

Abstract
Histone H4 undergoes extensive post-translational modifications (PTMs) at its N-terminal tail. Many of these PTMs profoundly affect the on and off status of gene transcription. The molecular mechanism by which histone PTMs modulate genetic and epigenetic processes is not fully understood. In particular, how a PTM mark affects the presence and level of other histone modification marks needs to be addressed and is essential for better understanding the molecular basis of histone code hypothesis. To dissect the interplaying relationship between different histone modification marks, we investigated how individual lysine acetylations and their different combinations at the H4 tail affect Arg-3 methylation in cis. Our data reveal that the effect of lysine acetylation on arginine methylation depends on the site of acetylation and the type of methylation. Although certain acetylations present a repressive impact on PRMT1-mediated methylation (type I methylation), lysine acetylation generally is correlated with enhanced methylation by PRMT5 (type II dimethylation). In particular, Lys-5 acetylation decreases the activity of PRMT1 but increases that of PRMT5. Furthermore, circular dichroism study and computer simulation demonstrate that hyperacetylation increases the content of ordered secondary structures at the H4 tail region. These findings provide new insights into the regulatory mechanism of Arg-3 methylation by H4 acetylation and unravel the complex intercommunications that exist between different the PTM marks in cis. The divergent activities of PRMT1 and PRMT5 with respect to different acetyl-H4 substrates suggest that type I and type II protein-arginine methyltransferases use distinct molecular determinants for substrate recognition and catalysis.
Keywords: Enzymes, Epigenetics, Gene Regulation, Histone Methylation, Histone Modification, Arginine Methylation, PRMT1, PRMT5
Introduction
The nucleosome is the fundamental structural unit of chromatin, in which 146 bp of DNA are wrapped around an octamer of two molecules of each of the four core histones (H2A, H2B, H3, and H4 proteins). Widespread post-translational modifications (PTMs)3 of the histone proteins have been identified, including acetylation, phosphorylation, methylation, ubiquitination, and ribosylation (1, 2). The types and sites of modifications and the specific functions of these PTM marks in modulating chromosomal remodeling and DNA function have been intensively studied in recent years. In particular, significant amounts of data have pointed out the functional correlation of histone modification with transcriptional regulation (3, 4). Although certain modifications are shown as representative marks of active transcription (e.g. H3K4 methylation, H3K36 methylation, H3 and H4 lysine acetylation, and H2B ubiquitylation), some others are correlated with transcriptional repression (e.g. H3K9 methylation, H3K27 methylation, H4K20 methylation, and H2A ubiquitylation) (5, 6). Of importance, many histone PTM marks co-occurring in the same histones can be synergistic or antagonistic with one another, forming complicated combinatorial histone modification patterns that have been proposed to function as a set of multivalent “histone codes” that promote or repress various chromosomal transactions that occur in the cell (1, 7, 8). Histone PTM patterns provide a biochemical index for individual cell types and disease states and correlate with particular biological phenomena of the cell (9–12). Given the abundance of histone modification marks and the dynamic changes of histone modification patterns in response to cell types and differentiation contexts, it remains a challenging biological theme to illuminate the molecular basis of how histone codes and code networks are biochemically created and manifest their downstream impacts on chromatin function (8).
The N-terminal tail of histone H4 is heavily modified at several sites, including Ser-1 phosphorylation, Arg-3 methylation, Lys-5, -8, -12, -16, and -20 acetylation (i.e. K5ac, K8ac, K12ac, and K16ac), and Lys-20 methylation (13–20). These modifications have been shown to be very dynamic, and distinct histone modification patterns have been observed in different cell types, different developmental stages of life, or in different phases of the cell cycle (9, 10, 21, 22). For example, Pesavento et al. (22) characterized H4 modification by using top-down mass spectrometric approaches coupled with two-dimensional liquid chromatography and identified 42 forms of H4 in HeLa cells each of which contain different modification patterns. Coon and co-workers (21) used mass spectrometry methods to identify 74 histone H4 isoforms in differentiating human embryonic stem cells. There is a strong need to understand how these combinatorial PTM patterns are established at the histone tail.
Acetylation represents one of the most frequent modifications at the H4 tail. The acetylation is introduced by several histone acetyltransferases, including p300/CBP (16, 23), Tip60 (24, 25), and yeast protein Esa1 (26). For example, both p300/CBP and Esa1 acetylate H4 at Lys-5, Lys-8, Lys-12, and Lys-16 in vitro and in the context of a nucleosome; acetylation of Lys-5 and Lys-8 by p300 is preferred (16, 23). Lys-16 is a preferred acetylation site by the MOF subunit of the MSL and NSL complexes (27, 28). Histone acetylation is reported to affect the assembly of higher order nucleosome structures (29) and is generally proposed to co-activate gene expression. Because there are four major acetylation sites at the H4 tail, i.e. Lys-5, Lys-8, Lys-12, and Lys-16, histone acetyltransferases catalysis can produce a total of 16 acetylated H4 isoforms. Effort is needed to address which acetylation site and which acetylation combination play predominant roles in determining the transcriptional status of the gene loci. H4K16 acetylation has been particularly shown to be a hallmark of open chromatin and transcriptional activation (30). It is noteworthy to point out that different acetylation patterns may be correlated with different functions. For example, Lys-5 and Lys-12 acetylations are known to be predeposition marks, highly enriched prior to chromatin assembly during S-phase (31, 32). Possibly, the acetylation event may affect gene expression by influencing or even determining other modifications at the histone tail. This is especially true given that histone acetylation has been demonstrated in several studies to be an early event and to occur upstream of other modifications (22, 33).
Here, we investigated how different H4 acetylations and their combinations affect methylation of H4 at Arg-3 (i.e. H4R3me) at the biochemical level. Protein arginine methylation is catalyzed by protein-arginine methyltransferases (PRMTs), which are S-adenosylmethionine (AdoMet)-dependent enzymes and are generally classified into type I and type II families. PRMT1 is the predominant member of type I PRMTs, and PRMT5 is a representative member of type II PRMTs in mammalian cells (Fig. 1). Type I PRMTs are able to transfer up to two methyl groups from the cofactor AdoMet to one single terminal nitrogen of the guanidino group of specific arginine residues in a protein substrate, resulting in ω-NG-monomethylarginine and ω-NG,NG-asymmetric dimethylarginine products (34–37). In contrast, type II PRMT enzymes place one methyl group on each of the two terminal guanidino nitrogens to form ω-NG-monomethylarginine and ω-NG,N′G-symmetric dimethylarginine products (37–39). Because of the lack of structural information, it is still poorly understood how the regiospecificity in methylated products is achieved. At the H4 tail, both PRMT1 and PRMT5 are able to methylate H4R3 in vivo. Of significance, the biological impact of H4R3 methylation by PRMT1 is opposite that of PRMT5; PRMT1-mediated dimethylation of Arg-3 is correlated with gene activation (15), but the methylation of H4R3 by PRMT5 is in many cases associated with gene repression (40). Our biochemical data show that PRMT1 recognizes and methylates H4 substrates that contain different acetyl marks in a very distinct manner from that of PRMT5, thus providing molecular insight into the mechanism of how PRMT1 and PRMT5 target the chromatin template and how acetylation contributes to the establishment of H4 modification patterns or codes by a cis-acting mechanism. This finding also unveils a clear distinction between type I PRMT and type II PRMT with respect to the mode of substrate specificity regulation.
FIGURE 1.

Methylation of H4R3 by PRMT1 and PRMT5.
