

Abstract
Helix pomatia agglutinin (HPA), the lectin from the albumen gland of the Roman snail, has been used in histochemical studies relating glycosylation changes to the metastatic potential of solid tumors. To facilitate the use of HPA in a clinical (diagnostic) setting, detailed analysis of the lectin, including cloning and recombinant production of HPA, is required. A combination of isoelectric focusing, amino acid sequence analysis, and cloning revealed two polypeptides in native HPA preparations (HPAI and HPAII), both consistent with GalNAc-binding lectins of the H-type family. Pairwise sequence alignment showed that HPAI and HPAII share 54% sequence identity whereas molecular modeling using SWISS-MODEL suggests they are likely to adopt similar tertiary structure. The inherent heterogeneity of native HPA highlighted the need for production of functional recombinant protein; this was addressed by preparing His-thioredoxin-tagged fusion products in Escherichia coli Rosetta-gami B (DE3) cells. The recombinant lectins agglutinated human blood group A erythrocytes whereas their oligosaccharide specificity, evaluated using glycan microarrays, showed that they predominantly bind glycans with terminal α-GalNAc residues. Surface plasmon resonance with immobilized GalNAc-BSA confirmed that recombinant HPAI and HPAII bind strongly with this ligand (Kd = 0.60 nm and 2.00 nm, respectively) with a somewhat higher affinity to native HPA (Kd = 7.67 nm). Recombinant HPAII also bound the breast cancer cells of breast cancer tissue specimens in a manner similar to native lectin. The recombinant HPA described here shows important potential for future studies of cancer cell glycosylation and as a reagent for cancer prognostication.
Keywords: Breast Cancer, Glycoprotein Structure, Lectin, Oligosaccharide, Protein Sequence, Protein Structure
Introduction
Changes in the glycosylation of proteins are a feature of tumorigenesis and correlate with aggressive cancer cell behavior (1, 2). Aberrant, cancer-associated, glycosylation can be detected using carbohydrate-binding proteins, with the lectin from the Roman snail, Helix pomatia (HPA)3 having been shown to be particularly useful in this regard (3). Studies with HPA have shown a correlation between poor prognosis breast cancer that is metastatic to the axillary lymph nodes and lectin binding (4, 5). Further work has demonstrated that HPA binding is associated with aggressive colorectal (6, 7) and cervical (8) and gastric carcinoma (9). These observations led to the hypothesis that changes in glycosylation detected by HPA may be the same across a range of solid tumors (3). The utility of HPA for prognostication of solid tumors appears to lie in its ability to recognize the cancer-associated Tn-antigen (GalNAcα-O-Ser/Thr) and other, as yet undefined, epitopes (9). Tn-antigen expression occurs in a diverse range of neoplastic lesions and may, in part, be due to a loss of function of the Cosmc gene encoding a molecular chaperone required for T-synthase formation (10). This is associated with a reduction in synthesis of the core 1 disaccharide of O-glycans which are formed by the addition of Gal to the Tn-antigen to generate the T-antigen: Galβ1–3GalNAcα1-Ser/Thr (11). Unfortunately, despite the apparent utility of HPA for cancer prognostication, the lectin has yet to find a place in clinical decision making.
An initial attempt to solve the three-dimensional structure of HPA was reported in 2000 (12), but the limited resolution (3.6 Å) and lack of amino acid sequence prevented the structure from being solved (12). In 2006, HPA crystals defracting to 1.3 Å allowed the sequence and structure to be determined (13). This confirmed earlier findings (14–17) that described HPA as a hexamer formed by disulfide bonds linking dimers, which then form a trimer of dimers by noncovalent interactions. The crystal structure showed the carbohydrate binding site is situated at the interface between two monomers. The novelty of the structure of the binding pocket led to the assignment of a new lectin family, the H-type, named after H. pomatia. At this time, another polypeptide sequence with a 54% sequence identity to the lectin was deposited in the EMBL DNA sequence data base (accession number: AJ639657 (Uniprot accession number: Q575S3)) by Gerlach and Schmidt who described this polypeptide as H. pomatia agglutinin. However, because it only showed 54% sequence identity to HPA, Sanchez et al. (13) suggested that this new protein may have derived from a different snail species. A further possibility was that this protein represented a second lectin in the HPA preparation.
