

Abstract
Actinomycetes, such as Mycobacterium species, are Gram-positive bacteria that utilize the small molecule mycothiol (MSH) as their primary reducing agent. Consequently, the enzymes involved in MSH biosynthesis are targets for drug development. The metal-dependent enzyme N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) catalyzes the hydrolysis of N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside to form 1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside and acetate, the fourth overall step in MSH biosynthesis. Inhibitors of metalloenzymes typically contain a group that binds to the active site metal ion; thus, a comprehensive understanding of the native cofactor(s) of metalloenzymes is critical for the development of biologically effective inhibitors. Herein, we examined the effect of metal ions on the overall activity of MshB and probed the identity of the native cofactor. We found that the activity of MshB follows the trend Fe2+ > Co2+ > Zn2+ > Mn2+ and Ni2+. Additionally, our results show that the identity of the cofactor bound to purified MshB is highly dependent on the purification conditions used (aerobic versus anaerobic), as well as the metal ion content of the medium during protein expression. MshB prefers Fe2+ under anaerobic conditions regardless of the metal ion content of the medium and switches between Fe2+ and Zn2+ under aerobic conditions as the metal content of the medium is altered. These results indicate that the cofactor bound to MshB under biological conditions is dependent on environmental conditions, suggesting that MshB may be a cambialistic metallohydrolase that contains a dynamic cofactor. Consequently, biologically effective inhibitors will likely need to dually target Fe2+-MshB and Zn2+-MshB.
Keywords: Enzyme Catalysis, Enzyme Kinetics, Hydrolases, Metalloenzymes, Metalloprotease, Metalloproteins
Introduction
Actinomycetes, such as Mycobacterium species, do not have glutathione. Instead, these organisms use the small molecule mycothiol (MSH)2 as their primary reducing agent and in xenobiotic metabolism for the detoxification of drugs and other toxins (1–4). MSH is likely to be critical for the survival of mycobacteria inside activated macrophages, where the mycobacteria are subjected to oxidative bursts. Consequently, the enzymes involved in MSH biosynthesis and detoxification (Fig. 1A), including the metalloenzymes N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) and MSH-conjugate amidase, are targets for the development of antibiotics for the treatment of diseases such as tuberculosis (5–10).
FIGURE 1.

A, reaction catalyzed by MshB. B, active site of MshB (Protein Data Bank code 1Q74) containing a catalytic zinc ion.
The enzyme MshB catalyzes the hydrolysis of N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside (GlcNAc-Ins) to form 1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside (GlcN-Ins) and acetate, the fourth overall step in MSH biosynthesis (rate-limiting step) (11). MshB is an attractive drug target because it is a metalloenzyme; there are past successes in targeting metalloenzymes, including inhibitors of carbonic anhydrase, matrix metalloproteases, and angiotensin-converting enzyme (12–15). Inhibitors of metalloenzymes typically contain a group that binds to the catalytic metal ion. Consequently, a comprehensive understanding of metalloenzyme cofactor preferences is necessary for the development of potent and specific metalloenzyme inhibitors.
MshB was previously identified as a Zn2+-dependent enzyme based on the observations that the enzyme copurifies with Zn2+ (Fig. 1B) and that the enzyme activity is reversibly inhibited by treatment with 1,10-phenanthroline (16–18). On the basis of the structure of the enzyme active site, MshB is thought to catalyze the hydrolysis of GlcNAc-Ins via one of two potential chemical mechanisms using general acid-base catalysis (GABC) (19). One possible mechanism uses a single bifunctional GABC to facilitate the hydrolysis of GlcNAc-Ins, whereas the other uses a GABC pair to carry out this reaction. However, Fe2+ was not examined as a potential cofactor in these experiments. Furthermore, MshB was purified using zinc immobilized metal ion affinity chromatography (IMAC) under aerobic conditions, which is biased toward zinc incorporation into metalloenzymes (16). Purified MshB contains nickel (0.82 eq) when purified using nickel IMAC (aerobic conditions) (16). There have been several examples over the last decade of Fe2+-enzymes being misidentified as exclusive Zn2+-enzymes, including peptide deformylase, S-ribosylhomocysteinase (LuxS), UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC), and possibly histone deacetylase-8 (HDAC8) (20–27). In all these enzymes, the Fe2+ cofactor is either exclusively preferred or is preferred over Zn2+ under certain environmental conditions. The incorporation of Zn2+ into these enzymes that led to the initial characterizations as zinc-enzymes is attributed to purification of the enzymes under aerobic conditions, which leads to oxidation of Fe2+, dissociation of Fe3+ from the enzyme, and replacement with Zn2+. The finding that the zinc ion observed in the crystal structure of MshB is a 5-coordinate metal ion (28) suggests that Fe2+ could also possibly serve as a cofactor for MshB because Fe2+ prefers coordination numbers of 5–6 (19). Consequently, we have examined whether Fe2+ can serve as an efficient cofactor for MshB.
Herein, we demonstrate that the activity of MshB is ∼3-fold higher with Fe2+ as a cofactor compared with Zn2+. The activity of Fe2+-MshB is air-sensitive, whereas the activity of Zn2+-MshB is stable under aerobic conditions. Additionally, we demonstrate that MshB preferentially binds Fe2+ over Zn2+ when purified under anaerobic conditions and when purified under aerobic conditions in the absence of added Zn2+. Although MshB exhibits a higher affinity for Zn2+ over Fe2+, it is likely that higher free [Fe2+] accounts for the cofactor preferences that are observed in these experiments. These results suggest that MshB likely uses Fe2+ and Zn2+ as biological cofactors under different environmental conditions. These results may have important biological implications in light of the dynamic changes in zinc and iron concentrations that occur during the course of infection and suggest that biologically effective inhibitors will need to dually target Fe2+-MshB and Zn2+-MshB.
