, left axes) are plotted along the NS5A-D3 (JFH-1) (top) and the NS5A-D3 (Con1) (bottom) sequences that are aligned as shown in Fig. 1. ▴, Δ13Cα; ×, Δ13Cβ; □, Δ13CO. The CSI analyses of 13Cα, 13Cβ, and 13CO values resulted in consensus helical segments that are indicated as thick black bars on the residue axes. The 3JHN-Hα coupling constants (Hz) (right axes), which are related to the backbone dihedral angle Φ (see “Experimental Procedures”) are plotted along the NS5A-D3 sequences as gray bars. B, overview of the NOE connectivities in NS5A-D3 (JFH-1) (top) and Con1 (bottom) that have been recorded using three-dimensional 1H,15N,1H NOESY-HSQC experiments. Only short sequential (i, i + 1) and medium range (i, i + 2), (i, i + 3), and (i,i + 4) NOE connectivities were detected in NS5A-D3 in the presence of 50% TFE. Typical NOE connectivities for an α-helix include sequential dNN(i, i + 1) and medium range dNN(i,i + 2), dαN(i,i + 3), and dαN(i,i + 4) connectivities.
NMR Structure Model of NS5A-D3 in 50% TFE
Using the combined experimental NMR data collected in 50% TFE as a starting point, we performed three-dimensional structure calculations on NS5A-D3 (JFH-1) and NS5A-D3 (Con1) using the CNS software (46). 13C chemical shift assignments, 3JHN-Hα coupling constant values, and a list of NOE cross-peaks (see supplemental Table 2) were used as experimental restraints for the calculations. Using the CNS dynamic annealing protocol, ∼500 structures were calculated for NS5A-D3 (JFH-1) and for NS5A-D3 (Con1) and led to a final set of 20 low energy structures that fulfill all of the experimental restraints (see statistics in supplemental Table 2). The absence of convergence during calculation of a global root mean square deviation over the Cα atoms of either NS5A-D3 (JFH-1 or Con1) reflects their predominant unfolded nature even in 50% TFE (see supplemental Fig. 3). Reflecting our previous observations, however, two different NS5A-D3 segments were found to adopt a regular α-helical conformation. The segment α1 corresponds to the longer one and extends from residue Ser360 to Gly377 in NS5A-D3 (JFH-1) and from residue Glu365 to Gly381 in NS5A-D3 (Con1). Following the superimposition of the 20 lowest energy structures over α1, the backbone (N, Cα, CO atoms) root mean square deviation for this segment is 2.00 Å for NS5A-D3 from JFH-1 and 1.84 Å for the domain from Con1 (Fig. 8, A–C, and supplemental Fig. 4). The α2 element is shorter and located at the extreme C terminus of NS5A-D3. In fact, α2 comprises the extreme C-terminal residues of NS5A-D3 and even a few residues coming from the purification tag. In the JFH-1 construct, α2 corresponds to residues Asp461–Val464 of domain 3 plus 3 residues from the tag. In the Con1 construct, α2 is slightly longer and comprises NS5A-D3 residues Ala440–Val445 and 6 additional residues from the tag. The backbone root mean square deviation of α2, over the 20 lowest energy structures, is 1.81 Å for NS5A-D3 from JFH-1 and 1.72 Å for the domain from Con1 (Fig. 8, B and D). Helices α1 of NS5A-D3 of JFH-1 and of Con1 are quite similar in length and correspond to the same region within domain 3. As shown in Fig. 8, E and F, the α1 helix has remarkable asymmetric distribution of hydrophobic residues and thus exhibits an amphipathic character. Save for Ile373 of JFH1, all hydrophobic residues of α1 are clustered on one side of the helix and are identical or conserved between JFH-1 and Con1. No particular structural features were observed for the genotype 2-specific 20-residue insertion (residues 431–450 of NS5A-D3 of JFH-1).
FIGURE 8.

NMR structure model of α1 and α2 helices of NS5A-D3. Shown are superimpositions of the backbone atoms (N, Cα, and CO atoms) of the 20 final structures of NS5A-D3 from JFH-1 HCV strain (A and B) and of NS5A-D3 from Con1 HCV strain (C and D). Superimpositions of the final NS5A-D3 structures were done over the backbone atoms of either the α1 helix (A and C) or the α2 helix (B and D). The amphipathic character of the α1 helix from NS5A-D3 (JFH-1) and from NS5A-D3 (Con1) is shown in E and F, respectively. The average α1 helix structures from both HCV strains are represented as a schematic and colored according to the Kite and Doolittle hydrophobicity scale (80) (−4.5 (green) to +4.5 (red)). The molecular surfaces are shown in the right panels and are colored using the same criteria as described above.