Our initial analysis of HPA suggested that native HPA was more complex than a single polypeptide, so we set out to identify the composition of the material. In addition, we recognized that further analysis of the sequence and structure of HPA would provide important insights into the carbohydrate-binding properties of the lectin and that this information could be used to prepare a recombinant form of the polypeptide, the availability of which might lead to the development of new tools for cancer diagnosis and, potentially, therapy.
EXPERIMENTAL PROCEDURES
Preparation of HPA
H. pomatia snails were obtained in May 2005 with the kind assistance of Dr. Anthony Leathem, Royal Free and University College Medical School, London, from a wood in Oxfordshire, England. Snails were maintained in the Cancer Glycobiology Laboratory at the University of Westminster in a screened terrarium, in artificial laboratory daylight, at 20 °C and were fed lettuce and water ad libitum. The snails were anesthetized and deshelled, and the albumen gland was dissected. HPA was purified from the albumen gland using the method of Vretblad et al. (18). In brief, albumen glands from 10 snails were minced in 10 ml of 10 mm sodium phosphate buffer, pH 7.2, containing 150 mm NaCl (PBS) per gram of albumen gland, and the lectin HPA was purified on a 1-ml N-acetylgalactosamine (GalNAc)-agarose affinity chromatography column eluted with 68 mm N-acetylglucosamine (GlcNAc).
SDS-PAGE
SDS-PAGE was performed using a Tris-Tricine-SDS electrophoresis buffer system (19) with step gradients of 15, 10, and 5% polyacrylamide; or with the Tris-glycine-SDS system for analysis of rHPAI-Trx/rHPAII-Trx which was performed according to Ref. 20. Samples were boiled for 5 min or treated with 50 mm dithiothreitol (DTT) where appropriate. After electrophoresis, the proteins were visualized by staining with 0.025% (w/v) Coomassie Brilliant Blue R-250.
Two-dimensional Electrophoresis
To separate the individual subunits of HPA a combination of isoelectric focusing and SDS-PAGE was used (21). 1 mg of HPA lectin was solubilized in 100 μl of a solution containing 8 m urea and 400 mm NH4HCO3, pH 8.1. An equal volume of 50 mm DTT was added and the mixture incubated for 15 min at 20 °C followed by addition of 9 μl of 100 mm iodoacetamide with a further incubation for 15 min. The protein solution was separated on a 7-cm linear immobilized pH gradient strip at 300 V for 30 min, 600 V for 45 min, and 3500 V for 2.45 h, at 20 °C. After the isoelectric focusing step the strip was embedded on the top of an SDS-polyacrylamide gel using molten agarose (1% w/v in Tris-Tricine-SDS cathode buffer) and subjected to electrophoresis as described previously.
MS/MS
Mass spectrometry of the protein spots from the 2-DE was performed using an Applied Biosystems 4700 Proteomics Analyzer (MALDI-TOF-TOF) by commercial arrangement with Dr. J. Thomas, School of Biology, University of York, UK. Protein spots were excised and digested in-gel with trypsin, and a mixture of the resulting peptides and α-cyano-4-hydroxycinnamic acid (3.6 mg/ml) in 10 mm ammonium citrate was spotted onto a MALDI plate and allowed to air dry. MS measurements were collected using a 4700 Proteomics Analyzer Discovery System (Applied Biosystems).
mRNA Extraction and Cloning of HPA Genes
RNA was isolated from the freshly excised albumen gland of H. pomatia using a combination of TRIzol Reagent (22) and PolyATtract mRNA Isolation System (Promega); reverse transcription of RNA to cDNA was performed using an Omniscript reverse transcriptase kit (Qiagen). RT-PCR using the cDNA prepared above and degenerate oligonucleotide primers designed following analysis of the protein sequences determined by MS/MS (see above) was used to amplify the HPA gene(s), and the amplicons were then ligated into the pGEM-T vector (Promega), transformed into E. coli DH5α T1 cells, and recombinants were selected using isopropyl 1-thio-β-d-galactopyranoside (100 mm) and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (20 μg/ml) selection. Putative positive colonies were evaluated using PCR and sequenced at the Advanced Biotechnology Centre, Imperial College, London. To obtain the full-length genes for the HPA proteins the positive clones were extended using dGTP and terminal transferase, and the product was amplified by PCR using the Phusion system (New England Biolabs) with ∼10 ng of template DNA, cloned into pGEM-T, and resequenced.
