

Abstract
Precise targeting and maintenance of axonal domains in myelinated axons is essential for saltatory conduction. Caspr and Caspr2, which localize at paranodal and juxtaparanodal domains, contain binding sites for the cytoskeletal adaptor protein 4.1B. The exact role of 4.1B in the organization and maintenance of axonal domains is still not clear. Here, we report the generation and characterization of 4.1B-null mice. We show that loss of 4.1B in the PNS results in mislocalization of Caspr at paranodes and destabilization of paranodal axoglial septate junctions (AGSJs) as early as postnatal day 30. In the CNS, Caspr localization is progressively disrupted and ultrastructural analysis showed paranodal regions that were completely devoid of AGSJs, with axolemma separated from the myelin loops, and loops coming off the axolemma. Most importantly, our phenotypic analysis of previously generated 4.1B mutants, used in the study by Horresh et al. (2010), showed that Caspr localization was not affected in the PNS, even after 1 year; and 4.1R was neither expressed, nor enriched at the paranodes. Furthermore, ultrastructural analysis of these 4.1B mutants showed destabilization of CNS AGSJs at ∼1 year. We also discovered that the 4.1B locus is differentially expressed in the PNS and CNS, and generates multiple splice isoforms in the PNS, suggesting 4.1B may function differently in the PNS versus CNS. Together, our studies provide direct evidence that 4.1B plays a pivotal role in interactions between the paranodal AGSJs and axonal cytoskeleton, and that 4.1B is critically required for long-term maintenance of axonal domains in myelinated axons.
Introduction
A fundamental characteristic of myelinated axons is their organization into distinct molecular domains that allows for saltatory action potential propagation. These domains include nodes, where voltage-gated sodium channels are enriched; paranodes, where myelin loops establish axoglial septate junctions (AGSJs) with the axolemma; and juxtaparanodes, where delayed-rectifier potassium channels are clustered (Bhat, 2003; Salzer, 2003; Thaxton and Bhat, 2009; Thaxton et al., 2011). Genetic ablation of Caspr resulted in loss of paranodal AGSJs and disorganization of the paranodal axonal cytoskeleton (Bhat et al., 2001; Garcia-Fresco et al., 2006). Disruption of paranodal AGSJs in Cgt, Caspr, Cont, and NfascNF155 mutants permitted juxtaparanodal components to move alongside nodal sodium channels, indicating that AGSJs serve a fence function at paranodes (Rosenbluth, 1988; Dupree et al., 1999; Bhat et al., 2001; Boyle et al., 2001; Pillai et al., 2009). Another member of the Caspr family, Caspr2, is required for the organization of the juxtaparanodal domain and localization of potassium channels (Poliak et al., 2003). The stabilization of Caspr and Caspr2, and their associated complexes, is thought to depend on cytoskeletal adaptor proteins that link these complexes with axonal and glial cytoskeleton for their long-term stability.
It was reported that the extracellular domain of Caspr is sufficient for membrane targeting, whereas the intracellular domain is required for Caspr stabilization at the paranodes (Gollan et al., 2002). Both Caspr and Caspr2 contain 4.1-binding sequences, suggesting that their interactions may be physiologically relevant (Girault et al., 1998; Denisenko-Nehrbass et al., 2003). Several members of the 4.1 protein family are expressed in the nervous system (Yamakawa et al., 1999; Ohara et al., 2000; Parra et al., 2000). Only 4.1B is enriched at paranodes and juxtaparanodes in myelinated axons (Ohara et al., 2000; Denisenko-Nehrbass et al., 2003). The presence of a 4.1/ezrin/radixin/moesin (FERM) and a spectrin-actin binding domain make 4.1B an ideal candidate to link Caspr and Caspr2-dependent complexes with the underlying axonal cytoskeleton. Disruption of paranodal AGSJs in Caspr and Cont mutants alters 4.1B localization (Gollan et al., 2002; Garcia-Fresco et al., 2006). However, Caspr2 mutants showed normal 4.1B localization (Traka et al., 2003). Recent studies showed that loss of 4.1B does not affect paranodal Caspr localization in sciatic nerves; and in vivo deletion of 4.1B-binding region in Caspr and Caspr2 did not affect Caspr localization, but affected Caspr2 localization (Horresh et al., 2010).