MATERIALS AND METHODS
General Procedures
All solutions were prepared using Milli-Q water. Primers were purchased from Integrated DNA Technologies. Genomic DNA was purchased from American Type Culture Collection. DNA sequencing was performed at the Virginia Bioinformatics Institute DNA Sequencing Facility (Virginia Tech). Plasmids and PCR products were purified using the Wizard Plus SV Minipreps DNA purification system and Wizard SV Gel and PCR Clean-up kits (Promega), respectively. All chemicals were purchased from Thermo Fisher Scientific, Sigma, and Gold Biotechnology. For all kinetic and thermodynamic experiments, solutions were prepared with reagents that did not contain extraneous metal ions and/or were treated with Chelex (Bio-Rad), and solutions were stored in “metal-free” plasticware. For Fe2+ experiments, a 400 mm FeCl2 stock was prepared in 10 mm dithionite and diluted to 100 mm with 1× assay buffer prior to incubation with apo-MshB. Similarly, a 100 mm ZnSO4 solution was prepared in 50 mm HEPES (pH 7.5) and diluted to 100 mm with 1× assay buffer prior to incubation with apo-MshB. To maintain anaerobic conditions, experiments were carried out either in an anaerobic chamber (Coy Laboratory Products, Grass Lake, MI) and/or using assay buffers containing 10 mm tris(2-carboxyethyl)phosphine (TCEP) and completed in <2 h to ensure that MshB was maximally active during the course of the assays. The concentrations of metal ions were measured by ion chromatography on a Dionex ICS-3000 system.
Cloning
The mshB genes from Mycobacterium smegmatis (Ms) and Mycobacterium tuberculosis (Mt) were cloned into expression vectors using Flexi® technology (Promega). The expression plasmids used in these studies yield recombinant proteins containing an N-terminal affinity tag linked to MshB via a tobacco etch virus (TEV) protease site: pVP55A (His tag) (29), pVP56K (His-maltose-binding protein (MBP) tag) (30), and pFN18K (HaloTag, Promega). The mshB genes were amplified from genomic DNA with PmeI and SgfI restriction sites at the 5′- and 3′-ends, respectively. PCR products were digested with Flexi enzyme blend (PmeI and SgfI) and ligated into Flexi-digested expression plasmids with T4 ligase (New England Biolabs). For MtMshB, which contains an internal SgfI site, the PCR product was first ligated into a pZeroBlunt vector (Stratagene), the internal restriction site was removed by introducing a silent mutation using the QuikChange Lightning site-directed mutagenesis kit (Stratagene), and the mshB gene was liberated by Flexi digest prior to ligation into the Flexi-digested pVP55A, pVP56K, and pFN18K vectors. The plasmid sequences were verified by DNA sequencing.
Protein Expression and Purification
For large-scale production of recombinant proteins, the pHis-MsMshB or pHisMBP-MtMshB plasmids were transformed into BL21(DE3) cells. Cells were grown in LB medium supplemented with ampicillin (100 μg/ml) or kanamycin (50 μg/ml) at 37 °C with shaking (250 rpm) until an A600 of ∼0.6 was reached. Protein expression was induced with the addition of 1 mm isopropyl β-d-thiogalactopyranoside, the temperature was decreased to 25 °C, and the cells were incubated with shaking overnight. After 4–14 h, cells were harvested by centrifugation, resuspended in Buffer A (30 mm HEPES, 150 mm NaCl, and 0.5 mm imidazole (pH 7.5)), and stored at −80 °C.
Cells were lysed using an EmulsiFlex-C3 high-pressure homogenizer (Avestin). The cell lysate was clarified by centrifugation (18,000 rpm, 4 °C) and loaded onto a pre-equilibrated (Buffer A) cobalt or nickel IMAC column (50 ml of chelating Sepharose (GE Healthcare) charged with NiCl2 or CoCl2). MshB was purified at 4 °C. The column was washed with 150 ml of Buffer A, and His-MshB or His-MBP-MshB was eluted using an imidazole step gradient (200 ml each of Buffer A + 10 mm imidazole, Buffer A + 25 mm imidazole, and Buffer A + 300 mm imidazole). Fractions containing His-MshB or His-MBP-MshB (via SDS-PAGE) were combined, concentrated (Amicon Ultra-15 centrifugal devices, Millipore), and dialyzed (SnakeSkin tubing, Mr cutoff of 10,000, Pierce) versus 2 × 4 liters of Buffer A overnight in the presence of His-TEV protease (300 μg/ml) to remove the His or His-MBP tag. The resulting TEV-cleaved protein was loaded onto a pre-equilibrated (Buffer A + 25 mm imidazole) cobalt or nickel IMAC column. His-MBP and His-TEV remain bound to the cobalt IMAC column, whereas MshB eluted in the flow-through fraction. Fractions containing MshB (via 12% SDS-PAGE) were combined, concentrated, and dialyzed versus 2 × 4 liters of 25 mm HEPES and 1.5 mm TCEP (pH 7.5) (Slide-A-Lyzer, Mr cutoff of 10,000, Pierce). Protein concentration was determined using the Bradford Assay (Pierce or Sigma). Protein aliquots were flash-frozen in liquid nitrogen and stored at −80 °C. Protein identity was confirmed via mass spectroscopy (by Keith Ray and Rich Helm, Virginia Tech) following digest with trypsin and peptide sequencing (MALDI-TOF/TOF). The resulting peptide sequences were analyzed using Matrix Science Mascot and confirmed the identity of MshB from M. smegmatis and M. tuberculosis, respectively, with peptide sequence coverage of 33% (MsMshB) and 44% (MtMshB). Sequences are provided in supplemental Fig. S1.