Production of Recombinant HPAI and HPAII Proteins
Recombinant HPAI and HPAII proteins were prepared as both histidine-tagged (rHPAI-His/rHPAII-His) and thioredoxin protein-tagged (rHPAI-Trx/rHPAII-Trx) fusion proteins using the pET-32a vector system (supplemental data 1), expressed in E. coli Rosetta-gami B (DE3) pLysS-competent cells (Novagen), supplemented with 100 μg/ml ampicillin, 34 μg/ml chloramphenicol, 15 μg/ml kanamycin, and 12.5 μg/ml tetracycline. Fresh grown colonies containing the recombinant plasmids were used to inoculate 50 ml of LB medium containing the appropriate antibiotics and grown overnight at 37 °C with shaking at 250 rpm, and 500 ml of prewarmed LB medium, containing the four antibiotics as before, was then inoculated with 25 ml of the overnight culture and grown at 37 °C until the A600 nm reached 0.5 after which the culture was cooled on ice for 30 min. Isopropyl 1-thio-β-d-galactopyranoside was then added to a final concentration of 1 mm, and the culture was grown at 18 °C for 18 h with shaking at 250 rpm. Cells were harvested by centrifugation and resuspended in 50 ml of 20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 10 mm imidazole, 0.1% v/v Triton X-100, 10 μg/ml lysozyme, and 0.1 mm phenylmethanesulfonyl fluoride (PMSF). The cell suspension was incubated on ice for 30 min, and the cells were lysed by sonication on ice (6 × 30 s (at 20 kHz at 40% power)) using a MS73 Status 200.
Recombinant proteins were purified in two steps, using nickel-agarose (GE Healthcare) and GalNAc-agarose columns (Sigma-Aldrich). Chromatography was carried out at 20 °C with buffer flow rates set to 1 ml/min. For nickel affinity chromatography, the E. coli cell lysate was loaded onto the column in 20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 20 mm imidazole, washed with 10 column volumes of the same buffer, and the recombinant protein was eluted with 5 column volumes of the same buffer containing 500 mm imidazole. For the second purification step, a GalNAc-agarose affinity column was loaded with the fractions containing the recombinant protein from the nickel chromatography step, unbound material was washed from the column with 5 column volumes of PBS, and recombinant protein was eluted from the column using 5 column volumes of PBS containing 250 mm GlcNAc. The purified proteins were dialyzed overnight against PBS at 4 °C, concentrated by centrifugation using the Centricon system using 5-kDa molecular mass cut-off (Millipore) and evaluated using SDS-PAGE (see above).
Agglutination Assays
Human erythrocytes with blood groups A, B, and O (National Blood Service, London, UK) were used as a 10% (v/v) suspension in PBS after washing three times by centrifuging 5 ml of blood at 1,500 × g for 5 min at 4 °C. Agglutination assays were conducted in 96-well round bottom plates (preblocked overnight at 4 °C with 1% (w/v) BSA in PBS). 5-μl aliquots of native HPA, rHPAI-Trx, or rHPAII-Trx solutions were mixed with 5 μl of PBS and 10 μl of 10% erythrocyte suspension in each test well. The 96-well plate was rocked at 20 °C for 1 h, and the extent of the agglutination was assessed using an inverted microscope and scored from (−) no agglutination to (+++) complete agglutination.
Analysis of Lectin Binding Using Surface Plasmon Resonance (SPR)
SPR analysis was carried out using a BIAcore 2000 using a CM5 biosensor chip (GE Healthcare). Ligands were immobilized onto the surface of the chip using an amine coupling reaction, as described in the instructions provided by the manufacturer (GE Healthcare), creating the following linkages: (i) reference cell (no ligand coupled); (ii) BSA; (iii) GalNAc linked to BSA via a 14-carbon linker spacer (Dextra Laboratories), (iv) GlcNAc linked to BSA via a 14-carbon linker spacer (Dextra Laboratories). In each case a solution of ∼500 μm ligand was injected over the sensor chip surface to give ∼3000 response units of immobilized ligand. Unbound sites on the chip were blocked with 1.0 m ethanolamine, pH 8.5, and the chip was washed with binding buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl) prior to use. Different concentrations of native HPA and rHPAI-Trx or rHPAII-Trx (0–35 nm) were injected over the chip surface at 20 °C in binding buffer at a flow rate of 20 μl/min. The injection time was 60 s, followed by a 3-min dissociation. The surface was regenerated by injection of 10 mm glycine, pH 2.2, after each run. BIAevaluation software, version 4.1 (GE Healthcare), was used for analysis of the SPR data.