Here, we report that 4.1B is essential for proper Caspr localization and maintenance of the paranodal and juxtaparanodal domains. We demonstrate that loss of 4.1B leads to destabilization of AGSJs at the paranodes and complete disorganization of the juxtaparanodal complexes. We also show that 4.1B mutants generated previously (Yi et al., 2005) have proper localization of paranodal proteins even after 1 year in the PNS, and that 4.1R is not involved in paranodal organization or rescue of 4.1B mutant paranodes. Together, our results establish an important role of 4.1B as a cytoskeletal adaptor protein that links axoglial junctional complexes with axonal cytoskeleton to ensure long-term stability and maintenance of axonal domains in myelinated axons.
Materials and Methods
Animals.
All animal experiments were performed according to University of North Carolina Institutional Animal Care and Use Committee-approved guidelines for ethical treatment of laboratory animals. 4.1B/Epb4.1l3 knock-out mice described previously (Yi et al., 2005) and transgenic mice expressing Cre recombinase under the ubiquitous β-Actin promoter (β-Actin-Cre) were purchased from The Jackson Laboratory.
Generation of 4.1B/Epb4.1l3 conditional mutants.
The 4.1B/Epb4.1l3 locus spans ∼133 kb of genomic DNA and generates several alternatively spliced mRNAs (see www.ensembl.org/Mus_musculus). To conditionally ablate the 4.1B gene, a targeting construct was generated in which loxP sites were targeted to flank exon 6. Using BAC recombineering technology, a BAC clone pBACe3.6 from 129 AB2.2 BAC library containing the 4.1B locus was targeted such that a neo cassette flanked by FRT, FRT-loxP sites was inserted in intron 5 and a second loxP site was inserted in intron 6. Before ES cell targeting, the functionality of the loxP sites, as well as the expected restriction enzyme digestion patterns in the targeting vector, were confirmed. The targeting construct was linearized with NotI followed by electroporation into ES cells. The targeted ES cells were screened by Southern blotting with a 500 bp probe upstream of the targeting vector in the 4.1B gene. Two independent targeted clones were used for generating 4.1B-floxed mice using standard procedures.
Generation of 4.1B antibodies.
cDNA sequences encoding a portion of the U3 domain of 4.1B (amino acids 655–793) were PCR amplified from a mouse cDNA library, cloned into pET28(a+) vector (Novagen), and expressed in Escherichia coli BL21(DE3). The recombinant protein was purified over a Ni-Sepharose column and used as an antigen to immunize rabbits and guinea pigs, as described previously (Bhat et al., 2001).
Tissue preparation, antibodies, and immunostaining.
The following antisera were previously described: guinea pig and rabbit anti-Caspr (Bhat et al., 2001; Pillai et al., 2009; Thaxton et al., 2010), guinea pig anti-NF186 and rat anti-pan Neurofascin (Pillai et al., 2009; Thaxton et al., 2010), rat anti-Ankyrin G (AnkG) and rabbit anti-Caspr2 (Thaxton et al., 2010), rabbit polyclonal anti-4.1R and 4.1N (Kang et al., 2009), and rabbit and guinea pig anti-4.1B antibodies (this study). Other primary antibodies used include the following: mouse anti-Kv1.1 (University of California Davis/NIH NeuroMab Facility; K20/78), mouse anti-Kv1.2 (University of California Davis/NIH NeuroMab Facility; K14/16), mouse anti-PSD95 (University of California Davis/NIH NeuroMab Facility; K28/43), and rabbit anti-α-Tubulin (Tub) (Cell Signaling). Secondary antibodies used for immunofluorescence were Alexa Fluor-488, -568, and -647 conjugated (Invitrogen). The HRP-conjugated secondary antibodies were purchased from Jackson ImmunoResearch.