Interaction between Cyclophilin A and NS5A-D3
Because mutations in NS5A-D3 have been shown to confer resistance to cyclophilin inhibitors (19, 21) and because of the high number of proline residues in this domain, we tested for a direct interaction between NS5A-D3 and CypA. In a first step, we mixed [15N]CypA with unlabeled NS5A-D3 either from the JFH-1 or Con1 HCV strain. In the presence of NS5A-D3, only a limited subset of CypA resonances was affected. The residues of CypA involved in the interaction with NS5A-D3 are similar to the ones we previously showed to be involved in the interaction with NS5A-D2 (JFH-1) (35). These correspond to residues centered around the PPIase active site of CypA that also match with the CsA binding site of the cyclophilin.
In a second step, we wanted to monitor these interactions on the NS5A-D3 side. We therefore mixed equimolar concentrations (i.e. 100 μm) of either [15N]NS5A-D3 (JFH-1) or [15N]NS5A-D3 (Con1) with unlabeled CypA and acquired 1H-15N HSQC spectra. By comparison with the NS5A-D3 spectra recorded alone, we identified the residues involved in interaction with CypA based on chemical shift perturbations and/or peak broadening. Most of the perturbed resonances belong to residues in the C-terminal half of NS5A-D3 (from Ser415 to Asp461 for JFH-1 and from Ser414 to Val445 for Con1) (Fig. 1). Numerous NMR peaks in the 455TCSEEDD461 motif from the JFH-1 strain or in the corresponding one from Con1 (435TVSEEAS441) were indeed broadened beyond detection in the presence of CypA. Interestingly, the insertion specific to JFH-1 is also involved in interaction with CypA, because 11 of these 20 residues have their corresponding peak shifted and/or broadened. Except for the C-terminal motifs mentioned above, almost all NS5A-D3 residues that are affected by CypA are located close to proline residues. Moreover, many but not all of the less intense peaks in the HSQC spectrum assigned to residues in close proximity of a cis proline were perturbed (Fig. 1, underlined). This indicates that CypA interacts with some proline residues in the cis conformation of NS5A-D3. The addition of cyclosporin A in the NS5A-D3·CypA samples completely abolished the interaction between NS5A-D3 and CypA. Indeed, in the presence of the cyclophilin inhibitor, all the NS5A-D3 1H,15N resonances recovered their full intensities and chemical shifts comparable with NS5A-D3 alone. These data led us to investigate the PPIase activity of CypA toward NS5A-D3.
NS5A-D3 Is a Substrate for the PPIase Activity of CypA
In order to assess the potential PPIase activity of CypA toward NS5A-D3, we used heteronuclear 15N z-exchange NMR experiments (47, 48) in the presence of catalytic amounts of CypA (i.e. with a 10:1 molecular ratio). In 15N z-exchange experiments, the isomerization processes between cis and trans conformers are characterized by exchange peaks connecting the major and minor 1H,15N amide peaks of a given residue, which is located close to a proline. In the presence of CypA and with mixing times in the 25–400 ms range, several exchange peaks were detected for both NS5A-D3 domains (Fig. 9, A and B). These observations led us to the conclusion that CypA has in vitro PPIase activity toward NS5A-D3 of both HCV strains. Residues on which a CypA-catalyzed PPIase activity has been detected are shown along the NS5A-D3 sequences in Fig. 9C. In NS5A-D3 (Con1), CypA PPIase activity was detected for 2 of 6 prolines, Pro400 and Pro418, based on the exchange peaks for residues Gln399, Ser401, and Leu419. Concerning NS5A-D3 from the JFH-1 HCV strain, CypA activity was also detected on the basis of 8 residues, Gln378, Ser401, Glu404, Ala406, Met416, Val444, Ala445, and Gly447, located in the close proximity of 6 proline residues, Pro379, Pro402, Pro405, Pro407, Pro417, and Pro446. CypA is, however, specific for some of the peptidyl-prolyl bonds of NS5A-D3 because no PPIase activity was observed around several proline residues. For most of these, we could exclude spectral overlap as the culprit because minor and major 1H,15N peaks had been assigned for surrounding residues. In accordance with the higher number of proline residues in NS5A-D3 from JFH1 (14 versus 6 prolines in NS5A-D3 from Con1), we observed more exchange peaks in the NS5A-D3 (JFH1) z-exchange spectra than in the ones of Con1. However, CypA showed in vitro PPIase activity toward only 1 (Pro446) of 4 prolyl bonds in the genotype 2-specific insertion, with no detectable activity at the other 3 prolines clustered in the Pro3 motif (Fig. 9C). From the buildup curves of the exchange peaks as a function of the z-exchange mixing time, reliable CypA-catalyzed exchange rates (kexch) were extracted for Gln399 in NS5A-D3 (Con1) and for Ser401, Glu404, Ala406, and Ala445 in NS5A-D3 (JFH-1) (Fig. 9, D–F), The strongest PPIase activity of CypA, with a kexch = 30.9 s−1, was measured on Ala445 located in the insertion of NS5A-D3 (JFH-1). The addition of cyclosporin A in the assays inhibited the PPIase activity of CypA toward NS5A-D3 because exchange peaks were no longer detectable.