Glycan Array Analysis
Native HPA, rHPAI-Trx, and r-HPAII-Trx were labeled with biotin (EZ-link biotinylation kit; Piercenet, Thermo Scientific) and detected using either Alexa Fluor 488 TFP or Cy5 reagent on the printed glycan array version 3 as described by Blixt et al. (23). Analysis was undertaken using the Core H of the Consortium for Functional Glycomics. In each case, 200 μg/ml lectin was applied to the array.
Histochemistry Using HPAI
Native HPA (10 μg/ml) and rHPAI-His (20 μg/ml) were prepared as above, labeled with biotin, and used to stain serial sections of paraffin wax-embedded breast cancer tissue (AccuMax, ref A202III; ISU Abxis, Seoul, Korea) as described by Brooks and Wilkinson (24) using Harris hematoxylin as the nuclear counterstain. Tissues samples were collected from patients with informed consent and according to national laws and local regulations.
Bioinformatics Analyses
BlastP was used to identify unknown amino acid sequences. ClustalW was used for multiple sequence alignment of DNA and protein sequences. HPAI structure predication was carried out using SWISS-MODEL in automated mode, and PyMOL X11Hybrid (DeLano Scientific LLC) was used for the visualization of the model structures; DaliLite was used for the structural alignment of HPAI and HPAII.
RESULTS
HPA Heterogeneity
We set out to analyze HPA because earlier studies hinted that the material may comprise a mixture of proteins (18, 25). HPA was purified from freshly dissected snail albumen glands using GalNAc-agarose resin, and the resultant preparation was analyzed using a Tris-Tricine SDS-PAGE system (Fig. 1A). In accordance with previous reports, a polypeptide of 79 kDa was observed (15, 18, 26). When the lectin was heated at 100 °C for 5 min the appearance of a band of 26 kDa consistent with the molecular mass of two monomers (each 13 kDa) was detected. Treatment of the preparation with DTT was expected to generate a single band consistent with the monomer on SDS-PAGE analysis. Somewhat surprisingly, two bands of ∼13 kDa as well as a band of 26 kDa were apparent. Initially, we thought that the two bands might represent glycosylated variants of the same peptide. Glycopeptides, however, tend to resolve poorly on SDS-PAGE due to the minor differences in the molecular mass of the species present. As the peptides we observed in the native HPA preparation separated into two sharp bands on SDS-PAGE, this was the first piece of evidence suggesting that the peptides probably derived from different amino acid sequences, as a dimer of identical monomers should not yield two similar but rather two identical species.
FIGURE 1.

Analysis of HPA lectin purified from the albumen gland of the Roman snail, H. pomatia, highlighted the complexity of the preparation. A, separation using Tris-Tricine SDS-PAGE at 180 V for 35 min. The lanes indicate native HPA, HPA after denaturation for 5 min at 100 °C, and HPA after treatment with 0.2 m DTT for 15 min at 20 °C. 25 μg of protein was loaded into each well. The protein was visualized by staining the gel with Coomassie Brilliant Blue. B, two-dimensional separation of 150 μg of HPA preparation using a 3–10 pH linear immobilized pH gradient strip and SDS-PAGE as for A. The spots indicated by the arrows and identified by Coomassie Brilliant Blue staining were used for MS/MS analysis. C, amino acid sequence data for spots 3, 4, and 14 as indicated.
When the HPA preparation was analyzed by two-dimensional electrophoresis, >20 polypeptide species were detected, ranging from 8 to 13 kDa (Fig. 1B). A similar observation was made using HPA purchased from Sigma with some differences in the relative spot intensities (data not shown). Peptide mass fingerprint analysis was performed with peptides derived from the isoelectric focusing-separated polypeptides, but no significant hits from the GenBank/EBI data bank search were obtained. When MS/MS analysis was used, four peptide sequences were obtained from the analysis of species 3 and 4 and three from the analysis of species 14 (Fig. 1C and supplemental data 2).
Cloning of HPAI and HPAII Genes
The peptide GEIDCGSDSSWP elucidated from sequencing species 14 was used to design degenerate oligonucleotide primers to allow the cloning of the HPA gene which was named HPAI. The sequence of HPAI was found to be identical to a sequence in the EMBL DNA sequence data base (accession number CAG26659) deposited by Gerlach and Schmidt. It become apparent that peptides from species 3 and 4 (Fig. 1B) did not originate from this sequence and that these might represent a new protein. To investigate this possibility, degenerate oligonucleotide primers corresponding to the CGNDAG sequence were prepared and used to clone a gene for a second HPA protein known as HPAII. The sequence of HPAII is identical to that deposited in GenBank by Sanchez et al. (accession number DQ341310) (13).