EXPERIMENTAL PROCEDURES
Design and Synthesis of Acetylated H4 Peptides
The N-terminal peptide of histone H4 containing the first 20 amino acid residues and different acetylation patterns were synthesized using Fmoc-based solid phase peptide synthesis (SPPS) protocols on a PS3 peptide synthesizer (Protein Technology. Tucson, AZ). Each amino acid was coupled to the solid phase by using 4 eq of amino acid/O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (Novabiochem). Fmoc was deprotected with 20% (v/v) piperidine/dimethylformamide. The N-terminal amino acid was acetylated with acetic anhydride. The peptide was cleaved from the Wang resin by incubating the resin in a cleavage solution consisting of 95% trifluoroacetic acid (TFA), 2.5% H2O, and 2.5% triisopropylsilane for 3 h. Peptides were precipitated in cold ether and then pelleted by a centrifuge at 3000 rpm for 10 min. After washing with ether, crude peptides were collected and purified using a Varian Prostar instrument equipped with a C-18 reversed phase (RP)-HPLC column. 0.05% TFA-containing water and 0.05% TFA-containing acetonitrile were two mobile phases used in gradient purification. The purity and identity of purified peptides were confirmed by analytical HPLC and MALDI-MS.
Calibration of Peptide Concentrations with NMR
The accurate concentration of each peptide was determined by measuring its one-dimensional 1H NMR spectra with external standard on a Bruker Avance 400 MHz instrument. For each NMR sample, a D2O solution was prepared containing 4.5 mm (weight-based) individual H4 peptides and 1 mm 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS), respectively. The 1H NMR spectra were collected at room temperature. The integration ratio between the proton peak of DSS at 0 ppm and the two proton peaks of the imidazole group of the histidine residue, i.e. His-18 (δ = 7–9 ppm), was measured to calculate the real concentration of each H4 peptide sample.
Protein Expression and Purification
Recombinant His-tagged rat PRMT1 was expressed in Escherichia coli and purified with nickel-charged His6 tag binding resin. The PRMT1-pET28b(+) plasmid was transformed into E. coli BL21(DE3) (Stratagene) by heat shock. Bacteria were incubated in LB media at 37 °C for growth and 16 °C for protein expression (induced by isopropyl 1-thio-β-d-galactopyranoside 0.3 mm). Cells were harvested by centrifuge and lysed by a French press. The supernatant containing PRMT1 protein was loaded to the nickel-charged His6 tag binding resin (Novagen) equilibrated with column buffer (25 mm Na-Hepes, pH 7.0, 300 mm NaCl, 1 mm PMSF, and 30 mm imidazole). Beads were washed thoroughly with column buffer and washing buffer (25 mm Na-Hepes, pH 7.0, 300 mm NaCl, 1 mm PMSF, and 70 mm imidazole) and eluted with elution buffer (25 mm Na-Hepes, pH 7.0, 300 mm NaCl, 1 mm PMSF, 100 mm EDTA, and 200 mm imidazole). Different eluents were checked by 12% SDS-PAGE. After dialysis and concentration, protein concentration was determined by the Bradford assay. Active FLAG-tagged human recombinant PRMT5/MEP50 was purchased from BPS Bioscience, Inc.
Radioactive Methylation Assays
The methylation assays of different H4 peptide substrates were performed using 14C-isotope labeled AdoMet at 30 °C. The typical reaction buffer contained 50 mm Hepes, pH 8.0, 10 mm NaCl, and 1 mm DTT. Peptide substrate and [14C]AdoMet were preincubated in the reaction buffer for 5 min prior to the initiation of methyl transfer reaction by the addition of PRMT. The reaction time was controlled under initial conditions so that typical reaction yields were within 15%. The reaction was quenched either by spreading the reaction mixture onto P81 filter paper disc (Whatman) or by mixing the reaction buffer with 5× protein-loading dye. For the filter-binding assay, the paper discs were washed with 50 mm NaHCO3, pH 9.0, and air-dried for 2 h, and then liquid scintillation was conducted to measure the amount of methylated products. For the gel-based assay, methylation mixtures were resolved on 16% SDS-PAGE, and the gel was dried and exposed to phosphor film for at least 36 h in the dark. The phosphorimage was scanned on a Typhoon 9400 scanner, and the amount of methylated products was quantitated with ImageQuant program (GE Healthcare). Data of Km and kcat values were obtained with the filter binding assay by measuring the initial velocity of reaction at varied concentrations of one substrate and fixed saturating concentration of the other substrate, and fitting the kinetic data with a Michaelis-Menten equation.
The methylation assays were also conducted for histone H4 protein and reconstituted nucleosomes to confirm the effect of H4 acetylation on its methylation by PRMT1 and PRMT5. Recombinant human core histones were purchased from New England Biolabs, and reconstituted nucleosomes were assembled using the EpiMark assembly kit (E5350S, New England Biolabs). The protein substrate was incubated with acetyl-CoA (20 μm) in the absence or presence of p300 or MOF (0.4 μm) in the reaction buffer (50 mm Hepes, pH 8.0, 50 mm NaCl, 1 mm EDTA, and 0.5 mm DTT) at 30 °C for 10 min. Then [14C]AdoMet (15 μm) and PRMT1 (0.01 μm) or PRMT5 (0.1 μm) was added to the reaction mixture to initialize H4 methylation. The methylated protein was separated on SDS-PAGE (15% for H4 protein and 17% for nucleosome) and visualized by storage phosphorimaging.
Circular Dichroism (CD) Measurement and Analysis
Secondary structures of unacetylated H4 peptide and tetraacetylated H4 peptide at different concentrations of TFE were studied with CD on a Jasco J-810 spectropolarimeter. A 1-mm CD cell with 200 μl of peptide sample (in 20 mm Tris buffer, pH 7.4) with 0 ∼ 80% TFE was loaded to the CD spectrophotometer. Nitrogen pressure was kept constant around 100 kilopascals. CD spectra of peptides were scanned from 260 to 190 nm with standard sensitivity, 0.5 nm data pitch, 1 nm bandwidth, 100 nm/min scanning speed, and 10× accumulation. The concentrations of peptides were selected to keep the negative peak value of ellipticity between −10 and −30 millidegrees. CD spectra were saved as a text file (Jasco 1.30) and submitted to Dichroweb, an on-line server for protein circular dichroism spectra deconvolution (41). Dichroweb incorporates five open source algorithms (Contin-LL, Selcon 3, CDSSTR, VARSSLC, and K2d) to calculate protein secondary structure content. Of these five algorithms, Provencher and Glockner Method (Contin-LL) and Self-Consistent Method (Selcon 3) were chosen to determine the percentage of secondary structural components (helix, strand, turn, and random coil) of each peptide sample.
Structure Simulation for the Unacetylated and Tetraacetylated H4 Peptides
The modeling was conducted on the Jaguar parallel machine at the Oak Ridge Leadership Computing Facility by using the AMBER suite of programs. The wild type and tetraacetylated H4 peptide were parameterized using the Antechamber (42) program with BCC charges. His-18 of the wild type peptide was recognized to be in the ϵ-protonation state through an H-bond network analysis carried out using the WHATIF server (43). The same protonation state was assigned to the histidine of the tetraacetylated peptide because the presence of nonstandard acetylated lysine residues prevented the use of WHATIF. Replica exchange (REMD) simulations have been performed with the Amber10 suite (44) of programs using the force field ff99SB (45) and the GB-neck solvent model (46) with a 0.2 m concentration of monovalent ions and a dielectric constant of 78.5 (corresponding to water). Both the wild type and the acetylated peptides underwent 200 steps of steepest descent minimization followed by 200 steps of conjugate gradient minimization. Twenty replicas of the minimized system were then created and gradually heated to their target temperatures in a short 1-ns run at constant pressure using Langevin dynamics with a collision frequency of 1.0 ps−1. The temperatures of the replicas were chosen in geometric progression to ensure uniform exchange probability for all pairs of temperatures (the values for the temperature T (in K) was set to 300, 314, 330, 345, 362, 380, 398, 417, 437, 458, 480, 503, 528, 553, 580, 608, 637, 668, 700, and 734). A 105-ns REMD simulation was then carried out; the first 5 ns of the simulation were discarded, and analysis was performed on a set of 1976 structures for the wild type and 1722 structures for the acetylated peptide, respectively. The structures, sampled at intervals of 50 ps along the T = 300 K trajectory, were then clustered using a quality threshold clustering algorithm (47). Secondary structure analysis was performed using the DSSP algorithm (48).