FIGURE 9.

Analysis of the PPIase enzymatic activity of CypA toward NS5A-D3. A and B, superimposition of a 1H,15N HSQC (in red) and a 1H,15N heteronuclear exchange spectrum (200-ms mixing time) that have been acquired on a [15N]NS5A-D3 sample in the presence of catalytic amount of CypA (1:10 ratio). A, NS5A-D3 (JFH-1); B, NS5A-D3 (Con1). The NMR resonances (trans, cis, and the two exchange peaks) of NS5A-D3 residues on which the PPIase enzymatic activity of CypA has been detected are connected by a dotted black box. C, residues on which a PPIase activity of CypA has been evidenced are shown in blue boldface type along the amino acid sequence of NS5A-D3 (JFH-1 (top) and Con1 (bottom)). Proline residues are highlighted in gray. Residues that gave both a major and a minor resonance in a 1H,15N HSQC spectrum are underlined. Residues Gly403 and Ser448 of NS5A-D3 (JFH-1) correspond to residues for which exchange peaks were tentatively assigned on the sole basis of 15N frequency because no minor peaks were assigned for these residues. D and E, determination of the CypA-catalyzed exchange rates (kexch, s−1) toward the peptide bonds Ala445-Pro446 in NS5A-D3 (JFH-1) (D) and Gln399-Pro400 in NS5A-D3 (Con1) (E). Taking into account the maximal intensity of the trans-cis exchange peak (Itc) and the trans diagonal peak (Itt), experimental data that have been measured for Itc/Itt (□, mean of triplicate experiments; bars, S.D. values in D and E) at different mixing times were fitted to the theoretical equation (Equation 2 in Ref. 35) (solid line) (see “Experimental Procedures”). F, CypA-catalyzed exchange rates (s−1; mean ± S.D.) that have been quantified using the procedure described above.
DISCUSSION
The JFH-1 (genotype 2a) strain, isolated from a Japanese patient with a fulminant hepatitis thus far is the only HCV strain that supports virus production in cell culture (59). In this study, we report the molecular characterization of its NS5A domain 3. The migration behavior of NS5A-D3 in SDS-PAGE, its elution profile in gel filtration at higher than expected molecular weight (see Fig. 2), the CD spectrum devoid of typical secondary structure element signature (see Fig. 5), and the limited dispersion of the amide proton NMR resonances (see Fig. 3A) all classify this protein as an intrinsically unstructured protein.
The amino acid sequences of NS5A-D3 of JFH-1 (2a) and Con1 (1b) have only 45% identity, and genotype 2a is characterized by an insertion of 20 amino acid residues (Asp431–Ser450) (see Fig. 1). The analyses of both JFH-1 and Con1 NS5A-D3 sequences with disorder predictors (53, 55) combined with detailed analyses of experimental Cα and Cβ 13C chemical shifts with the SSP software (56) led to the identification of two regions in each NS5A-D3 that have an intrinsic propensity to adopt an α-helical conformation in an aqueous environment (see Fig. 4). The existence of this small α-helical structural element around the N-terminal Glu366-Ala367-Leu368 tripeptide in the JFH-1 NS5A-D3 (Fig. 3B) is more pronounced than in the Con1 HCV strain, where 13C chemical shift values in the 369–379 region had merely pointed to a nascent helical tendency (39).
The comparative studies of NS5A-D3 from JFH-1 and Con1 by NMR spectroscopy in the presence of 50% TFE allowed us to further explore the putative structural conformations that these domains tend to adopt in aqueous media. All of the experimental data collected (i.e. the 1H,15N proton amide and 13Cα, 13Cβ, 13CO chemical shifts, the 3JHN-Hαcoupling, and finally the short sequential (i, i + 1) and medium range (i, i + 2), (i, i + 3), (i,i + 4) NOE contacts) show that in 50% TFE, the α-helical tendency observed mainly in the NS5A-D3 JFH-1 domain extends into a full-length helix (see Figs. 6 and 7). Predicted by secondary structure predictor programs to be helical in all genotypes (see supplemental Fig. 1), it is now also experimentally visible in Con1 NS5A-D3 when 50% TFE is used. The NMR-derived structural model for both NS5A-D3 from JFH-1 and Con1 HCV strains show two α-helices (see Figs. 7 and 8) whose lengths fit well with the helical content that was estimated by CD spectroscopy in 50% TFE. The α1 helix corresponds to residues Ser360–Gly377 in NS5A-D3 (JFH-1) and to residues Glu365–Gly381 in NS5A-D3 (Con1) and is well defined in both NMR models. The shorter α2 helix is located at the extreme C terminus of the domain but contains a few residues from the purification tag. Hence, we cannot exclude the possibility that the purification tag may influence the conformation of the C terminus of NS5A-D3, even if disorder predictions indeed suggested that the C terminus should be ordered (see Fig. 4).