The predicted molecular masses of HPAI and HPAII are 10,861 and 10,662 Da, respectively, and these corresponded broadly to the bands observed during SDS-PAGE analysis of the native protein (Fig. 1A). The slight differences in molecular mass are potentially attributable to post-translational modifications such as glycosylation. The isoelectric points for HPAI and HPAII are predicted to be 6.77 and 8.47, respectively; again, these were within the range of the isoelectric points observed for the HPA polypeptides that were obtained during the two-dimensional electrophoresis analysis (Fig. 1B).
Pairwise sequence alignment of HPAI and HPAII showed that the two sequences share 54% sequence identity (Fig. 2A). Homology models of HPAI (based on PDB 2CE6, native HPAII and 2CCV, HPAII with GalNAc soaked into the crystal) were generated using SWISS-MODEL and allowed the structural similarity between the two proteins to be assessed in silico (Fig. 2, B and C). The only apparent difference between the carbohydrate binding site of HPAI and HPAII is at position 24 with asparagine in HPAI and glycine in HPAII. This substitution would not be expected to interfere with the carbohydrate binding properties of the lectin because only the nitrogen of the main chain of the amino acid participates in hydrogen bonding with the O of the acetyl group of the carbohydrate. Analysis of the amino acid sequence for HPA-I indicated that, in contrast to HPA-II, it does not possess an N-linked glycosylation consensus sequence at Asn55.
FIGURE 2.

Pairwise sequence alignment and structure prediction of HPAI and HPAII. A, pairwise alignment of the amino acid sequences for HPAI and HPAII using ClustalW. Identical amino acids are indicated with an asterisk, amino acids that form the carbohydrate-binding site are shown in bold, and amino acids that form hydrogen bonds during trimer formation are shown in italics. The amino acid residues identified by MS/MS are underlined. B, ribbon representations of HPAI (blue) and HPAII (green) viewed from the top. GalNAc is shown in red. C, surface representation of the HPAI and HPAII GalNAc binding site. The binding pocket is formed at the interface of the two monomeric domains, shown in blue and green. GalNAc is shown in red.
Functional Analysis of HPAI and HPAII
HPAI and HPAII were successfully prepared as His and Trx fusion proteins (Fig. 3A). To check whether the addition of the Trx tag affected the structure of the protein, the migration of rHPAI-Trx/rHPAII-Trx was evaluated by separation on PAGE (Fig. 3B). In this analysis, both rHPAI-Trx and rHPAII-Trx were observed as trimeric proteins, held together with intermonomer disulfide bridges, that could be broken by heating to boiling for 5 min or by the addition of DTT.
FIGURE 3.

Preparation of recombinant Trx fusion proteins of HPA and analysis of agglutination of human erythrocytes of blood group A. A, data for rHPAI-Trx. Lane 1,cell pellet after induction of HPA expression. Lane 2, Ni affinity column flow through. Lane 3, elution of rHPAI-Trx from the nickel column by addition of 500 mm imidazole. Lane 4, elution of rHPAI-Trx from GalNAc-agarose column using 250 mm GlcNAc in PBS. In each case 10 μl of the chromatography fraction was prepared in sample buffer and loaded into the well, and the proteins were separated for 35 min at 180 V and visualized using Coomassie Brilliant Blue. The recombinant protein band is indicated by the black arrow. B, semi-native SDS-PAGE separation of rHPAI-Trx/rHPAII-Trx before (lane 1) and after treatment at 100 °C for 5 min (lane 2) or 50 mm DTT (lane 3). C, agglutination profile of human erythrocytes of blood group A using rHPAI-Trx, rHPAII-Trx, and native lectin. Agglutination was scored as full agglutination (+++), weak agglutination (++) or (+), and no agglutination (−). Inhibition of agglutination of red blood cells (blood group A) using native and rHPAI-Trx/rHPAII-Trx incubated in the presence of GalNAc and GlcNAc in a range of concentrations is shown.
The rHPAI-Trx and rHPAII-Trx fusion proteins agglutinated human erythrocytes of blood group A in a similar manner to native HPA (Fig. 3C). Concentrations of lectin <2.5 μg/ml were insufficient to result in agglutination, even after several hours of incubation, whereas at ≥12.5 μg/ml, agglutination was observed within seconds. GalNAc and GlcNAc competitively inhibited both lectin agglutination reactions when used at ≥25 mm (Fig. 3C). rHPAI-Trx and rHPAII-Trx did not agglutinate erythrocytes of blood group B or O (data not shown).