Molecular Weight Determination
The solution molecular weight of MshB was examined using size exclusion chromatography (supplemental Fig. S2) (31). Purified enzyme was loaded onto a Superdex 200 10/300 GL column (GE Healthcare) pre-equilibrated with 50 mm sodium phosphate and 150 mm NaCl (pH 7.5). The elution volumes were used to calculate the Kav values (Kav = (Ve − V0)/(Vt − V0), where V0 is the void volume of the column, Vt is the total volume of the column, and Ve is the elution volume of the protein). A standard curve was prepared by plotting log Mr versus Kav using the following protein standards (GE Healthcare): aprotinin (6.5 kDa), ribonuclease A (13.7 kDa), carbonic anhydrase (29 kDa), ovalbumin (44 kDa), and conalbumin (75 kDa).
Preparation of Apo-MshB and Metal Reconstitution
As purified, MshB contains bound metal ions, and the metal ion content of purified MshB varies depending on the nature of the purification used (supplemental Table S1). Consequently, for our studies, we prepared apo-MshB and then selectively reconstituted the enzyme with the desired metal ions. For the preparation of apo-MshB, purified protein (≤100 μm) was incubated with 10 mm HEPES, 20 mm dipicolinic acid, and 250 μm EDTA (pH 7.5) on ice. After 1 h, the protein solution was concentrated, washed (diluted with buffer and then concentrated) with 3 × 15 ml of 25 mm HEPES and 1.5 mm TCEP (pH 7.5), and run over a desalting column to remove residual dipicolinic acid/EDTA. Metal ion concentrations were determined using the ICS-3000 system. Apo-MshB samples contained ≤10% metal/protein (supplemental Table S2).
Prior to activity measurements, apo-MshB (≤10 μm) was incubated with a stoichiometric concentration of the desired metal ion (CoCl2, FeCl2, FeCl3, MnCl2, NiCl2, or ZnSO4) and incubated on ice for 30 min. For experiments examining the optimal metal/protein ratio, apo-MshB was incubated with various concentrations of the desired metal ion (0–20 μm) for 30 min on ice prior to activity assay.
MshB Deacetylase Activity
MshB deacetylase activity was measured with the substrate GlcNAc (Sigma) using a fluorescamine-based assay (32). In general, assay mixtures containing 50 mm HEPES, 50 mm NaCl (pH 7.5), and 0–150 mm GlcNAc were pre-equilibrated at 30 °C, and the reactions were initiated by the addition of enzyme (1 μm). For pH dependence experiments, the following buffers (all 50 mm containing 1 mm TCEP) were used: MES (pH 6–6.8), MOPS (pH 6.5–7.5), HEPES (pH 7.3–8.8), N,N-bis(2-hydroxyethyl)glycine (pH 8–9), borate (pH 9–10), and carbonate (pH 10–11). After incubation for various times, reactions aliquots (30 μl) were quenched by the addition of 20% TCA (10 μl), and the cleared supernatant (25 μl) was transferred into a 96-well plate, diluted with 75 μl of 1 m borate (pH 9), and reacted with fluorescamine (30 μl in CH3CN, Invitrogen). After 10 min, the fluorescence was measured (excitation, 395 nm; emission, 485 nm) using a SpectraMax M5 plate reader (Molecular Devices). The initial rates of product formation (<10%) were determined from these data. Equation 1 was fit to the pH-rate profile, where k1 represents V/K at the pH optimum, and Ka1 and Ka2 represent the dissociation constants describing the two ionizations.
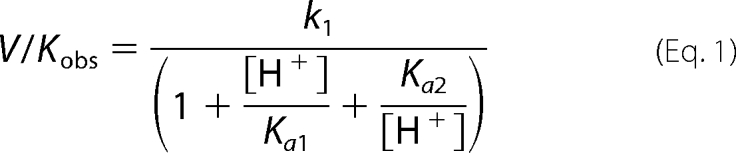 |
For determination of the steady-state parameters, deacetylase activity was measured at six to eight different concentrations of GlcNAc (0–150 mm), and the parameters kcat, Km, and kcat/Km were obtained by fitting the Michaelis-Menten equation to the initial linear velocities using the curve-fitting program KaleidaGraph (Synergy Software), which also calculates the asymptotic standard errors.
UV-visible Spectrophotometry
Apo-MsMshB (1 μm) was incubated with 1 μm FeCl2 or ZnSO4 in an anaerobic glove box in 50 mm HEPES and 10 mm TCEP (pH 7.5) for 30 min on ice to reconstitute the holoenzyme. The enzyme solutions were transferred to sealed anaerobic cuvettes (Precision Cells), and the UV-visible spectrum was recorded on an Agilent 8453 UV-visible spectrophotometer. The spectrum of the Zn2+-MshB sample was subtracted from Fe2+-MshB to account for background absorbance attributed to the protein. The absorbance difference spectrum for Fe2+-MshB is shown in supplemental Fig. S4.