Regions in intrinsically disordered proteins that have residual structure in aqueous solution may undergo disorder-to-order transition upon interaction with relevant biological partner(s) and have therefore been described as molecular recognition elements (60). The α1 helix in NS5A-D3, identified in this study, displays a pronounced asymmetrical distribution of the hydrophobic residues, rendering it amphipathic (see Fig. 8). Moreover, all of the hydrophobic residues in α1, except for Ile373 in JFH-1, are strictly conserved or have the same physicochemical properties among all reference sequences of the different HCV genotypes (see supplemental Fig. 1), strongly suggesting an important structural and/or functional role of this helix formation for some step(s) in the viral replication cycle. The amphipathic nature of α1 in NS5A-D3 may constitute a crucial feature for the proper establishment of an interaction with a biological partner. Numerous protein-protein interactions have been reported for NS5A, including viral (61–65) or host cell proteins (reviewed in Ref. 66). The unfolded nature of D2 and D3 could contribute to their hublike character for protein interactions with high specificities and low affinities (67). Potential interaction candidates for the helices in NS5A-D3 are the host cell apolipoprotein E (ApoE) and the HCV capsid protein (Core). ApoE has been reported to interact with NS5A and to be required for the production of infectious particles (68, 69). The ApoE-NS5A interaction appeared to be strongly affected by deletion of NS5A-D3 (68). ApoE is reported to interact with NS5A via its C-terminal α-helix domain (residues 205–280) (70), which corresponds to the lipid- or lipoprotein-binding region and is equally predicted to form an amphipathic α-helix (71). ApoE hence constitutes a possible candidate to interact with the amphipathic α1 helix in NS5A-D3. However, when we tested the interaction, by NMR, between both NS5A-D3 (JFH-1) and NS5A-D3 (Con1) and ApoE3 N-terminal domain (72) or even full-length monomeric mutant ApoE3-J5 (73), we did not observe any interaction (data not shown). There are several possibilities to explain our results: (i) domain 3 is required but not sufficient to promote the interaction of NS5A with ApoE; (ii) the mutations in the monomeric ApoE3-J5 mutant interfere with the NS5A-D3/ApoE interaction; (iii) a specific conformation of ApoE is required for interaction with NS5A; or (iv) another cofactor (protein, lipids, etc.) is mandatory to the proper establishment of the NS5A-D3/ApoE interaction that has, so far, only been observed in HCV-infected cells or in HCV particles. The other candidate, the HCV Core protein, is crucial for the production of infectious viruses (74, 75), a process for which domain 3 of NS5A is also a key element (26, 38). Several studies have pointed out NS5A-D3 as being essential for the NS5A/Core interaction (26, 64). However, both the membrane anchoring and the oligomerization properties of Core as well as its highly basic nature (76) render the investigation of the NS5A-D3/Core interaction very challenging and have prevented so far in vitro analyses by NMR.
Because mutations that confer resistance to cyclophilin inhibitors not only map to the D2 domain but equally to NS5A-D3 (19, 21), we investigated by NMR spectroscopy a possible direct interaction between this domain and CypA. For two different genotypes, we found a direct interaction between NS5A-D3 and CypA via residues of the PPIase active site that coincides with the CsA binding pocket. Residues of NS5A-D3 that are involved in the interaction with CypA reside mainly in the second half of D3 (Fig. 1) that is located just N-terminal of the α2 helix. Interestingly, numerous residues of the 20-residue insertion in JFH-1 are also involved in CypA interaction. Disappearance of the minor cis-related cross-peaks in the HSQC spectra of NS5A-D3 (see Fig. 1) suggests that especially the cis prolines are targeted. Using 1H,15N heteronuclear z-exchange spectroscopy (47, 48), we show that CypA has in vitro PPIase activity toward NS5A-D3 (see Fig. 9). However, in contrast to what we observed previously with NS5A-D2 (35), CypA exhibits a certain degree of enzymatic specificity toward given peptidyl-prolyl bonds, with an important in vitro PPIase activity toward the peptide bond preceding the Pro446 that is located in the JFH-1 insertion. The exchange rates (kexch) measured for the NS5A-D3 residues are in the 1–30 s−1 range and are similar to the ones measured toward NS5A-D2 (14–61 s−1) (35) and also toward the Gly221-Pro222 HIV capsid bond (15 s−1).