RESULTS
Design, Synthesis, and Characterization of Modified H4 Peptide Library
The N-terminal tail of H4 is subject to many PTMs, including Arg-3 methylation and acetylation at Lys-5, -8, -12, and -16 sites. To quantitatively evaluate the in cis effect of acetylation marks on Arg-3 methylation, we designed a library of H4 peptides containing the first 20 amino acids that incorporate all 16 possible acetylation combinations, including 1 unacetylated, 4 monoacetylated, 6 diacetylated, 4 triacetylated, and 1 tetraacetylated forms (Table 1). All peptides are N-terminally capped with acetic anhydride because virtually all H4 proteins are N-terminally acetylated in vivo (10, 21, 22, 49). These peptides are used as substrates of PRMT1 and PRMT5 to evaluate how different acetylation combinations affect the methylation of Arg-3 catalyzed by these two enzymes. It is worthwhile to stress that our data and others have fully determined that the N-terminal H4 peptide sequence represents an authentic substrate of PRMT1, and its catalytic properties are very similar to that of the full-length H4 protein (50). All the peptides were synthesized using the standard Fmoc SPPS protocols, purified on C-18 reversed phase HPLC column, and confirmed with MALDI-MS as described previously (51).
TABLE 1.
Sequences of synthetic H4 peptides
The ac symbol indicates acetyl group.
| Abbreviation | Sequence | Mr calculated | Mr measured |
|---|---|---|---|
| A. WT H4 | ac-SGRGKGGKGLGKGGAKRHRK | 2034.2 | 2034.1 |
| B. H4K5ac | ac-SGRGKacGGKGLGKGGAKRHRK | 2076.2 | 2076.1 |
| C. H4K8ac | ac-SGRGKGGKacGLGKGGAKRHRK | 2076.2 | 2076.2 |
| D. H4K12ac | ac-SGRGKGGKGLGKacGGAKRHRK | 2076.2 | 2076.2 |
| E. H4K16ac | ac-SGRGKGGKGLGKGGAKacRHRK | 2076.2 | 2076.3 |
| F. H4K5ac8ac | ac-SGRGKacGGKacGLGKGGAKRHRK | 2118.2 | 2118.1 |
| G. H4K5ac12ac | ac-SGRGKacGGKGLGKacGGAKRHRK | 2118.2 | 2118.4 |
| H. H4K5ac16ac | ac-SGRGKacGGKGLGKGGAKacRHRK | 2118.2 | 2118.2 |
| I. H4K8ac12ac | ac-SGRGKGGKacGLGKacGGAKRHRK | 2118.2 | 2118.5 |
| J. H4K8ac16ac | ac-SGRGKGGKacGLGKGGAacKRHRK | 2118.2 | 2118.1 |
| K. H4K12ac16ac | ac-SGRGKGGKGLGKacGGAKacRHRK | 2118.2 | 2118.3 |
| L. H4K5ac8ac12ac | ac-SGRGKacGGKacGLGKacGGAKRHRK | 2160.2 | 2160.6 |
| M. H4K5ac8ac16ac | ac-SGRGKacGGKacGLGKGGAKacRHRK | 2160.2 | 2160.7 |
| N. H4K5ac12ac16ac | ac-SGRGKacGGKGLGKacGGAKacRHRK | 2160.2 | 2160.3 |
| O. H4K8ac12ac16ac | ac-SGRGKGGKacGLGKacGGAKacRHRK | 2160.2 | 2160.6 |
| P. H4K5ac8ac12ac16ac | ac-SGRGKacGGKacGLGKacGGAKacRHRK | 2202.2 | 2202.4 |
Because synthetic peptides typically contain varying amounts of TFA counter ions as a result of HPLC purification, we sought to determine the accurate concentration of each H4 peptide prior to enzymatic analysis. The NMR spectra of H4 peptides show two very characteristic peaks at 7.4 and 8.7 ppm, which come from the imidazole side chain of His-18 (Fig. 2). We used the integration ratio between these two peaks and the methyl peak of standard reagent DSS to calibrate concentration of each H4 peptide. A typical one-dimensional 1H NMR spectrum used for calibration of H4 peptide concentration is shown in Fig. 2. The use of NMR calibration to obtain the accurate concentration of H4 peptides is technically critical for accurately quantifying and comparing the effect induced by individual acetylation marks. As a matter of fact, varying degrees of difference were observed between weight-based concentrations and NMR-calibrated concentrations (data not shown).
FIGURE 2.

A typical NMR spectrum used for calibration of H4 peptide concentration. The NMR solution contained 4.5 mm H4K16ac peptide and 1 mm DSS in D2O.
Impact of H4 Acetylation on PRMT1-catalyzed R3 Methylation
Arginine 3 of H4 is methylated by PRMT1. However, the potential effect of acetylation at Lys-5, -8, -12, and -16 on Arg-3 methylation is not clear. The four acetylations at Lys-5, -8, -12, and -16 generate multiple combinations. It remains to be determined whether individual combination marks affect Arg-3 methylation differentially or in a similar manner and which acetyl mark predominantly modulates Arg-3 methylation.
With the purified H4 library peptides, we tested PRMT1-catalyzed methylation of individual H4 peptides each of which contains a unique acetylation pattern. The methylation reaction was composed of 0.1 μm PRMT1, 20 μm [14C]AdoMet, and 100 μm of each substrate, and the reaction was allowed to proceed for 10 min. The reaction mixtures were resolved on 16% SDS-PAGE, and the methylated products were visualized by phosphorimaging and quantitated using ImageQuant software (Fig. 3). At first glance of the experimental data, it is apparent that some acetylation combinations decrease but some others increase the level of Arg-3 methylation. In particular, Lys-5 acetylation is detrimental to Arg-3 methylation, which alone decreases Arg-3 methylation by 30%. In combination with other lysine acetylations, e.g. with K8ac or K8ac and K12ac, the repressive effect of K5ac is even stronger; the degree of repression reaches 3- and 5-fold, respectively. On the other hand, K16ac leads to a positive impact on Arg-3 methylation. Lys-16 acetylation alone increases the PRMT activity by 30%. The majority of H4 peptides that contain the K16ac mark (except M and P) are slightly better substrates of PRMT1. For instance, if K16ac co-exists with K5ac, K8ac, or K12ac, the activation effect is dominant. Compared with K5ac and K16ac, the effect of K8ac and K12ac in regulating Arg-3 methylation seems quite marginal by itself and is influenced by the presence of the K5ac or K16ac mark. For example, a repressive effect is observed when K8ac co-presents with K5ac, but a positive effect is observed when K8ac co-presents with K16ac. Overall, these data support that K5ac and K16ac are two counteractive modification marks that affect Arg-3 methylation in opposite ways; K5ac is the predominant factor that negatively impacts Arg-3 methylation and K16ac positively modulates Arg-3 methylation by PRMT1.