Tissues used for immunostaining were prepared as previously described (Thaxton et al., 2010). Briefly, sciatic nerves were removed from anesthetized littermate wild type and mutants of either sex and fixed in 4% paraformaldehyde in PBS for 30 min at 4°C. The nerves were washed with PBS several times and either stored at 4°C or immediately teased. The nerves were teased into individual fibers in PBS, mounted on glass slides, and dried overnight at room temperature, followed by storage at −80°C or immediate processing by treatment with acetone at −20°C for 20 min and several washes with PBS, followed by immunostaining. For spinal cord sections, wild type and mutants of either sex were deeply anesthetized and transcardially perfused with saline buffer followed by ice-cold 4% paraformaldehyde in PBS. The spinal cord was dissected out and postfixed in 4% paraformaldehyde overnight at 4°C. The tissues were rinsed several times in PBS and sectioned to 30 μm using a Vibratome (Leica). The spinal cord sections were then immediately immunostained as previously described (Garcia-Fresco et al., 2006; Pillai et al., 2009).
RNA preparation and RT-PCR.
DRGs from β-Act-Cre;4.1BFlox, 4.1B−/−, and control littermate mice of either sex were collected and simultaneously processed in RNAlater Stabilization Reagent (QIAGEN). Total RNA was isolated using QIAshredder columns (QIAGEN) and RNeasy Mini Kit (QIAGEN). RNA concentration was estimated and equal amounts of RNA were used for reverse transcription followed by PCR amplification using the MyTaq One-Step RT-PCR kit (Bioline). Primers used for PCR were as follows: 4.1B, 5′-CTGGTTGGACCCTGCTAAGG-3′ and 5′-CTTGGCATGGTGTAAGTCC-3′; 4.1R, 5′-AAGAGTTTAGCGGCTGAAGC-3′ and 5′-CGGGCTTCTGGGAGGCTT-3′; and Actin, 5′-GCTCCGGCATGTGCAA-3′ and 5′-AGGATCTTCATGAGGTAGT-3′.
Immunoblotting and immunoprecipitation.
Sciatic nerves from littermate wild type and mutants of either sex were excised and crushed using a glass homogenizer in ice-cold lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mm EDTA, 1% Triton X-100, 1% SDS, and a protease mixture tablet). The lysate was incubated on ice for 30 min and centrifuged at 16,000 × g for 20 min at 4°C. The supernatant was used for protein estimation and immunoblotting.
Spinal cords from littermate wild type and mutants of either sex were excised and either directly processed or frozen at −80°C. Spinal cords were homogenized using a glass mortar and pestle on ice with 1 ml of ice-cold homogenization buffer (50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 320 mm sucrose, 10 mm EDTA, pH 8.0, and a protease inhibitor mixture tablet). The homogenate was centrifuged at 1000 × g for 10 min at 4°C. The supernatant was collected and subjected to additional centrifugation at 100,000 × g for 30 min at 4°C. The resulting pellet was then resuspended in lysis buffer (50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 10 mm EDTA, 1% Triton X-100, and a protease inhibitor mixture tablet) and incubated for 30 min at 4°C with occasional trituration followed by centrifugation at 100,000 × g for 30 min at 4°C and the supernatant was collected as the final lysate. Protein concentrations of the lysate were determined using the Lowry assay (BC assay; Bio-Rad).
For immunoprecipitation (IP), immobilized protein A-Sepharose beads (Thermo Fisher Scientific) were washed with PBS and incubated with either primary antibodies for 2–4 h at 4°C (antibody-bound beads) or 500 μg of total protein for 1–2 h at 4°C (preclear beads). The antibody-bound beads were washed once, and the lysate from the preclear beads was added and rotated overnight at 4°C. After brief centrifugation, the supernatant was collected as the post-IP material. The preclear and IP beads were washed three times with wash buffer (50 mm Tris-HCl, pH 7.5, 50 mm NaCl). Lysate, preclear beads, IP beads, and post-IP material were resolved by SDS-PAGE and transferred onto nitrocellulose membranes, followed by immunoblotting procedures described above.
Transmission electron microscopy.
Transmission electron microscopy of age-matched wild-type, β-Act-Cre;4.1BFlox, and 4.1B−/− mutant mice of either sex was performed essentially as described previously (Garcia-Fresco et al., 2006; Pillai et al., 2009; Thaxton et al., 2010).