The oligosaccharide binding properties of the recombinant lectins were analyzed further on the printed glycan microarray of the Consortium for Functional Glycomics, which comprised 320 different immobilized glycan structures (Fig. 4 and supplemental data 3). In this system rHPAI-Trx and rHPAII-Trx recognized almost identical glycan residues and showed a preference for glycans containing terminal α-linked GalNAc; with GalNAcα1–3Gal > GalNAcα1–4Gal. rHPAI-Trx and rHPAII-Trx did not bind oligosaccharides containing sialic acid, but in all other respects the native and recombinant material showed the same glycan binding on the array. The native HPA preparation recognized the same oligosaccharides (although with different intensities) as have previously been reported by Sanchez et al. (13).
FIGURE 4.

Glycan microarray analysis of native HPA, rHPAI-Trx, and rHPAII-Trx as measured by fluorescence intensity. Results over 10,000 relative fluorescence units were taken as significant.
The affinity of interaction between the different lectins and GalNAc-BSA was analyzed by SPR using the BIAcore system. All three proteins (native HPA, rHPAI-Trx, and rHPAII-Trx) bound tightly to immobilized GalNAc in a concentration-dependent manner (Fig. 5A). Using the observed steady-state binding (response units at equilibrium (RUeq)), it was possible to estimate the dissociation constants (Kd) for the binding of each protein to GalNAc, which showed that all three lectins bind with approximately the same affinity (Fig. 5, B and C) with Kd values in the nanomolar range.
FIGURE 5.

SPR analysis of binding of native HPA, rHPAI-Trx, and rHPAII-Trx to GalNAc-BSA immobilized on a CM5 Biosensor chip surface using the BIAcore 2000 system. Approximately 3000 response units of ligand was immobilized on the sensor chip surface while the lectin concentration varied between 0 and 35 nm (as indicated). The flow rate was maintained at 20 μl/min, the injection time was typically 120 s followed by 3-min dissociation. A shows the binding of native HPA to immobilized GalNAc. B shows a plot of response units at equilibrium versus lectin concentration for the binding of HPAII to GalNAc, data which can, in turn, be used to determine the Kd value (C).
The binding of rHPAI-His to breast cancer tissue was evaluated using a breast cancer tissue array and compared with native HPA and showed that rHPAI-His binds to cancer tissue samples in a manner similar to that observed with the native lectin (Fig. 6).
FIGURE 6.
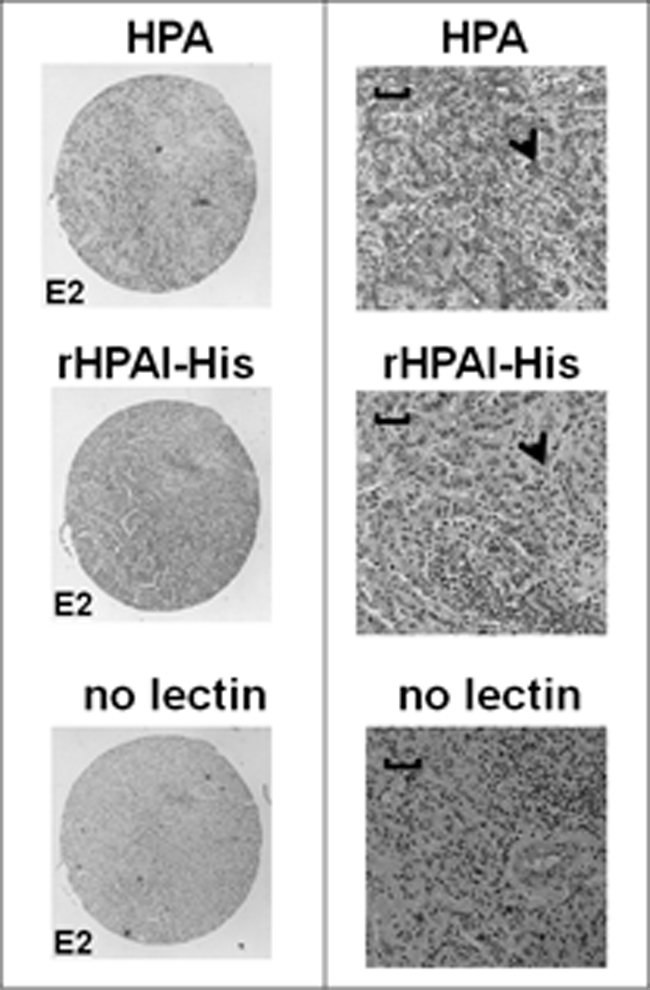
Light micrographs of serial sections of an infiltrating breast carcinoma tissue stained with native and rHPAI-His and counterstained with hematoxylin. The sections were incubated with either biotinylated native HPA, biotinylated rHPAI-His, or processed without addition of lectin, as indicated. The left panel shows the low magnification view (×4 objective), and the right panel shows a higher magnification view (×20 objective). Scale bars, 100 μm. Lectin binding was visualized following incubation with streptavidin-peroxidase by the addition of the chromogenic substrate diaminobenzidine to give a brown-colored precipitate (shown by the arrowheads). Nuclei were stained with Harris hematoxylin.