FIGURE 3.

Effects of lysine acetylation on Arg-3 methylation by PRMT1. The reaction buffer contained 50 mm Hepes, pH 8.0, 10 mm NaCl, and 1 mm DTT. The concentrations of PRMT1, [14C]AdoMet, and H4–20 were 0.1, 20, and 100 μm, respectively. The reaction time was 10 min.
To understand the mechanism by which K5ac and K16ac affect Arg-3 methylation by PRMT1, we measured the steady-state kinetic parameters of unacetylated and several acetylated H4 peptides. The data shown in Table 2 reveal that for the methylation of H4K5ac peptide, the kcat decreases by half, but there is little change in Km. These data suggest that acetylation of Lys-5 does not cause explicit changes in the substrate binding and recognition by PRMT1. Likely, the positively charged Lys-5 is required for a methyl transfer step in the catalytic pathway of PRMT1, and acetylation of Lys-5 blocks this key step in the methyl transfer reaction as reflected in its effect on kcat. However, acetylation of Lys-16 affects Km more than kcat values, and V/K is increased from 1.78 to 3.22 min− 1μm−1. Thus, K16ac appears to increase the affinity of enzyme-substrate association but has little effect on the catalytic step.
TABLE 2.
Steady-state kinetic parameters of PRMT1 catalysis
The methylation of each peptide by PRMT1 was tested with the radioactive filter binding assay. Varied concentrations of peptide (0–20 μm) and 15 μm of [14C]AdoMet were incubated at 30 °C for 5 min in the reaction buffer (50 mm HEPES, pH 8.0, 10 mm NaCl, and 1 mm DTT). The reaction was initiated with PRMT1. The methylated products were purified on to P81 filter paper and quantified by liquid scintillation. Calculated methylation rate was plotted as a function of peptide concentration, and the data were fitted with Michaelis-Menten equation.
| Substrates | Km | kcat | V/K |
|---|---|---|---|
| μm | min−1 | min−1 μm−1 | |
| A. WT H4–20 | 0.34 ± 0.05 | 0.64 ± 0.13 | 1.78 ± 0.69 |
| B. H4(1–20)K5ac | 0.37 ± 0.06 | 0.32 ± 0.01 | 0.86 ± 0.14 |
| C. H4(1–20)K8ac | 0.58 ± 0.10 | 0.50 ± 0.02 | 0.86 ± 0.15 |
| D. H4(1–20)K12ac | 0.27 ± 0.05 | 0.47 ± 0.01 | 1.74 ± 0.32 |
| E. H4(1–20)K16ac | 0.23 ± 0.05 | 0.74 ± 0.06 | 3.22 ± 0.75 |
| H. H4(1–20)K5acK16ac | 3.29 ± 0.53 | 1.02 ± 0.07 | 0.31 ± 0.05 |
| J. H4(1–20)K8acK16ac | 1.11 ± 0.17 | 1.33 ± 0.06 | 0.84 ± 0.19 |
| K. H4(1–20)K12acK16ac | 0.38 ± 0.07 | 0.77 ± 0.03 | 2.06 ± 0.38 |
Impact of H4 Acetylation on PRMT5-mediated Arg-3 Methylation
In addition to asymmetric dimethylation by PRMT1, H4R3 can be symmetrically dimethylated by PRMT5 in vivo. Of great interest is that these two types of dimethylation in many cases are correlated with opposite functions in gene transcriptional regulation; PRMT1 activates gene expression, but PRMT5 represses transcription (15, 40). To understand the molecular basis of such functional oppositeness, we have examined how H4 acetylation affects symmetric dimethylation of H4R3 by PRMT5. We subjected the 16-mer H4 library peptides to PRMT5 catalysis under similar conditions as the methylation by PRMT1. Concentrations of PRMT5, [14C]AdoMet, and H4 peptide were maintained at 0.1, 30, and 200 μm, respectively, and the reaction proceeded for 1 h. The methylated products were resolved on SDS-PAGE and analyzed by phosphorimaging. As shown in Fig. 4, intriguingly, most acetylated H4 peptides are better methylated by PRMT5 than the unacetylated H4. This is in stark contrast with the methylation catalyzed by PRMT1, in which the fully acetylated H4 is a poor substrate. The preference of acetylated H4 by PRMT5 is further validated by the data of kcat and Km measurements (Table 3). In particular, K5ac up-regulates the value of kcat, suggesting that this modification mark provides favorable contact with the active site of PRMT5 to facilitate methyl transfer. Also, K16ac increases Km, suggesting that this distal modification mark weakens the binding affinity of H4-PRMT5. Thus, the impacts of K5ac and K16ac are opposite that of PRMT1 catalysis.
FIGURE 4.

Effects of acetylation on Arg-3 methylation catalyzed by PRMT5. Reaction buffer contained 50 mm Hepes, pH 8.0, 10 mm NaCl, and 1 mm DTT. The concentrations of PRMT5, [14C]AdoMet, and H4 peptide were 0.1, 30, and 200 μm respectively. The reaction time was 1 h.
TABLE 3.
Kinetic parameters of PRMT5 catalysis
The catalytic activity of PRMT5 on each peptide was tested with the radioactive filter binding assay. Varied concentrations of peptide (0–10 μm) and 30 μm of [14C]AdoMet were incubated at 30 °C for 5 min in the reaction buffer (50 mm HEPES (pH 8.0), 50 mm NaCl, 1 mm EDTA and 0.5 mm DTT) prior to the addition of PRMT5. The methylated products were loaded onto P81 filter paper and quantified by liquid scintillation. Calculated methylation rate was plotted as a function of peptide concentration, and the data were fitted with Michaelis-Menten equation.
| Substrates | Km | kcat | V/K |
|---|---|---|---|
| μm | min−1 | min−1 μm−1 | |
| A. WT H4–20 | 0.63 ± 0.11 | 0.043 ± 0.002 | 0.068 ± 0.012 |
| B. H4(1–20)K5ac | 0.38 ± 0.08 | 0.079 ± 0.003 | 0.21 ± 0.044 |
| C. H4(1–20)K8ac | 0.66 ± 0.14 | 0.042 ± 0.003 | 0.064 ± 0.014 |
| D. H4(1–20)K12ac | 0.52 ± 0.13 | 0.049 ± 0.003 | 0.094 ± 0.024 |
| E. H4(1–20)K16ac | 1.20 ± 0.16 | 0.048 ± 0.002 | 0.040 ± 0.006 |
Impact of H4 Acetylation on PRMT1 and PRMT5 Activity Using H4 Protein and Nucleosome as Substrates
So far, we used a 16-mer H4 peptide library consisting of different acetylation patterns to dissect the detailed effects of H4 acetylation on the activity of PRMT1 and PRMT5. To further confirm these results, we investigated methylation of H4 at the protein level by these two PRMT members. In the experiment, acetylation of recombinant H4 protein was first introduced by incubation with acetyl-CoA and histone acetyltransferase proteins p300 or MOF. Next, the acetylated H4 protein was subjected to PRMT1 and PRMT5 catalysis using [14C]AdoMet as the methyl donor. The reaction mixtures were then resolved on SDS-PAGE, and the methylated H4 band was visualized by storage phosphorimaging. As seen in Fig. 5, it is clear that acetylation on H4 protein inhibits its methylation by PRMT1 but promotes its methylation by PRMT5. These data coincide well with the peptide methylation data showing that hyperacetylation of H4 inhibited PRMT1 activity but potentiated PRMT5 activity. We also attempted to test such effects by using nucleosomal substrates. The reconstituted nucleosome was assembled from the recombinant core histone proteins and a 208-bp 5 S rDNA by using the EpiMark protocol (supplemental Fig. S1). However, we found that H4 protein in the context of nucleosome or even in the presence of DNA was not appreciably methylated by either PRMT1 or PRMT5 (supplemental Fig. S2). This important observation implicates that DNA poses a physical barrier between nucleosomal histone and PRMTs, preventing the N-terminal tails of the core histones from being methylated by PRMTs. Quite likely, arginine methylation of the chromatin template by PRMTs would require additional cofactors or accessory proteins.