Several research teams have shown that CypA is a host factor that plays a key role in the HCV replication process (17–20). Inhibitors of CypA, such as CsA (15, 77) or non-immunosuppressive analogues (8, 10–12), thereby exert anti-HCV activity. The D3 domain of NS5A, despite its minor contribution for RNA replication (27, 78, 79) but major role for assembly (26, 38) is a site for mutations conferring resistance against CsA or even Debio-025, a structural analog. The mutations have been identified at the extreme C terminus of the domain (i.e. V448A in a genotype 1b subgenomic replicon (21) and V464A in a JFH-1 (2a) subgenome (19)). Interestingly, in the latter case, the mutation slows down proteolytic cleavage at the NS5A/B site, suggesting an indirect relationship between NS5A-D3 and CypA. Our results indicate a direct interaction of CypA with the C-terminal region of NS5A-D3, reinforcing the idea that cyclophilins might engage in multiple functional interactions with the viral proteins, most notably with NS5A.
In conclusion, we report here a comparative structure-function study between the NS5A-D3 proteins from two different HCV strains. Importantly, although intrinsically unfolded, NS5A-D3 exhibits localized helical propensity, which might be crucial for molecular interactions with host or other viral proteins. Our results also point out a direct molecular relationship between NS5A-D3 and the host chaperone CypA. Because the binding and PPIase activity of CypA are tightly linked, additional experiments are required to determine which one operates in vivo during the HCV replication cycle.
Supplementary Material
Acknowledgments
We gratefully acknowledge RD-Biotech (Besançon, France) for the cloning and the initial expression and purification tests for NS5A-D3 and Jennifer Molle for technical assistance. CD experiments were performed on the platform “Production et Analyse de Protéines” of the IFR 128 BioSciences Gerland-Lyon Sud. We gratefully acknowledge Prof. Jianjun Wang and Dr. Jianglei Chen (Wayne State University, Detroit, MI) for providing the purified ApoE3-J5 protein.
This work was supported by the French Centre National de la Recherche Scientifique and Universities of Lille and Lyon, and grants from the French National Agency for Research on AIDS and Viral Hepatitis and from the European Commission (VIRGIL Network of Excellence on Antiviral Drug Resistance). The NMR facility used in this study was funded by the Région Nord-Pas de Calais (France), CNRS, the Universities of Lille 1 and Lille 2, and the Institut Pasteur de Lille.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2 and Figs. 1–4.
- HCV
- hepatitis C virus
- aa
- amino acid(s)
- CsA
- cyclosporin A
- CypA
- CypB, and Cyp40, cyclophilin A, B, and 40, respectively
- HSQC
- heteronuclear single quantum correlation
- NS5A-D3 (JFH-1)
- recombinant protein representing aa 355–464 of NS5A from JFH1 HCV strain with an N-terminal methionine and a C-terminal LQHHHHHH extension
- NS5A-D3 (Con1)
- recombinant protein representing aa 359–445 of NS5A from Con1 HCV strain with an N-terminal methionine and a C-terminal LQHHHHHH extension
- PPIase
- peptidyl-prolyl cis/trans-isomerase
- TFE
- 2,2,2-trifluoroethanol
- CSI
- chemical shift index.