DISCUSSION AND CONCLUSIONS
In this study the lectin HPA was found to be a heterogeneous mixture of polypeptides coded for by separate genes expressed in the albumen gland of H. pomatia. It has been proposed that the main physiological function of HPA in the snail is to protect the eggs by binding and agglutinating pathogens on their surface (26, 27). HPA has also been shown to enhance phagocytosis of foreign cells by H. pomatia hemocytes (25). Consistent with these findings are the diverse range of glycan structures that HPA bound on the glycan microarray. In addition, the analysis showed that although native HPA recognizes many glycosylated epitopes (including α-GalNAc/α-GlcNAc and sialic acid), the recombinant material only bound glycans terminating in α-GalNAc/α-GlcNAc. It is possible that native HPA binds to NeuNAc-containing oligosaccharides on the CFG array by virtue of a contaminant in the native lectin preparation. The structural analysis by Sanchez et al. (13) discounted the possibility of a secondary binding site on the lectin. A further possibility is the presence of another polypeptide in the HPA preparation capable of binding sialylated residues (28).
In silico structural analysis, through a comparison of the x-ray crystallography data of HPAII (13) and molecular modeling, have enabled the putative three-dimensional structure of HPAI to be determined. These investigations show that HPAI belongs to the H-type lectin family and that GalNAc will fit in the binding pocket with the same orientation in both polypeptides, HPAI and HPAII.
Recombinant forms of both of the polypeptides have been produced, and analysis confirmed HPAI and HPAII as GalNAc-binding lectins. Both HPAI and HPAII bind to GalNAc with approximately equal affinity and in a similar order of magnitude as that reported by Oyelaran et al. (29) who studied the interaction between HPA with GalNAc (at various coating densities). The BiaCore data were also consistent with the results of the “end-point” agglutination assay where both proteins performed in a similar manner. Clearly, further work is required to fully characterize the proteins more extensively, but the data obtained thus far suggest similar binding characteristics for the two polypeptides.
Our initial analysis using a breast cancer tissue array showed that rHPAI-His binds breast cancer cells. The focus of future work will be identification of the cancer-associated glycans and glycoproteins recognized by HPA. Studies focused on the binding of recombinant HPA to cancer tissues will be an important next step in driving forward method development efforts directed toward the practical use of recombinant HPAI and HPAII for cancer diagnosis and prognostication.
Supplementary Material
Acknowledgments
We thank Dr. Peter Moody, University of Leicester, for assistance with the BiaCore analysis; Andrew Jenks for help with the histochemistry; and Dr. Anthony Leathem, Royal Free and University College Medical School, for constructive comments.

The on-line version of this article (available at http://www.jbc.org) contains supplemental data 1–3.
- HPA
- Helix pomatia agglutinin
- SPR
- surface plasmon resonance
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- Trx
- thioredoxin.