FIGURE 5.

Acetylation on H4 protein inhibits its methylation by PRMT1, although it promotes its methylation by PRMT5. A, p300 acetylation inhibits H4 methylation by PRMT1. H4 protein was incubated with acetyl-CoA in the absence or presence of p300 before submission to PRMT1 methylation with [14C]AdoMet. Methylated H4 bands were separated by 15% SDS-PAGE and visualized by storage phosphor scan, which is the same method as for B–D. B, p300 acetylation promotes H4 methylation by PRMT5. H4 protein was incubated with acetyl-CoA in the absence or presence of p300 before submission to PRMT5 methylation. C, MOF acetylation inhibits H4 methylation by PRMT1. H4 protein was incubated with acetyl-CoA in the absence or presence of MOF before submission to PRMT1 methylation. D, MOF acetylation promotes H4 methylation by PRMT5. H4 protein was incubated with acetyl-CoA in the absence or presence of MOF before submission to PRMT5 methylation.
Structural Changes of H4 Induced by Acetylation
It is of great interest to understand the structural basis of the distinct effect of lysine acetylation on Arg-3 methylation, especially K16ac, which is remote from the methylation site. Thus far, there is no information available on whether and how lysine acetylation changes the structure of the histone tails in the nucleosome. It could be possible that acetylation causes changes in the secondary structure of the H4 tail that affect its interaction with PRMTs. To examine whether there are any structural changes in H4 following lysine acetylation, we measured CD spectra of wide type unacetylated H4 peptide and the tetraacetylated H4 peptide. In particular, the CD spectra were measured at different concentrations of TFE (0–80%) to determine whether hydrophobicity of the solvent environment affects the secondary structures (Fig. 6, A and B). The collected CD spectra were analyzed by Dichroweb, an on-line server for protein CD spectra deconvolution (41) to calculate secondary structure contents (helix, strand, turn, and random coil) for each peptide sample. As shown in Fig. 6C, the contents of random coils occupied a high percentage in both the unacetylated and tetraacetylated H4 peptide. This is not surprising given that the N-terminal sequence of H4 is invisible in the crystal structures of nucleosome (52), suggesting it is largely in disordered states. However, we clearly noticed that for the H4 peptide with tetraacetyl marks (i.e. tetraacetylated H4 peptide), the content of helix and strand became appreciably higher than that of the unacetylated H4, namely 36 versus 13%. This suggests that lysine acetylation renders the H4 peptide in more structured states. Furthermore, it was observed that as TFE concentration increased, the amount of ordered secondary structures appeared to go higher (Fig. 6D), implicating that hydrophobic environment favors formation of ordered structures in H4 N-terminal region. Overall, these data demonstrate that lysine acetylation promotes the tendency of H4 tail to form ordered secondary structures.
FIGURE 6.

Secondary structure analysis of unacetylated H4 peptide (A) and tetraacetylated H4 peptide (P). CD spectra of unacetylated H4 (A) and tetraacetylated H4 (B) were measured at different concentrations of TFE (0–80%) in 20 mm Tris-HCl buffer, pH 7.4. Black, 0% TFE; blue, 5% TFE; orange, 20% TFE; green, 40% TFE; red, 80% TFE. The CD data were analyzed by Dichroweb to calculate secondary structure compositions. C, column graph showing the distribution of secondary structures for the two peptides at 20% of TFE. D, changes in the composition of ordered structures (i.e. helix and strand) for each peptide with TFE concentration.
To further confirm the structural impact of lysine acetylation on the H4 sequence, we conducted a simulation analysis of the unacetylated H4 and tetraacetylated H4 peptides. The modeling was conducted on the Jaguar parallel machine by using the AMBER suite of programs. The simulations show that a dramatic structural change occurs upon lysine acetylation. The first evidence for the structuring effect of tetra-acetylation is provided by the distribution of the radius of gyration plot (Fig. 7a). This distribution appears relatively flat and broad (average, 12.16 Å; variance, 6.06 Å2) in the case of the wild type peptide but undergoes a shift toward lower values when the peptide is acetylated. This observation is suggestive of a transition from a population of extended, predominantly random coil peptides to a population of compact and more globular structures. Fig. 7b shows that in the wild type peptide, the distribution of the pairwise backbone root mean square deviation had an average of 7.67 Å and a variance of 4.20 Å2. These values shift to 4.93 Å and 2.36 Å2, respectively, for the acetylated peptide suggesting that acetylation creates a more homogeneous population with a smaller number of structural families. This prediction is confirmed by clustering analysis where a cutoff as large as 9.0 Å was necessary to group the wild type population into 14 clusters, whereas a cutoff of 6.0 Å was sufficient to generate 18 clusters in the case of the acetylated peptide.
FIGURE 7.

Simulated structural changes upon tetraacetylation of the N-terminal H4 tail. a, probability distribution of the radius of gyration of the unacetylated and tetra-acetylated H4 peptides. b, probability distribution of the pairwise backbone root mean square deviation (RMSD) of the structures of the equilibrium population.
The clusters of the equilibrium population of the unacetylated peptide roughly belong to three main structural groups whose representative conformations can be seen in Fig. 8. The first group (50% of structures) is mainly populated by extended conformations where lysine and arginine side chains stick out from the axis of the molecule to minimize electrostatic repulsion. The second group (24% of structures) is populated by distorted hairpin-like conformations where the standard β-hairpin is replaced by a swollen bubble-like loop arising from the repulsion between lysines and arginines. Finally, the third group (26% of structures) includes extended conformations featuring a small loop at one or both ends that can be regarded as intermediates in the interconversion pathway between the structures of the first two groups.
FIGURE 8.

Clustering of the equilibrium population of the unacetylated (left column) and tetraacetylated (right column) H4 peptides. Rows a–c show the representative conformations of clusters 1–3, respectively. The color code represents different secondary structural elements as identified throuh a DSSP analysis (48). Purple, α-helix; blue, 310-helix; green, turn; orange, random coil.
Three major classes can also be detected in the equilibrium population of the tetraacetylated H4 peptide based on the results of the clustering analysis (Fig. 8). The motif of the first class, including 59% of structures, is represented by three 310 helices orthogonal to each other and is therefore somewhat reminiscent of the hairpin-like conformations of group 2 of the unacetylated peptide. It can therefore be suggested that acetylation induces the stabilization and structuring of a conformation that was already present in the population of the unacetylated species. As a final remark, we note that this motif brings the N and C termini of the H4 tail peptide close to one another thus suggesting a possible mechanism through which the acetylation of Lys-16 affects the methylation propensity of Arg-3 from a distal region of the molecule. The second class, amounting to 24% of the structures, features extended conformations composed by two or three 310 helices linked by turns. The third class (17% of structures) is characterized by an L-shaped motif composed by two orthogonal 310 helices linked by a central turn region, which, similarly to the case of the unacetylated peptide, can be considered as intermediates between the conformations of the former two classes. In fact, in conditions of dynamic equilibrium, it can be suggested that the extended conformations of class 2 can bend in the L-shaped conformation of class 3, which finally completes the folding in the orthogonal arrangement of helices of class 1.