HaloTag Pulldown Experiments
BL21(DE3) cells were transformed with pHalo-MshB and grown in chemically defined medium (100 ml) (33) supplemented with kanamycin (50 μg/ml) at 37 °C with shaking (250 rpm) until an A600 of ∼0.6 was reached. Protein expression was induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside along with the addition of no added metals, 20 μm ZnSO4, 20 μm ferric ammonium citrate, or both metal supplements, and the cells were incubated overnight (12–14 h) with shaking (250 rpm) at 25 °C. Cells were harvested by centrifugation and washed with 1 × 10 ml of 5 mm CaCl2 and 2 × 10 ml of 10 mm MOPS (pH 7). Cell pellets were resuspended in 3 ml of pulldown buffer (40 mm MOPS, 150 mm NaCl, and 10 mm TCEP (pH 7.5)) and lysed by incubation with lysozyme (1 mg/ml) at room temperature for 15–30 min. For anaerobic experiments, washed cell pellets were transferred into an anaerobic chamber prior to resuspension in pulldown buffer. The cell lysate was cleared by centrifugation (15,000 rpm, 25–30 min) and then incubated with 150 μl of HaloLinkTM resin (pre-equilibrated in pulldown buffer, Promega) for 30 min. HaloLinkTM resin was washed with pulldown buffer (5 × 500 μl), resuspended in 250 μl of pulldown buffer containing TEV protease (5 units/μl), and incubated at 37 °C for 45–60 min. The TEV protease expressed and purified in our laboratory was dialyzed against an EDTA-containing buffer and therefore has a low metal content (<0.001 metal ion/protein). Cleaved MshB was separated from the resin by centrifugation (13,200 rpm, 2 min). The concentrations of metal ions were determined using the ICS-3000 system, and the concentration of MshB was determined using the Bradford assay.
Metal Ion Affinity Experiments
The affinity of MshB for Zn(II) was determined using ultrafiltration as described for LpxC deacetylase (27). Briefly, apo-MshB (1 μm) was incubated in a metal ion and pH buffer containing 1 mm nitrilotriacetic acid, 5 mm MOPS (pH 7), and 0–0.5 mm total zinc (0–3.3 nm free zinc) at 30 °C for ≥25 min. Free and bound metal ions were separated by centrifugation (1500 relative centrifugal force, 5 min), and the concentration of metal ions (filtrate and retentate) was determined using ion chromatography. The KDZn(II) value was determined by fitting a binding isotherm to these data.
The affinity of MshB for Fe(II) was determined by measuring the catalytic activity of MshB in the presence of varying concentrations of free Fe(II). Apo-MshB (1 μm) was incubated in a metal ion and pH buffer containing 1 mm nitrilotriacetic acid, 5 mm MOPS (pH 7), and 0–950 μm total Fe(II) (0–2.6 μm free Fe(II)) at 30 °C for ≥30 min in an anaerobic chamber. Activity assays were carried out as described above using 50 mm GlcNAc and 1 μm MshB. The KDFe(II) value was determined by fitting a binding isotherm to these data.
Metal Ion Dissociation Rate Constants
The first-order rate constant for metal ion dissociation from MshB was measured by determining the time-dependent loss of activity upon incubation of MshB with the chelator EDTA. Zn2+-MshB or Fe2+-MshB (50 μm) was diluted into assay buffer (50 mm HEPES, 50 mm NaCl, and 1 mm TCEP (pH 7.5)) containing 1 mm EDTA and incubated at 30 °C. After various times (0–200 min), an aliquot of enzyme was diluted 100-fold into assay buffer containing 20 mm GlcNAc, and the activity was measured as described above. A single exponential decay was fit to the initial rates as a function of time to obtain the koff value.
RESULTS
MshB Is a Mononuclear Metalloenzyme
MshB was previously shown to undergo reversible inhibition by treatment with 1,10-phenanthroline, demonstrating that MshB is a metalloenzyme (16, 28). However, determination of whether MshB is a mononuclear or binuclear metalloenzyme was not examined. Because Zn2+ is proposed to be the native cofactor for MshB, we examined the effect of varying the Zn2+/MshB ratio on catalytic activity (Fig. 2A). The results from these experiments demonstrate that MshB was maximally active with ∼1 Zn2+ ion/MshB, indicating that MshB is a mononuclear metalloenzyme. This finding is consistent with the crystal structure of MshB that reveals a single bound zinc ion in the active site (Fig. 1B) (28). Experiments on the titration of Fe2+ and Co2+ with MshB are also consistent with a mononuclear metalloenzyme (supplemental Fig. S3).
FIGURE 2.

Catalytic activity of MshB. A, activation of apo-MshB with Zn2+ (○). Deacetylase activity was measured as a function of zinc/MshB stoichiometry. Apo-MshB was incubated with 0–2 eq of Zn2+. After 30 min, the enzyme was diluted into assay buffer containing the substrate GlcNAc (50 mm), and the resulting deacetylase activity was measured at 30 °C as described under “Materials and Methods.” B, pH dependence of the Zn2+-MshB-catalyzed reaction. The V/K values were measured at 30 °C using subsaturating concentrations of GlcNAc (5 mm) as described under “Materials and Methods.” pKa values of 7.3 and 10.4 were determined by fitting Equation 1 to these data. C, steady-state turnover catalyzed by metal-substituted MshB: apo-MshB (▿), Co2+ (■), Zn2+ (○), Fe2+ (●), and Fe3+ (△). Apo-MshB was incubated with stoichiometric amounts of metal ions. After 30 min, the enzyme was diluted into assay buffer containing substrate, and the initial rates for the deacetylation of GlcNAc (0–150 mm) were measured at 30 °C as described under “Materials and Methods.” The steady-state parameters kcat, Km, and kcat/Km (Table 1) were obtained by fitting the Michaelis-Menten equation to the initial rates. D, Fe2+-MshB activity is air-sensitive. Apo-MshB was reconstituted with Fe2+ or Zn2+, and the resulting deacetylase activity was measured at 30 °C at t = 0 (anaerobic; black bars). The enzyme was then either exposed to aerobic conditions for 3 h (gray bars) or kept under anaerobic conditions for 3 h (white bars), and the resulting deacetylase activity was measured using the substrate GlcNAc (50 mm) as described under “Materials and Methods.”