REFERENCES
- 1. National Institutes of Health (2002) Hepatology 36, Suppl. 1, S2–S20 [Google Scholar]
- 2. Appel N., Schaller T., Penin F., Bartenschlager R. (2006) J. Biol. Chem. 281, 9833–9836 [DOI] [PubMed] [Google Scholar]
- 3. Moradpour D., Penin F., Rice C. M. (2007) Nat. Rev. Microbiol. 5, 453–463 [DOI] [PubMed] [Google Scholar]
- 4. Barik S. (2006) Cell Mol. Life Sci. 63, 2889–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fischer G., Wittmann-Liebold B., Lang K., Kiefhaber T., Schmid F. X. (1989) Nature 337, 476–478 [DOI] [PubMed] [Google Scholar]
- 6. Handschumacher R. E., Harding M. W., Rice J., Drugge R. J., Speicher D. W. (1984) Science 226, 544–547 [DOI] [PubMed] [Google Scholar]
- 7. Nakagawa M., Sakamoto N., Tanabe Y., Koyama T., Itsui Y., Takeda Y., Chen C. H., Kakinuma S., Oooka S., Maekawa S., Enomoto N., Watanabe M. (2005) Gastroenterology 129, 1031–1041 [DOI] [PubMed] [Google Scholar]
- 8. Coelmont L., Kaptein S., Paeshuyse J., Vliegen I., Dumont J. M., Vuagniaux G., Neyts J. (2009) Antimicrob. Agents Chemother. 53, 967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Landrieu I., Hanoulle X., Bonachera F., Hamel A., Sibille N., Yin Y., Wieruszeski J. M., Horvath D., Wei Q., Vuagniaux G., Lippens G. (2010) Biochemistry 49, 4679–4686 [DOI] [PubMed] [Google Scholar]
- 10. Paeshuyse J., Kaul A., De Clercq E., Rosenwirth B., Dumont J. M., Scalfaro P., Bartenschlager R., Neyts J. (2006) Hepatology 43, 761–770 [DOI] [PubMed] [Google Scholar]
- 11. Ma S., Boerner J. E., TiongYip C., Weidmann B., Ryder N. S., Cooreman M. P., Lin K. (2006) Antimicrob. Agents Chemother. 50, 2976–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hopkins S., Scorneaux B., Huang Z., Murray M. G., Wring S., Smitley C., Harris R., Erdmann F., Fischer G., Ribeill Y. (2010) Antimicrob. Agents Chemother. 54, 660–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaither L. A., Borawski J., Anderson L. J., Balabanis K. A., Devay P., Joberty G., Rau C., Schirle M., Bouwmeester T., Mickanin C., Zhao S., Vickers C., Lee L., Deng G., Baryza J., Fujimoto R. A., Lin K., Compton T., Wiedmann B. (2010) Virology 397, 43–55 [DOI] [PubMed] [Google Scholar]
- 14. Goto K., Watashi K., Inoue D., Hijikata M., Shimotohno K. (2009) Cancer Sci. 100, 1943–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishii N., Watashi K., Hishiki T., Goto K., Inoue D., Hijikata M., Wakita T., Kato N., Shimotohno K. (2006) J. Virol. 80, 4510–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Watashi K., Ishii N., Hijikata M., Inoue D., Murata T., Miyanari Y., Shimotohno K. (2005) Mol. Cell. 19, 111–122 [DOI] [PubMed] [Google Scholar]
- 17. Yang F., Robotham J. M., Nelson H. B., Irsigler A., Kenworthy R., Tang H. (2008) J. Virol. 82, 5269–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chatterji U., Bobardt M., Selvarajah S., Yang F., Tang H., Sakamoto N., Vuagniaux G., Parkinson T., Gallay P. (2009) J. Biol. Chem. 284, 16998–17005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaul A., Stauffer S., Berger C., Pertel T., Schmitt J., Kallis S., Zayas M., Lopez M. Z., Lohmann V., Luban J., Bartenschlager R. (2009) PLoS Pathog. 5, e1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu Z., Yang F., Robotham J. M., Tang H. (2009) J. Virol. 83, 6554–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernandes F., Poole D. S., Hoover S., Middleton R., Andrei A. C., Gerstner J., Striker R. (2007) Hepatology 46, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 22. Puyang X., Poulin D. L., Mathy J. E., Anderson L. J., Ma S., Fang Z., Zhu S., Lin K., Fujimoto R., Compton T., Wiedmann B. (2010) Antimicrob. Agents Chemother. 54, 1981–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robida J. M., Nelson H. B., Liu Z., Tang H. (2007) J. Virol. 81, 5829–5840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chatterji U., Bobardt M. D., Lim P., Gallay P. A. (2010) J. Gen. Virol. 91, 1189–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ciesek S., Steinmann E., Wedemeyer H., Manns M. P., Neyts J., Tautz N., Madan V., Bartenschlager R., von Hahn T., Pietschmann T. (2009) Hepatology 50, 1638–1645 [DOI] [PubMed] [Google Scholar]