DISCUSSION
It has become increasingly recognized that multiple PTM marks at the N-terminal tails of the core histones intercommunicate with one another to fundamentally regulate DNA functions such as transcription, replication, recombination, and damage repair (1, 53). At the molecular level, how individual PTM patterns or codes are created and how they affect downstream molecular events are poorly defined. More studies are needed to address the communicational relationship between functionally related histone modification marks. Acetylation of H4 at its N-terminal tail is commonly seen in many cell types. With 16 possible acetylation combinations, cells may benefit by utilizing such combinatorial modification tricks to fine tune and/or maximize multivalent readouts for diversified functionality. In this study, we investigated in detail the impact of individual acetylation marks at Lys-5, Lys-8, Lys-12, Lys-16, and their different combinations on type I and type II methylation at site Arg-3. It was previously shown that H4 acetylation reduced PRMT1-mediated Arg-R3 methylation (15). However, it is not clear how individual acetylations combinatorially affect Arg-3 methylation and which acetylation site plays a predominant role in affecting Arg-3 methylation. Furthermore, Arg-3 can be either asymmetrically dimethylated or symmetrically dimethylated. It remains unknown whether lysine acetylation affects these two types of methylation in the same or a distinct manner. To answer these mechanistic questions, we created a library of H4 peptides containing all the possible acetylated isoforms. The concentration of each peptide was calibrated with NMR to obtain the accurate concentration prior to the enzymatic methylation experiments. It is known that synthetic peptides usually contain varying amounts of counter ions (especially trifluoroacetate), and thus weight-based concentration has different degrees of uncertainty. This calibration is particularly critical for accurate quantitation of the impact of individual acetylation marks on Arg-3 methylation.
Our biochemical data reveal that the impact of H4 acetylation on PRMT1-mediated Arg-3 methylation depends on the individual pattern of acetylation combination. The clear observation is that K5ac is the predominant modification mark that negatively impacts on Arg-3 methylation by PRMT1 (Fig. 3). The lower kcat of H4K5ac methylation with regard to that of the wild type H4 suggests that the acetyl group on Lys-5 produces a steric or hydrophobic hindrance in the active site, reducing the ability of PRMT1 in methylating H4R3. Acetylation at Lys-8 and Lys-12 seems to have minimal effects on Arg-3 methylation, but their presence can augment the impact of K5ac or K16ac. Interestingly, K16ac enhances Arg-3 methylation, and to some degree antagonizes the effect of Lys-5 acetylation. This is slightly different from the result of a previous study showing that all forms of H4 acetylation repress Arg-3 methylation (15). The difference is likely caused by the methods used in peptide concentration determination. In our experiments, the peptide concentrations were accurately calibrated by NMR technique, but in the previous study peptide concentrations were weight-based. Despite this technical difference, the repressive impact of H4 acetylation on type I methylation of Arg-3 was clearly observed in several acetylated H4 forms, e.g. B, F, L, and P (all containing the K5ac mark). It is important to mention that our biochemical results coincide well with the in vivo data that methylated Arg-3 was found to be present with K16ac mark in many H4 isoforms (21, 22) and co-existence of K16ac with R3me was observed in higher histone H4 population than any other acetylated H4 isoforms (22).
The observed acetylation effect also suggests valuable clues about PRMT1-H4 interaction. It seems reasonable that K16ac affects Km, but not kcat, given the remoteness of this residue to Arg-3 along the H4 backbone chain. The prominent impact of Lys-16 acetylation on Arg-3 methylation is in good agreement with a previous study showing that the amino acid residues on the H4 tail distal from Arg-3 (i.e. amino acids 16–20) interact with PRMT1 and contribute to substrate recognition (54). The crystal structure of PRMT1 highlights that an acidic area exists on the surface of PRMT1 (55) and suggests that positive charges of substrates are needed for binding to the enzyme. Here, our data demonstrate that electrostatic interaction does not seem to be the sole factor determining the PRMT1-substrate interaction because Lys-16 acetylation favors the methylation at Arg-3. This is also supported by the fact that in H2A, a PRMT1 substrate with similarity to H4, threonine is placed at the equivalent position of H4K16.
Our data about the divergent impacts of acetylation on Arg-3 methylation, e.g. Lys-5 acetylation represses whereas Lys-16 acetylation enhances Arg-3 methylation, also provide insights into the function of histone acetyltransferases. Many acetylated H4 isoforms, especially the one containing K16ac, favor Arg-3 methylation by PRMT1. Therefore, certain H4 acetylation marks are compatible with type I methylation of Arg-3. Recent MS analysis of H4 protein in differentiating human embryonic stem cells revealed that Lys-16 was the most abundant acetylated residue, and K16ac and K5ac rarely occurred on the same histone H4 molecule, indicating that, to some extent, these two acetylations are mutually exclusive and likely have distinct functions (21). This is consistent with previous notions that Lys-16 acetylation has a distinct function from the acetylation of other lysine residues at the H4 tail, including K5ac (56, 57). Given that PRMT1-mediated Arg-3 methylation is associated with gene activation, it may be that Lys-5 acetylation has repressive function and Lys-16 acetylation has activating function in gene regulation. Although histone acetylations are generally considered as gene activation marks, they might also be able to exhibit repressive function under certain contexts by fine-tuning its site specificity at the histone tail by balanced usage of counteractive PTM marks such as K5ac and K16ac. Although a quantitative correlation of the exact linkage of individual H4 acetylation marks to transcriptional on/off status needs to be investigated in the future, a few studies indeed suggest that histone acetylation may possess repressive function in gene expression regulation in certain contexts (58–61).
In addition to type I methylation by PRMT1, H4R3 is also a cellular target of PRMT5, a type II PRMT enzyme. This raises an interesting question: does H4 acetylation affect PRMT5 activity in H4R3 methylation in a similar manner as that of PRMT1? To answer this question, we tested the methylation of the 16-mer H4 peptide library by PRMT5. The observed effects of acetylation on type II methylation of Arg-3 are strikingly different from PRMT1-mediated Arg-3 methylation (summarized in Fig. 9). The acetylation, overall, has an activating impact on PRMT5 activity. This is clearly seen in that the fully tetraacetylated H4 only shows background methylation level in the PRMT1 catalysis, but the tetraacetylated H4 has a higher methylation level than that of the wild type H4 in PRMT5 catalysis (Fig. 4). The acetylation effect on PRMT5 activity was also confirmed with H4 protein as the substrate (Fig. 5). The most effective acetylation seems to be on Lys-5 as all the peptides contacting the K5ac mark are better methylated by PRMT5. The steady-state kinetic study shows that V/K of H4K5ac is 3-fold that of WT-H4, supporting that H4K5ac is a better substrate of PRMT5. However, the acetylation of Lys-16 slightly represses Arg-3 methylation by PRMT5 by increasing its Km value.
FIGURE 9.