The pH Dependence of the MshB-catalyzed Reaction Is Bell-shaped
The MshB-catalyzed reaction (V/K conditions) exhibits a bell-shaped dependence on pH, indicating that there are two ionizations that are important for maximal catalytic activity (Fig. 2B) with observed pKa values of 7.3 and 10.4 for Zn2+-MshB. These results are consistent with MshB proceeding through either a single bifunctional GABC or a GABC pair mechanism, as observed for other metal-dependent deacetylases (19).
Fe2+-MshB Exhibits the Highest Activity
In previous experiments, the steady-state parameters for Zn2+-MshB were determined using the substrates GlcNAc-Ins, monobromobimane S-conjugated MSH, and bimane S-conjugated Cys-GlcN-Ins (16). Although it was stated that the activity of apo-MshB could be restored with Zn2+, Ni2+, Mn2+, or Co2+, no specific values for the rate enhancements observed were reported, and the steady-state parameters for MshB activity were provided only for Zn2+-MshB. Because information about the chemical mechanism can be gained from the relative changes in activity upon reconstitution with different metal ions and Fe2+ had not been previously examined as a potential cofactor, we determined the steady-state parameters for MshB substituted with different metal ions (Fig. 2C and Table 1). We chose to use the commercially available substrate GlcNAc in these experiments. GlcNAc has a weakened affinity for MshB compared with GlcNAc-Ins; however, the group that undergoes hydrolysis in these two substrates is the same (16, 32).
TABLE 1.
Catalytic activity of metal-substituted MshB
Apo-MshB was incubated with stoichiometric metal for 45 min prior to activity measurement. The substrate used was GlcNAc.
| Km | kcat | kcat/Km | |
|---|---|---|---|
| mm | s−1 | m−1s−1 | |
| Apo-MshB | 36 ± 7 | 0.13 ± 0.01 | 3.5 ± 0.3 |
| Co2+-MshB | 34 ± 5 | 1.1 ± 0.08 | 32 ± 2 |
| Fe2+-MshB | 41 ± 5 | 2.1 ± 0.15 | 52 ± 3 |
| Fe3+-MshB | 56 ± 13 | 0.11 ± 0.01 | 1.8 ± 0.3 |
| Mn2+-MshB | 54 ± 13 | 0.38 ± 0.04 | 7.0 ± 1.0 |
| Ni2+-MshB | 53 ± 14 | 0.27 ± 0.03 | 5.3 ± 0.8 |
| Zn2-MshBa | 38 ± 4 | 0.77 ± 0.04 | 20 ± 1 |
The relative ability of various metal ions to activate MshB was measured. In these experiments, apo-MshB was reconstituted (stoichiometric) with the metal ion of interest, and the initial rates of product formation were measured at different substrate concentrations. Interestingly, we observed that MshB exhibited the highest activity with Fe2+ (Fig. 2C), with the overall trend Fe2+ > Co2+ > Zn2+ > Mn2+ > Ni2+. A closer examination of the steady-state parameters (Table 1) revealed that changes to the identity of the catalytic cofactor had only a minor effect on Km (<2-fold), with much larger effects on kcat (8-fold) and kcat/Km (∼10-fold).
Characterization of Fe2+-MshB
To confirm that the activation of MshB was specific for Fe2+ and not Fe3+, we measured the ability of Fe3+ to activate apo-MshB. The results in Fig. 2C indicate that the activity of Fe3+-MshB was comparable with that of apo-MshB (at stoichiometric metal/enzyme), suggesting that Fe3+ is not an efficient catalyst. Additionally, we examined the effect of oxygen on the activity of Fe2+-MshB. Results from these experiments (Fig. 2D) show that there was a time-dependent loss of activity for Fe2+-MshB under aerobic conditions, whereas the activity of Fe2+-MshB under anaerobic conditions was stable over a similar period of time. In contrast, the activity of Zn2+-MshB was stable under aerobic conditions. We also examined the absorbance spectrum of Fe2+-MshB. The Fe2+-MshB difference spectrum has a broad peak at 362 nm (supplemental Fig. S4), consistent with an iron-containing enzyme.
The Cofactor Bound to MshB Is Dependent on Environmental Conditions
Native cofactor identification is defined in large part by the metal ion that copurifies with the enzyme (19). Therefore, we developed a method that would allow us to rapidly purify MshB under various conditions using the HaloTag® technology. The HaloTag does not bind metal ions, and its rapid nature should prevent re-equilibration or switching of bound cofactors during the purification process.
Because previous experiments examining the identity of the cofactor bound to MshB relied on nickel or zinc IMAC purification (16), which artificially introduce metal ions, we initially expressed MshB in LB medium and examined the cofactor bound to purified MshB under aerobic conditions using the HaloTag approach (Fig. 3 and supplemental Table S3). Under these conditions, MshB was isolated with Zn2+ as the predominant bound cofactor, consistent with what has been observed following zinc IMAC purification (16).
FIGURE 3.

Metal ion content of recombinant MshB isolated from Escherichia coli in HaloTag pulldown experiments. pHalo-MshB-transformed E. coli BL21(DE3) cells were grown in LB medium or chemically defined medium (CDM) and induced with 1 mm isopropyl β-d-thiogalactopyranoside with or without iron or zinc supplementation (20 μm). MshB was purified in HaloTag pulldown experiments under aerobic and anaerobic conditions as described under “Materials and Methods.” A, purification of MshB using HaloTag. An aliquot of protein at each step of the purification was analyzed on a 12% SDS-polyacrylamide gel. Lane 1, Mr marker; lanes 2 and 6, cleared cell lysates containing HaloTag fusion proteins (MsMshB and MtMshB, respectively); lane 3, positive control (MshB); lanes 4 and 7, pulldown supernatant after TEV cleavage (*; MsMshB and MtMshB, respectively); lane 5, TEV-only control. B, the iron/zinc ratio of metal ions bound to MshB following aerobic (black bars) and anaerobic (gray bars) purification was determined by ion chromatography as described under “Materials and Methods.”