Image analysis.
Confocal images were captured with a Bio-Rad Radiance 2000 laser-scanning system attached to a Zeiss Axioplan2 microscope. Scanning parameters were optimized for wild-type tissues and maintained for scanning the mutant tissues. The immunofluorescence images shown are Z stacks of four to eight sections with a scan step of 0.25 μm. Adobe Photoshop was used for assembling all figures.
Results
Developmental expression of 4.1B and generation of 4.1B-null mutants
Members of the 4.1 protein family are cytoskeletal adaptors that link membrane proteins to the underlying cytoskeleton (Peters et al., 1998; Hoover and Bryant, 2000). 4.1B, a member of this family, is expressed in the nervous system and is highly enriched at the paranodal and juxtaparanodal domains in myelinated axons (Ohara et al., 2000). Based on 4.1B subcellular localization and its binding with Caspr and Caspr2 (Ohara et al., 2000; Denisenko-Nehrbass et al., 2003), it was hypothesized that 4.1B links axoglial junctions or scaffolds with the axonal cytoskeleton. The primary structure of 4.1B contains a FERM domain, a spectrin/actin binding domain (SAB), a C-terminal domain (CTD), and three unique regions U1–U3 (Fig. 1A). We generated antibodies to the U3 unique region of 4.1B (Fig. 1A, black bar) and performed immunostaining of teased sciatic nerve fibers (PNS) and spinal cord sections (CNS) to establish the subcellular localization of 4.1B in the PNS and CNS (Fig. 1B,C). As shown in Figure 1B, 4.1B is enriched at the paranodes and juxtaparanodes (Fig. 1Ba, red arrows) and overlaps with the paranode-specific protein, Caspr (Fig. 1Bb). A merged image (Fig. 1Bc) clearly shows that 4.1B is enriched in the juxtaparanodal region (red arrow) and is not expressed at the nodes (Fig. 1Bc, arrow). Similarly, in the CNS, 4.1B is enriched at the paranodes and juxtaparanodes but is not present at the nodes (Fig. 1Ca–c). To determine when 4.1B expression begins during postnatal development, we performed immunoblot analysis of 4.1B from postnatal day 2 (P2) to P24 (Fig. 1D). This revealed that two major isoforms of 4.1B (145 and 125 kDa) are expressed in the spinal cord throughout early postnatal development. We immunoblotted the lysates against Caspr, which revealed that Caspr is also expressed throughout postnatal development, as has been reported previously (Bhat et al., 2001).
Figure 1.
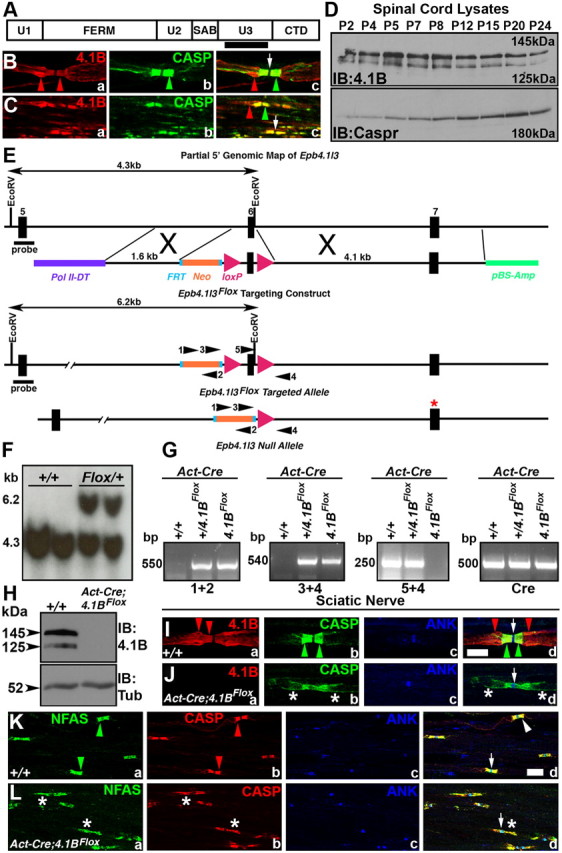
Primary structure and subcellular localization of 4.1B and generation of 4.1BFlox mice. A, Schematic of the primary structure of mouse 4.1B (deduced from GenBank accession number AF152247) (Parra et al., 2000) shows a FERM domain, a SAB domain, a CTD, and three unique (U1, U2, U3) domains that are specific to each 4.1 protein. The black bar represents a region in the U3 domain of 4.1B that was used to generate 4.1B antibodies. B, C, Wild-type teased sciatic nerve fibers (B) and spinal cord sections (C) immunostained with anti-4.1B (red) and Caspr (green) showing enrichment of 4.1B at the paranodes and the juxtaparanodes (Ba, Ca, red arrowheads) and