- 26. Appel N., Zayas M., Miller S., Krijnse-Locker J., Schaller T., Friebe P., Kallis S., Engel U., Bartenschlager R. (2008) PLoS Pathog. 4, e1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tellinghuisen T. L., Foss K. L., Treadaway J. C., Rice C. M. (2008) J. Virol. 82, 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang L., Hwang J., Sharma S. D., Hargittai M. R., Chen Y., Arnold J. J., Raney K. D., Cameron C. E. (2005) J. Biol. Chem. 280, 36417–36428 [DOI] [PubMed] [Google Scholar]
- 29. Foster T. L., Belyaeva T., Stonehouse N. J., Pearson A. R., Harris M. (2010) J. Virol. 84, 9267–9277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Penin F., Brass V., Appel N., Ramboarina S., Montserret R., Ficheux D., Blum H. E., Bartenschlager R., Moradpour D. (2004) J. Biol. Chem. 279, 40835–40843 [DOI] [PubMed] [Google Scholar]
- 31. Tellinghuisen T. L., Marcotrigiano J., Rice C. M. (2005) Nature 435, 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Love R. A., Brodsky O., Hickey M. J., Wells P. A., Cronin C. N. (2009) J. Virol. 83, 4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gao M., Nettles R. E., Belema M., Snyder L. B., Nguyen V. N., Fridell R. A., Serrano-Wu M. H., Langley D. R., Sun J. H., O'Boyle D. R., 2nd, Lemm J. A., Wang C., Knipe J. O., Chien C., Colonno R. J., Grasela D. M., Meanwell N. A., Hamann L. G. (2010) Nature 465, 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang F., Robotham J. M., Grise H., Frausto S., Madan V., Zayas M., Bartenschlager R., Robinson M., Greenstein A. E., Nag A., Logan T. M., Bienkiewicz E., Tang H. (2010) PLoS Pathog. 6, e1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hanoulle X., Badillo A., Wieruszeski J. M., Verdegem D., Landrieu I., Bartenschlager R., Penin F., Lippens G. (2009) J. Biol. Chem. 284, 13589–13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang Y., Ye H., Kang C. B., Yoon H. S. (2007) Biochemistry 46, 11550–11558 [DOI] [PubMed] [Google Scholar]
- 37. Fernandes F., Ansari I. U., Striker R. (2010) PLoS One 5, e9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tellinghuisen T. L., Foss K. L., Treadaway J. (2008) PLoS Pathog. 4, e1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hanoulle X., Verdegem D., Badillo A., Wieruszeski J. M., Penin F., Lippens G. (2009) Biochem. Biophys. Res. Commun. 381, 634–638 [DOI] [PubMed] [Google Scholar]
- 40. Combet C., Blanchet C., Geourjon C., Deléage G. (2000) Trends Biochem. Sci. 25, 147–150 [DOI] [PubMed] [Google Scholar]
- 41. Combet C., Garnier N., Charavay C., Grando D., Crisan D., Lopez J., Dehne-Garcia A., Geourjon C., Bettler E., Hulo C., Le Mercier P., Bartenschlager R., Diepolder H., Moradpour D., Pawlotsky J. M., Rice C. M., Trépo C., Penin F., Deléage G. (2007) Nucleic Acids Res. 35, D363–D366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y. H., Yang J. T., Chau K. H. (1974) Biochemistry 13, 3350–3359 [DOI] [PubMed] [Google Scholar]
- 43. Verdegem D., Dijkstra K., Hanoulle X., Lippens G. (2008) J. Biomol. NMR 42, 11–21 [DOI] [PubMed] [Google Scholar]
- 44. Wang A. C., Bax A. (1995) J. Am. Chem. Soc. 117, 1810–1813 [Google Scholar]
- 45. Vuister G. W., Bax A. (1993) J. Am. Chem. Soc. 115, 7772–7777 [Google Scholar]
- 46. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 47. Farrow N. A., Zhang O., Forman-Kay J. D., Kay L. E. (1994) J. Biomol. NMR 4, 727–734 [DOI] [PubMed] [Google Scholar]
- 48. Kern D., Eisenmesser E. Z., Wolf-Watz M. (2005) Methods Enzymol. 394, 507–524 [DOI] [PubMed] [Google Scholar]
- 49. Huang L., Sineva E. V., Hargittai M. R., Sharma S. D., Suthar M., Raney K. D., Cameron C. E. (2004) Protein Expr. Purif. 37, 144–153 [DOI] [PubMed] [Google Scholar]
- 50. Receveur-Bréchot V., Bourhis J. M., Uversky V. N., Canard B., Longhi S. (2006) Proteins 62, 24–45 [DOI] [PubMed] [Google Scholar]
- 51. Tompa P. (2002) Trends Biochem. Sci. 27, 527–533 [DOI] [PubMed] [Google Scholar]
- 52. Wishart D. S., Sykes B. D. (1994) J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 53. Li X., Romero P., Rani M., Dunker A. K., Obradovic Z. (1999) Genome Inform. Ser. Workshop Genome Inform. 10, 30–40 [PubMed] [Google Scholar]