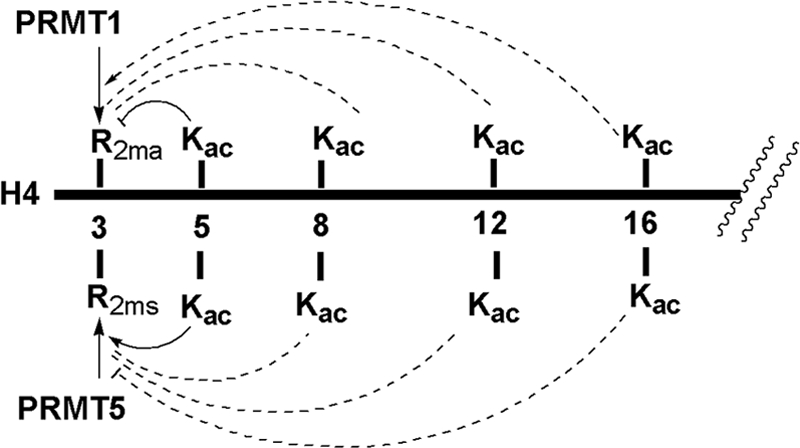
Summary of the effects of lysine acetylation on Arg-3 methylation in H4. Acetylation of the N-terminal H4 tail reciprocally affects PRMT1-mediated Arg-3 methylation and PRMT5-mediated Arg-3 methylation. A solid line means that the effect of acetylation is appreciably strong, and a dotted line means that the effect of acetylation is relatively weak.
The finding that H4 acetylation affects Arg-3 methylation by PRMT5 in a way opposite from that of PRMT1 catalysis has important biological implications. It provides evidence that the interplay between Arg-3 methylation and lysine acetylation that acts in cis in the H4 tail is not simply a linear relationship. Instead, the exact effect of acetylation on Arg-3 methylation is dependent on the specific acetylation pattern and methylation type. In particular, K5ac suppresses PRMT1 activity but enhances PRMT5 activity. K16ac enhances PRMT1 activity but slightly decreases PRMT5 activity. The effects of K8ac and K12ac seem to be marginal and depend largely on whether K5ac or K16ac is present. These data also offer biochemical insights for understanding substrate recognition and methylation by type I and type II PRMTs. Thus far, no structural information is available about how PRMT1 and PRMT5 recognize and methylate H4R3. Based on the finding that H4 acetylation affects PRMT1 activity in a distinct manner from PRMT5, the contacts of PRMT1 with H4 substrate should be very different from that of PRMT5. Possibly, PRMT5 has a larger active site space than does PRMT1 to accommodate the acetyl group on H4K5. Thus, although both PRMT proteins methylate the same substrate at the same site, the substrate specificity regulation is very different. In addition, the opposite effect of H4 acetylation on type I and type II arginine methylation offers a molecular insight into the function of PRMT1 and PRMT5 in transcriptional regulation. For gene activation events in which acetylation occurs upstream of Arg-3 methylation during transcription, PRMT1 recruitment may occur in an early stage of gene expression, likely through binding to specific activation marks, e.g. K16ac. PRMT1-mediated R3 methylation then further opens chromatin structures for active transcription. However, PRMT5-mediated H4R3 methylation may occur at a final stage of transcription. Hyperacetylation of H4 promotes the association of PRMT5 with the chromatin template. PRMT5 writes the inactive type II methylation mark on Arg-3, which subsequently recruits repressive proteins such as histone deacetylases to erase acetylation marks on the H4 tail, thus closing the chromatin structure for gene silencing. These hypotheses need to be addressed in the future.
To provide structural information on how lysine acetylation affects Arg-3 methylation, we measured the CD spectra of unacetylated and tetraacetylated H4 peptides to examine whether there was any structural change induced by multiple acetylations. Indeed, analysis of the CD data showed that the contents of structured motifs were higher after acetylation (Fig. 6). These data are in good agreement with the in silico simulation studies. The molecular dynamics simulations provide atomic level insights into the structural effects of the tetraacetylation. The observed dramatic decrease both in the average value and variance of the radius of gyration marks a shift from extended to compact and more structured conformations. The population also becomes more homogeneous upon acetylation, with fewer but better defined folds, as evidenced by the decrease in the average value and variance of the pairwise root mean square deviation. These predictions were confirmed and complemented by the results of clustering analysis of the molecular dynamics trajectories. For the unacetylated H4 peptide, extended conformations appeared to dominate the equilibrium population (∼50%), with the loop motif representing a minority group of structures (∼25%). This situation was essentially reversed in the case of the tetraacetylated peptide with more compact conformations becoming dominant. Therefore, acetylation appears to stabilize the distorted loop motif that is not only more populated but also better structured compared with the unacetylated H4 peptide. From a physicochemical point of view, the conformational transition induced by acetylation has a straightforward interpretation. In the unacetylated H4 tail, electrostatic repulsion of the positively charged lysine residues forces the peptide into an extended conformation. Acetylation, however, eliminates this electrostatic repulsion. The structuring effect of acetylation is possibly even more pronounced in vivo where the high positive charge of lysines induces a strong interaction between the unacetylated H4 tail and water or nucleosomal DNA. An important implication of the loop structures that are populated in the tetraacetylated H4 is that they enable a close contact between the Arg-3 site and distal regions in the H4 tail (e.g. residues 16–19), possibly explaining the influence of K16ac on the methylation of Arg-3. Another likely consequence of the conformational transition induced by acetylation is a change in the shape complementarity between the H4 tail and the active site of the methyltransferases PRMT1 and PRMT5. This hypothesis could be tested by docking representatives of the most populated clusters of both the unacetylated and tetraacetylated H4 peptides with PRMTs by further simulation studies.
Supplementary Material
Acknowledgments
Computational resources were provided through an INCITE allocation (to I. I.) at the Oak Ridge Leadership Computing Facility, supported by the Department of Energy under Contract DE-AC05-00OR22725.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM086717 (to Y. G. Z.). This work was also supported by American Heart Association Grant 09BGIA2220207 (to Y. G. Z.) and by Georgia State University Molecular Basis of Disease Program (to S. A. and L. H.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- PTM
- post-translational modification
- TFE
- 2,2,2-trifluoroethanol
- DSS
- 4,4-dimethyl-4-silapentane-1-sulfonic acid
- AdoMet
- S-adenosylmethionine
- PRMT
- protein-arginine methyltransferase
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl.