We also probed what happened to the identity of the cofactor bound to MshB when the protein was expressed under more stringent (metal ion) conditions. In these experiments, MshB was expressed in chemically defined medium with or without zinc and/or iron supplementation to examine the effect that metal ion availability in the surrounding environment has on the identity of the bound cofactor, and MshB was purified under anaerobic and aerobic conditions to probe the effect of oxygen on the bound cofactor. Results from these experiments (Fig. 3B) clearly indicate a strong preference for the Fe2+ cofactor under anaerobic conditions, regardless of the metal ion content of the medium. Additionally, these results suggest that the preferred cofactor changes between Zn2+ and Fe2+ depending on the metal ion content of the medium when MshB is purified under aerobic conditions. Because we used ion chromatography to measure the metal ions bound to MshB, we know that the iron bound to MshB is Fe2+ and not Fe3+, as these ions elute at different retention times (supplemental Fig. S5).
Zn2+ Has a Higher Affinity than Fe2+ for MshB
To determine whether the preference we observed in the pulldown experiments could be explained by a higher affinity of iron for MshB, we measured the affinity of Zn2+ and Fe2+ for MshB. The results from these experiments (Fig. 4 and Table 2) show that Zn2+ has an ∼5300-fold higher affinity for MshB compared with Fe2+, with KDmetal values of 0.02 and 106 nm for Zn2+ and Fe2+, respectively.
FIGURE 4.

Properties of metal ion binding to MshB. A, metal ion affinity of MshB for Zn2+ (○) and Fe2+ (●). Apo-MshB was equilibrated with buffered metal ion solutions (5 mm MOPS and 1 mm nitrilotriacetic acid (pH 7)) as described under “Materials and Methods”. The fraction-bound zinc was determined by ultrafiltration and ion chromatography analysis, and the fraction-bound iron was determined by enhancement of MshB deacetylase activity using GlcNAc substrate (50 mm; see “Materials and Methods”). A binding isotherm was fit to the resulting data to obtain KDZn(II) and KDFe(II) values (Table 2). B, metal ion dissociation from MshB. Holo-MshB reconstituted with Zn2+ (○) or Fe2+ (●) was incubated in assay buffer (50 mm HEPES, 50 mm NaCl, and 1 mm TCEP (pH 7.5)) containing 1 mm EDTA or 5 mm EDTA (♦; Zn2+). At various times, the enzyme was diluted into assay buffer containing GlcNAc (50 mm), and the deacetylase activity was measured at 30 °C as described under “Materials and Methods.” The koff values (Table 2) were obtained by fitting a single exponential decay equation to these data. EM/ETotal, fraction of enzyme containing a bound metal ion; EM, enzyme-metal complex; ETotal, enzyme total.
TABLE 2.
Metal binding properties of MshB
| KDa | koff | konb | |
|---|---|---|---|
| nm | min−1 | m−1s−1 | |
| Fe2+-MshB | 106 ± 15 | 0.032 ± 0.006 | 5.0 × 103 |
| Zn2+-MshB | 0.02 ± 0.004 | 0.033 ± 0.001 | 2.8 × 107 |
a The affinity of MshB for Zn2+ and Fe2+ was determined using ultrafiltration or activity measurements in metal ion-buffered solutions at 30 °C as described under “Materials and Methods.”
b The association rate constants (kon values) for Zn2+ and Fe2+ were calculated from the KD and koff values.
To gain insights into whether the observed differences in metal ion affinity for MshB were due to differences in metal ion association and/or dissociation, we measured the dissociation constants for Zn2+ and Fe2+ from MshB (Table 2). The results from these experiments indicate that the dissociation of Zn2+ and Fe2+ from MshB was comparable (∼0.03 min−1). Consequently, the differences in the KD values are due to the faster association rate constant for Zn2+ compared with Fe2+, which were calculated (KD = koff/kon) to be 2.8 × 107 and 5.0 × 103 m−1 s−1, respectively.
DISCUSSION
MshB Is Most Active with the Fe2+ Cofactor
The data presented here indicate that MshB functions as a mononuclear metallohydrolase (Fig. 2A). The activity of MshB follows the trend Fe2+ > Co2+ > Zn2+ > Mn2+ and Ni2+, which is inversely related to the Lewis acidity of the metals (with the exception of Mn2+). The finding that the highest activity was with Fe2+-MshB is the first evidence to suggest that MshB may not be an exclusive Zn2+-dependent enzyme as previously reported and raises the possibility that Fe2+ may function as a biologically relevant cofactor, thereby making MshB a cambialistic enzyme. Consistent with these data, we observed that MshB copurified with Fe2+ and Zn2+ when expressed in chemically defined medium (lacking metal ion supplementation).
We measured the steady-state parameters for MshB reconstituted with various metal ions. The results from these experiments indicate that there is no significant effect on Km, which may suggest that substrate binding is not affected by changes in the identity of the metal ion. Significantly larger effects were observed on kcat and kcat/Km, suggesting that the rate of the chemical step may be altered by changes to the catalytic metal ion, as expected for a metallohydrolase.
Fe2+-MshB May Be Subject to Redox Regulation
The activity observed upon activation with iron is due to Fe2+, not Fe3+, for the following reasons. 1) Fe3+ could not restore activity to apo-MshB (Fig. 2C); 2) a time-dependent decrease in activity was observed for Fe2+-MshB under aerobic conditions (Fig. 2D), consistent with oxidation of Fe2+ to Fe3+; and 3) MshB copurified with Fe2+, not Fe3+ (Fig. 3B). These results suggest that the biological activity of Fe2+-MshB may be regulated by redox changes in the surrounding environment. This possibility may have biological implications for pathogenic mycobacteria that reside inside macrophages, where they are subject to oxidative bursts upon (macrophage) activation.