- 54. Romero P., Obradovic Z., Li X., Garner E. C., Brown C. J., Dunker A. K. (2001) Proteins 42, 38–48 [DOI] [PubMed] [Google Scholar]
- 55. Ishida T., Kinoshita K. (2008) Bioinformatics 24, 1344–1348 [DOI] [PubMed] [Google Scholar]
- 56. Marsh J. A., Singh V. K., Jia Z., Forman-Kay J. D. (2006) Protein. Sci. 15, 2795–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cammers-Goodwin A., Allen T. J., Oslick S. L., McClure K. F., Lee J. H., Kemp D. S. (1996) J. Am. Chem. Soc. 118, 3082–3090 [Google Scholar]
- 58. Jasanoff A., Fersht A. R. (1994) Biochemistry 33, 2129–2135 [DOI] [PubMed] [Google Scholar]
- 59. Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H. G., Mizokami M., Bartenschlager R., Liang T. J. (2005) Nat. Med. 11, 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vacic V., Oldfield C. J., Mohan A., Radivojac P., Cortese M. S., Uversky V. N., Dunker A. K. (2007) J. Proteome. Res. 6, 2351–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dimitrova M., Imbert I., Kieny M. P., Schuster C. (2003) J. Virol. 77, 5401–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lai C. K., Jeng K. S., Machida K., Lai M. M. (2008) J. Virol. 82, 8838–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin C., Wu J. W., Hsiao K., Su M. S. (1997) J. Virol. 71, 6465–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Masaki T., Suzuki R., Murakami K., Aizaki H., Ishii K., Murayama A., Date T., Matsuura Y., Miyamura T., Wakita T., Suzuki T. (2008) J. Virol. 82, 7964–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shirota Y., Luo H., Qin W., Kaneko S., Yamashita T., Kobayashi K., Murakami S. (2002) J. Biol. Chem. 277, 11149–11155 [DOI] [PubMed] [Google Scholar]
- 66. de Chassey B., Navratil V., Tafforeau L., Hiet M. S., Aublin-Gex A., Agaugué S., Meiffren G., Pradezynski F., Faria B. F., Chantier T., Le Breton M., Pellet J., Davoust N., Mangeot P. E., Chaboud A., Penin F., Jacob Y., Vidalain P. O., Vidal M., André P., Rabourdin-Combe C., Lotteau V. (2008) Mol. Syst. Biol. 4, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Uversky V. N., Oldfield C. J., Dunker A. K. (2008) Annu. Rev. Biophys. 37, 215–246 [DOI] [PubMed] [Google Scholar]
- 68. Benga W. J., Krieger S. E., Dimitrova M., Zeisel M. B., Parnot M., Lupberger J., Hildt E., Luo G., McLauchlan J., Baumert T. F., Schuster C. (2010) Hepatology 51, 43–53 [DOI] [PubMed] [Google Scholar]
- 69. Jiang J., Luo G. (2009) J. Virol. 83, 12680–12691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cun W., Jiang J., Luo G. (2010) J. Virol. 84, 11532–11541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hatters D. M., Peters-Libeu C. A., Weisgraber K. H. (2006) Trends Biochem. Sci. 31, 445–454 [DOI] [PubMed] [Google Scholar]
- 72. Monteilhet C., Lachacinski N., Aggerbeck L. P. (1993) Gene 125, 223–228 [DOI] [PubMed] [Google Scholar]
- 73. Zhang Y., Vasudevan S., Sojitrawala R., Zhao W., Cui C., Xu C., Fan D., Newhouse Y., Balestra R., Jerome W. G., Weisgraber K., Li Q., Wang J. (2007) Biochemistry 46, 10722–10732 [DOI] [PubMed] [Google Scholar]
- 74. Miyanari Y., Atsuzawa K., Usuda N., Watashi K., Hishiki T., Zayas M., Bartenschlager R., Wakita T., Hijikata M., Shimotohno K. (2007) Nat. Cell. Biol. 9, 1089–1097 [DOI] [PubMed] [Google Scholar]
- 75. Shavinskaya A., Boulant S., Penin F., McLauchlan J., Bartenschlager R. (2007) J. Biol. Chem. 282, 37158–37169 [DOI] [PubMed] [Google Scholar]
- 76. Boulant S., Vanbelle C., Ebel C., Penin F., Lavergne J. P. (2005) J. Virol. 79, 11353–11365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nakagawa M., Sakamoto N., Enomoto N., Tanabe Y., Kanazawa N., Koyama T., Kurosaki M., Maekawa S., Yamashiro T., Chen C. H., Itsui Y., Kakinuma S., Watanabe M. (2004) Biochem. Biophys. Res. Commun. 313, 42–47 [DOI] [PubMed] [Google Scholar]
- 78. Appel N., Pietschmann T., Bartenschlager R. (2005) J. Virol. 79, 3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Moradpour D., Evans M. J., Gosert R., Yuan Z., Blum H. E., Goff S. P., Lindenbach B. D., Rice C. M. (2004) J. Virol. 78, 7400–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kyte J., Doolittle R. F. (1982) J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.