REFERENCES
- 1. Dwek M. V., Brooks S. A. (2004) Curr. Cancer Drug Targets 4, 425–442 [DOI] [PubMed] [Google Scholar]
- 2. Brockhausen I. (2006) EMBO Rep. 7, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brooks S. A. (2000) Histol. Histopathol. 15, 143–158 [DOI] [PubMed] [Google Scholar]
- 4. Leathem A. J., Brooks S. A. (1987) Lancet 1, 1054–1056 [DOI] [PubMed] [Google Scholar]
- 5. Brooks S. A., Leathem A. J. C. (1991) Lancet 338, 71–74 [DOI] [PubMed] [Google Scholar]
- 6. Schumacher U., Higgs D., Loizidou M., Pickering R., Leathem A., Taylor I. (1994) Cancer 74, 3104–3107 [DOI] [PubMed] [Google Scholar]
- 7. Schumacher U., Adam E. (1997) Histochem. J. 29, 677–684 [DOI] [PubMed] [Google Scholar]
- 8. Terasawa K., Furumoto H., Kamada M., Aono T. (1996) Cancer Res. 56, 2229–2232 [PubMed] [Google Scholar]
- 9. Okuyama T., Maehara Y., Kakeji Y., Tsuijitani S., Korenaga D., Sugimachi K. (1998) Cancer 82, 1468–1475 [DOI] [PubMed] [Google Scholar]
- 10. Ju T., Lanneau G. S., Gautam T., Wang Y., Xia B., Stowell S. R., Willard M. T., Wang W., Xia J. Y., Zuna R. E., Laszik Z., Benbrook D. M., Hanigan M. H., Cummings R. D. (2008) Cancer Res. 68, 1636–1646 [DOI] [PubMed] [Google Scholar]
- 11. Kudo T., Iwai T., Kubota T., Iwasaki H., Takayma Y., Hiruma T., Inaba N., Zhang Y., Gotoh M., Togayachi A., Narimatsu H. (2002) J. Biol. Chem. 277, 47724–47731 [DOI] [PubMed] [Google Scholar]
- 12. Lisgarten J. N., Chattopadhyay T. K., Pitts J. E., Palmer R. A., Reynolds C. D., Dao-Thi M. H., van Driessche E., Beeckmans S. (2000) Acta Crystallogr. D Biol. Crystallogr. 56, 109–109 [DOI] [PubMed] [Google Scholar]
- 13. Sanchez J. F., Lescar J., Chazalet V., Audfray A., Gagnon J., Alvarez R., Breton C., Imberty A., Mitchell E. P. (2006) J. Biol. Chem. 281, 20171–20180 [DOI] [PubMed] [Google Scholar]
- 14. Uhlenbruck G., Kim Z., Prokop O. (1967) Nature 213, 76–77 [Google Scholar]
- 15. Hammarström S., Kabat E. A. (1969) Biochemistry 8, 2696–2705 [DOI] [PubMed] [Google Scholar]
- 16. Hammarström S., Westöö A., Björk I. (1972) Scand. J. Immunol. 1, 295–309 [DOI] [PubMed] [Google Scholar]
- 17. Hammarström S. (1974) Ann. N.Y. Acad. Sci. 234, 183–197 [DOI] [PubMed] [Google Scholar]
- 18. Vretblad P., Hjorth R., Låås T. (1979) Biochim. Biophys. Acta 579, 52–61 [DOI] [PubMed] [Google Scholar]
- 19. Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 20. Taylor K. E., van den Berg C. W. (2007) Immunology 120, 404–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Görg A., Obermaier C., Boguth G., Weiss W. (1999) Electrophoresis 20, 712–717 [DOI] [PubMed] [Google Scholar]
- 22. Chomczynski P., Mackey K. (1995) Anal. Biochem. 225, 163–164 [DOI] [PubMed] [Google Scholar]
- 23. Blixt O., Head S., Mondala T., Scanlan C., Huflejt M. E., Alvarez R., Bryan M. C., Fazio F., Calarese D., Stevens J., Razi N., Stevens D. J., Skehel J. J., van Die I., Burton D. R., Wilson I. A., Cummings R., Bovin N., Wong C. H., Paulson J. C. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 17033–17038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brooks S. A., Wilkinson D. (2003) Acta Histochem. 105, 205–212 [DOI] [PubMed] [Google Scholar]
- 25. Renwrantz L., Marquart A., Mock A., Richards E. (2009) J. Molluscan Studies 5 75, 41–49 [Google Scholar]
- 26. Takatsu A., Mukaida M., Ishiyama I. (1971) Jpn. J. Exp. Med. 41, 411–421 [PubMed] [Google Scholar]
- 27. Hammarström S., Lindberg A. A., Robertsson E. S. (1972) Eur. J. Biochem. 25, 274–282 [DOI] [PubMed] [Google Scholar]
- 28. Markiv A. (2008) Identification, Cloning and Functional Characterisation of Lectins from the Albumen Gland of the Roman Snail Helix pomatia. Ph.D. thesis, University of Westminster, London, UK [Google Scholar]
- 29. Oyelaran O., Li Q., Farnsworth D., Gildersleeve J. C. (2009) J. Proteome Res. 8, 3529–3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}