Cofactor Preferences Are Determined by Metal Ion Availability
We examined the cofactor preferences of MshB for the first time by expressing the protein under conditions of varying metal ion availability and purifying the protein under anaerobic conditions using a new purification protocol that relies on the HaloTag rather than IMAC purification. We observed that MshB preferred Fe2+ when isolated under anaerobic conditions, regardless of the metal ion content of the medium used to grow the cells (Fig. 3B). Additionally, we found that the cofactor bound to MshB under aerobic conditions switched between Zn2+ and Fe2+ depending on the metal ion availability of the medium used during the protein expression. Similar results were observed with the metal-dependent deacetylase LpxC (supplemental Table S4) (27). In light of the observed activity data for MshB and LpxC, these results imply that at least a subset of metallohydrolases may be cambialistic enzymes that utilize multiple cofactors in vivo and can use metal (cofactor) switching as a mechanism for regulation of enzyme activity in response to changing environmental conditions.
The affinity of MshB for Zn2+ was ∼5300-fold greater than for Fe2+ (Table 2), indicating that the observed metal ion preferences are not dictated solely by metal ion affinity. Instead, the observed preference for Fe2+ under anaerobic conditions appears to be dictated by the greater availability of free iron versus free zinc (estimated 10–400 pm Zn2+ and 0.2–6 μm Fe2+) (27). Interestingly, the dissociation rate constants for Fe(II) and Zn(II) are comparable (0.03 min−1), suggesting that the observed differences in affinity are attributed primarily to metal ion association with MshB. It should be pointed out that the dissociation of Zn(II) from MshB does not go to an end point of zero. Similar findings have been reported for carboxypeptidase A (34, 35), carbonic anhydrase (36), and, to some extent, LpxC (27) and may suggest that a fraction of the active site Zn(II) dissociates more rapidly. Although the association rate constant for Zn(II) with MshB approaches that of a diffusion-controlled process (2.8 × 107 m−1 s−1), the association rate constant for Fe(II) is much slower than a diffusion-controlled process (5 × 103 m−1 s−1) and is more consistent with a two-step binding mechanism. Two-step binding mechanisms have been used to describe Zn(II) binding to carbonic anhydrase (37) and Fe(II) binding to LpxC (27).
Significance of an Iron/Zinc Cambialistic MshB
The results suggesting that MshB may be a cambialistic enzyme that can utilize either Fe2+ or Zn2+ as a cofactor are also interesting in light of the changes in zinc and iron that occur in the mycobacterial environment (vacuoles of infected macrophages) during the course of infection. Metal imaging experiments were used to visualize changes in metal ion concentrations that occur in macrophages both following infection with mycobacteria and after activation of the infected macrophages (38). Results from these experiments indicate that the vacuoles (markers for the location of mycobacteria) of macrophages infected with M. tuberculosis contain a much higher concentration of iron (3 mm) compared with zinc (450 μm). Furthermore, the results from these experiments indicate that zinc and iron are the only divalent metal ions that undergo significant changes in concentration during the course of infection. Activation of macrophages infected with Mycobacterium avium by TNF-α leads to a significant increase in zinc (from 0.12 to 1.8 mm) and decrease in iron (from 1.2 to 0.27 mm) in the vacuoles. These dynamic changes to the iron/zinc concentrations in the mycobacterial environment during the course of infection may have a large impact on metalloenzymes whose activity is altered by changes in zinc and iron concentrations, such as MshB. Furthermore, the ability of MshB to function as an iron/zinc cambialistic enzyme would enable MshB to adapt to changing environmental conditions, such as those encountered in the macrophages, and allow the organism to continue producing MSH throughout the course of infection when large changes in iron/zinc concentrations occur. This could be one key factor that aids mycobacterial survival inside the macrophages. Additionally, these results suggest that biologically effective inhibitors will need to dually target Fe2+-MshB and Zn2+-MshB.
Supplementary Material
Acknowledgments
We gratefully acknowledge Prof. Pablo Sobrado (Virginia Tech) for the pVP55A and pVP56K plasmids and Prof. Rich Helm and Dr. Keith Ray (Virginia Tech) for mass spectroscopic analysis of proteins.
This work was supported by Jeffress Memorial Trust Grant J960 (to M. H.)

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and Tables S1–S4.
- MSH
- mycothiol
- GlcNAc-Ins
- N-acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside
- GlcN-Ins
- 1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside
- GABC
- general acid-base catalysis
- IMAC
- immobilized metal ion affinity chromatography
- TCEP
- tris(2-carboxyethyl)phosphine
- TEV
- tobacco etch virus
- MBP
- maltose-binding protein
- Ms
- M. smegmatis
- Mt
- M. tuberculosis.