REFERENCES
- 1. Strahl B. D., Allis C. D. (2000) Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 2. Kouzarides T. (2007) Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 3. Li B., Carey M., Workman J. L. (2007) Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 4. Berger S. L. (2007) Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 5. Lachner M., Jenuwein T. (2002) Curr. Opin. Cell Biol. 14, 286–298 [DOI] [PubMed] [Google Scholar]
- 6. Zheng Y. G., Wu J., Chen Z., Goodman M. (2008) Med. Res. Rev. 28, 645–687 [DOI] [PubMed] [Google Scholar]
- 7. Wang G. G., Allis C. D., Chi P. (2007) Trends Mol. Med. 13, 363–372 [DOI] [PubMed] [Google Scholar]
- 8. Ruthenburg A. J., Li H., Patel D. J., Allis C. D. (2007) Nat. Rev. Mol. Cell Biol. 8, 983–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shechter D., Nicklay J. J., Chitta R. K., Shabanowitz J., Hunt D. F., Allis C. D. (2009) J. Biol. Chem. 284, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nicklay J. J., Shechter D., Chitta R. K., Garcia B. A., Shabanowitz J., Allis C. D., Hunt D. F. (2009) J. Biol. Chem. 284, 1075–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cuomo A., Moretti S., Minucci S., Bonaldi T. (2010) Amino Acids, [DOI] [PubMed] [Google Scholar]
- 12. Lennartsson A., Ekwall K. (2009) Biochim. Biophys. Acta 1790, 863–868 [DOI] [PubMed] [Google Scholar]
- 13. Utley R. T., Lacoste N., Jobin-Robitaille O., Allard S., Côté J. (2005) Mol. Cell. Biol. 25, 8179–8190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheung W. L., Turner F. B., Krishnamoorthy T., Wolner B., Ahn S. H., Foley M., Dorsey J. A., Peterson C. L., Berger S. L., Allis C. D. (2005) Curr. Biol. 15, 656–660 [DOI] [PubMed] [Google Scholar]
- 15. Wang H., Huang Z. Q., Xia L., Feng Q., Erdjument-Bromage H., Strahl B. D., Briggs S. D., Allis C. D., Wong J., Tempst P., Zhang Y. (2001) Science 293, 853–857 [DOI] [PubMed] [Google Scholar]
- 16. Ogryzko V. V., Schiltz R. L., Russanova V., Howard B. H., Nakatani Y. (1996) Cell 87, 953–959 [DOI] [PubMed] [Google Scholar]
- 17. Zhang K., Williams K. E., Huang L., Yau P., Siino J. S., Bradbury E. M., Jones P. R., Minch M. J., Burlingame A. L. (2002) Mol. Cell. Proteomics 1, 500–508 [DOI] [PubMed] [Google Scholar]
- 18. Zhang L., Eugeni E. E., Parthun M. R., Freitas M. A. (2003) Chromosoma 112, 77–86 [DOI] [PubMed] [Google Scholar]
- 19. Schotta G., Lachner M., Sarma K., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T. (2004) Genes Dev. 18, 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beck H. C., Nielsen E. C., Matthiesen R., Jensen L. H., Sehested M., Finn P., Grauslund M., Hansen A. M., Jensen O. N. (2006) Mol. Cell. Proteomics 5, 1314–1325 [DOI] [PubMed] [Google Scholar]
- 21. Phanstiel D., Brumbaugh J., Berggren W. T., Conard K., Feng X., Levenstein M. E., McAlister G. C., Thomson J. A., Coon J. J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 4093–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pesavento J. J., Bullock C. R., LeDuc R. D., Mizzen C. A., Kelleher N. L. (2008) J. Biol. Chem. 283, 14927–14937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schiltz R. L., Mizzen C. A., Vassilev A., Cook R. G., Allis C. D., Nakatani Y. (1999) J. Biol. Chem. 274, 1189–1192 [DOI] [PubMed] [Google Scholar]
- 24. Doyon Y., Selleck W., Lane W. S., Tan S., Côté J. (2004) Mol. Cell. Biol. 24, 1884–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kimura A., Horikoshi M. (1998) Genes Cells 3, 789–800 [DOI] [PubMed] [Google Scholar]
- 26. Yamamoto T., Horikoshi M. (1997) J. Biol. Chem. 272, 30595–30598 [DOI] [PubMed] [Google Scholar]
- 27. Smith E. R., Cayrou C., Huang R., Lane W. S., Côté J., Lucchesi J. C. (2005) Mol. Cell. Biol. 25, 9175–9188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cai Y., Jin J., Swanson S. K., Cole M. D., Choi S. H., Florens L., Washburn M. P., Conaway J. W., Conaway R. C. (2010) J. Biol. Chem. 285, 4268–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kan P. Y., Caterino T. L., Hayes J. J. (2009) Mol. Cell. Biol. 29, 538–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shogren-Knaak M., Peterson C. L. (2006) Cell Cycle 5, 1361–1365 [DOI] [PubMed] [Google Scholar]
- 31. Chicoine L. G., Schulman I. G., Richman R., Cook R. G., Allis C. D. (1986) J. Biol. Chem. 261, 1071–1076 [PubMed] [Google Scholar]
- 32. Allis C. D., Chicoine L. G., Richman R., Schulman I. G. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 8048–8052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Daujat S., Bauer U. M., Shah V., Turner B., Berger S., Kouzarides T. (2002) Curr. Biol. 12, 2090–2097 [DOI] [PubMed] [Google Scholar]
- 34. Gary J. D., Clarke S. (1998) Prog. Nucleic Acid Res. Mol. Biol. 61, 65–131 [DOI] [PubMed] [Google Scholar]
- 35. Bedford M. T., Richard S. (2005) Mol. Cell 18, 263–272 [DOI] [PubMed] [Google Scholar]
- 36. Lee D. Y., Teyssier C., Strahl B. D., Stallcup M. R. (2005) Endocr. Rev. 26, 147–170 [DOI] [PubMed] [Google Scholar]
- 37. McBride A. E., Silver P. A. (2001) Cell 106, 5–8 [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y., Reinberg D. (2001) Genes Dev. 15, 2343–2360 [DOI] [PubMed] [Google Scholar]
- 39. Clarke S. (1993) Curr. Opin. Cell Biol. 5, 977–983 [DOI] [PubMed] [Google Scholar]
- 40. Pal S., Vishwanath S. N., Erdjument-Bromage H., Tempst P., Sif S. (2004) Mol. Cell. Biol. 24, 9630–9645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Whitmore L., Wallace B. A. (2004) Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J., Wang W., Kollman P. A., Case D. A. (2006) J. Mol. Graph. Model 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 43. Vriend G. (1990) J. Mol. Graph. 8, 52–56, 29 [DOI] [PubMed] [Google Scholar]
- 44. Case D. A., Darden T. A., Cheatham T. E., 3rd, Simmerling C. L., Wang J., Duke R. E., Luo R., Crowley M., Walker R. C., Zhang W., Merz K. M., Wang B., Hayik S., Roitberg A., Seabra G., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Yang L., Tan C., Mongan J., Hornak V., Cui G., Mathews D. H., Seetin M. G., Sagui C., Babin V., Kollman P. A. (2008) AMBER 10, University of California, San Francisco [Google Scholar]
- 45. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. (2006) Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mongan J., Simmerling C., McCammon J. A., Case D. A., Onufriev A. (2007) J. Chem. Theory Comput. 3, 156–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heyer L. J., Kruglyak S., Yooseph S. (1999) Genome Res. 9, 1106–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kabsch W., Sander C. (1983) Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 49. Song O. K., Wang X., Waterborg J. H., Sternglanz R. (2003) J. Biol. Chem. 278, 38109–38112 [DOI] [PubMed] [Google Scholar]
- 50. Obianyo O., Osborne T. C., Thompson P. R. (2008) Biochemistry 47, 10420–10427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xie N., Elangwe E. N., Asher S., Zheng Y. G. (2009) Bioconjug. Chem. 20, 360–366 [DOI] [PubMed] [Google Scholar]
- 52. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 53. Peterson C. L., Laniel M. A. (2004) Curr. Biol. 14, R546–R551 [DOI] [PubMed] [Google Scholar]
- 54. Osborne T. C., Obianyo O., Zhang X., Cheng X., Thompson P. R. (2007) Biochemistry 46, 13370–13381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang X., Cheng X. (2003) Structure 11, 509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vyskot B., Siroky J., Hladilova R., Belyaev N. D., Turner B. M. (1999) Genome 42, 343–350 [PubMed] [Google Scholar]
- 57. Dion M. F., Altschuler S. J., Wu L. F., Rando O. J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5501–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Turner B. M. (1989) Exp. Cell Res. 182, 206–214 [DOI] [PubMed] [Google Scholar]
- 59. Turner B. M., O'Neill L. P. (1995) Semin. Cell Biol. 6, 229–236 [DOI] [PubMed] [Google Scholar]
- 60. Ceol C. J., Horvitz H. R. (2004) Dev. Cell 6, 563–576 [DOI] [PubMed] [Google Scholar]
- 61. Kurdistani S. K., Tavazoie S., Grunstein M. (2004) Cell 117, 721–733 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.