REFERENCES
- 1. Newton G. L., Buchmeier N., Fahey R. C. (2008) Microbiol. Mol. Biol. Rev. 72, 471–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jothivasan V. K., Hamilton C. J. (2008) Nat. Prod. Rep. 25, 1091–1117 [DOI] [PubMed] [Google Scholar]
- 3. Rawat M., Av-Gay Y. (2007) FEMS Microbiol. Rev. 31, 278–292 [DOI] [PubMed] [Google Scholar]
- 4. Fan F., Vetting M. W., Frantom P. A., Blanchard J. S. (2009) Curr. Opin. Chem. Biol. 13, 451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gammon D. W., Steenkamp D. J., Mavumengwana V., Marakalala M. J., Mudzunga T. T., Hunter R., Munyololo M. (2010) Bioorg. Med. Chem. 18, 2501–2514 [DOI] [PubMed] [Google Scholar]
- 6. Gutierrez-Lugo M. T., Baker H., Shiloach J., Boshoff H., Bewley C. A. (2009) J. Biomol. Screen. 14, 643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gutierrez-Lugo M. T., Bewley C. A. (2008) J. Med. Chem. 51, 2606–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Metaferia B. B., Fetterolf B. J., Shazad-Ul-Hussan S., Moravec M., Smith J. A., Ray S., Gutierrez-Lugo M. T., Bewley C. A. (2007) J. Med. Chem. 50, 6326–6336 [DOI] [PubMed] [Google Scholar]
- 9. Nicholas G. M., Eckman L. L., Newton G. L., Fahey R. C., Ray S., Bewley C. A. (2003) Bioorg. Med. Chem. 11, 601–608 [DOI] [PubMed] [Google Scholar]
- 10. Bhave D. P., Muse W. B., 3rd, Carroll K. S. (2007) Infect. Disord. Drug Targets 7, 140–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Newton G. L., Av-Gay Y., Fahey R. C. (2000) J. Bacteriol. 182, 6958–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Supuran C. T. (2010) Bioorg. Med. Chem. Lett. 20, 3467–3474 [DOI] [PubMed] [Google Scholar]
- 13. Lia N. G., Shib Z. H., Tang Y. P., Duan J. A. (2009) Curr. Med. Chem. 16, 3805–3827 [DOI] [PubMed] [Google Scholar]
- 14. Drag M., Salvesen G. S. (2010) Nat. Rev. Drug Discov. 9, 690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. White R. J., Margolis P. S., Trias J., Yuan Z. Y. (2003) Curr. Opin. Pharmacol. 3, 502–507 [DOI] [PubMed] [Google Scholar]
- 16. Newton G. L., Ko M., Ta P., Av-Gay Y., Fahey R. C. (2006) Protein Expr. Purif. 47, 542–550 [DOI] [PubMed] [Google Scholar]
- 17. Steffek M., Newton G. L., Av-Gay Y., Fahey R. C. (2003) Biochemistry 42, 12067–12076 [DOI] [PubMed] [Google Scholar]
- 18. Nicholas G. M., Eckman L. L., Kovác P., Otero-Quintero S., Bewley C. A. (2003) Bioorg. Med. Chem. 11, 2641–2647 [DOI] [PubMed] [Google Scholar]
- 19. Hernick M., Fierke C. A. (2010) in Comprehensive Natural Products II (Mander L. N., Lui H.-W. B. eds) pp. 547–581, Elsevier Science Publishing Co., Inc., Oxford, UK [Google Scholar]
- 20. Groche D., Becker A., Schlichting I., Kabsch W., Schultz S., Wagner A. F. V. (1998) Biochem. Biophys. Res. Commun. 246, 342–346 [DOI] [PubMed] [Google Scholar]
- 21. Rajagopalan P. T. R., Yu X. C., Pei D. H. (1997) J. Am. Chem. Soc. 119, 12418–12419 [Google Scholar]
- 22. Rajagopalan P. T., Pei D. (1998) J. Biol. Chem. 273, 22305–22310 [DOI] [PubMed] [Google Scholar]
- 23. Ragusa S., Blanquet S., Meinnel T. (1998) J. Mol. Biol. 280, 515–523 [DOI] [PubMed] [Google Scholar]
- 24. Zhu J., Dizin E., Hu X., Wavreille A. S., Park J., Pei D. (2003) Biochemistry 42, 4717–4726 [DOI] [PubMed] [Google Scholar]
- 25. Gantt S. L., Gattis S. G., Fierke C. A. (2006) Biochemistry 45, 6170–6178 [DOI] [PubMed] [Google Scholar]
- 26. Hernick M., Gattis S. G., Penner-Hahn J. E., Fierke C. A. (2010) Biochemistry 49, 2246–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gattis S. G., Hernick M., Fierke C. A. (2010) J. Biol. Chem. 285, 33788–33796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maynes J. T., Garen C., Cherney M. M., Newton G., Arad D., Av-Gay Y., Fahey R. C., James M. N. G. (2003) J. Biol. Chem. 278, 47166–47170 [DOI] [PubMed] [Google Scholar]
- 29. Sobrado P., Goren M. A., James D., Amundson C. K., Fox B. G. (2008) Protein Expr. Purif. 58, 229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blommel P. G., Fox B. G. (2007) Protein Expr. Purif. 55, 53–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andrews P. (1964) Biochem. J. 91, 222–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang X., Hernick M. (2011) Anal. Biochem. 414, 278–281 [DOI] [PubMed] [Google Scholar]
- 33. Outten F. W., Outten C. E., Hale J., O'Halloran T. V. (2000) J. Biol. Chem. 275, 31024–31029 [DOI] [PubMed] [Google Scholar]
- 34. Chong C. R., Auld D. S. (2000) Biochemistry 39, 7580–7588 [DOI] [PubMed] [Google Scholar]
- 35. Chong C. R., Auld D. S. (2007) J. Med. Chem. 50, 5524–5527 [DOI] [PubMed] [Google Scholar]
- 36. Huang C. C., Lesburg C. A., Kiefer L. L., Fierke C. A., Christianson D. W. (1996) Biochemistry 35, 3439–3446 [DOI] [PubMed] [Google Scholar]
- 37. Kiefer L. L., Fierke C. A. (1994) Biochemistry 33, 15233–15240 [DOI] [PubMed] [Google Scholar]
- 38. Wagner D., Maser J., Lai B., Cai Z., Barry C. E., 3rd, Höner Zu Bentrup K., Russell D. G., Bermudez L. E. (2005) J. Immunol. 174, 1491–1500 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.