Caspr at the paranodes (Bb, Cb, green arrowheads). Note that 4.1B extends beyond the paranodal area (Bc, Cc, red arrowheads). D, Immunoblots of wild-type spinal cord lysates showing developmental expression of 4.1B (top panel) and Caspr (bottom panel). E, A partial genomic map of the 4.1B (Epb4.1l3) locus showing exons 5–7. A targeting construct with loxP sites flanking exon 6. Cre-mediated recombination of the loxP sites resulted in the deletion of exon 6, which inserted a premature stop codon in exon 7 (red asterisk). F, Southern blot analysis confirming the targeted 4.1BFlox allele. G, PCR amplification using mouse tail genomic DNA from wild-type (+/+), heterozygous Floxed (+/4.1BFlox), homozygous Floxed (4.1BFlox), and β-Act-Cre-expressing (Act-Cre) mice. Primer combinations used are shown in E. H, Immunoblot analysis of wild-type (+/+) and β-Act-Cre;4.1BFlox (4.1BAct-Flox) spinal cord lysates using anti-4.1B (top panel) and Tub (bottom panel). Note the loss of both isoforms of 4.1B in 4.1BAct-Flox mutants (right lane). I, Immunofluorescence of teased sciatic nerve fibers from wild-type (+/+) mice (Ia–Id) showing typical localization of 4.1B at the paranodes/juxtaparanodes (Ia, Id, red arrows), Caspr at the paranodes (CASP, green) (Ib, Id, green arrows), and AnkG at the nodes (Ic, Id, blue, arrow). J, Immunofluorescence of teased sciatic nerve fibers from 4.1BAct-Flox mutants (Ja–Jd) showing loss of 4.1B (red) (Ja) with disrupted localization of Caspr in the paranodal region (Jb, green asterisks) but normal AnkG localization at the node (Jc, Jd, arrow). K, Immunofluorescence of teased sciatic nerve fibers from wild-type (+/+) mice (Ka–Kd) showing typical localization of NFAS (which recognizes both NF155 and NF186) at the paranodes (green arrowheads) and nodes (Ia, Id, green), Caspr at the paranodes (Kb, Kd, red), and AnkG at the nodes (Kc, Kd, blue, arrow). In Kd, the arrowhead points to merge of NFAS and CASP at the paranodes and the arrow points to the node. L, Immunofluorescence of teased sciatic nerve fibers from 4.1BAct-Flox mutants (Ja–Jd) showing disrupted NFAS (Ja, green, asterisks) and Caspr localization at the paranodal region (Jb, red, asterisks). AnkG localization at the node is not affected (Lc, Ld). Note that localization of both paranodal proteins NFAS and CASP is disorganized. Scale bars: I–L, 10 μm.
4.1B mouse mutants have been previously generated using conventional methods (Yi et al., 2005). As these presumed 4.1B-null mutants did not reveal any developmental or behavioral deficits, concerns were raised whether some 4.1B protein isoforms might still be expressed from the disrupted 4.1B locus. To address this issue, we took advantage of the detailed 4.1B genomic information to generate a floxed allele of 4.1B that would essentially ablate all isoforms of 4.1B on Cre-mediated recombination (Fig. 1E) (see Materials and Methods). A targeting construct was generated in which exon 6 (which encodes a portion of the FERM domain) was flanked with loxP sites (Fig. 1E) and deletion of exon 6 would cause a frameshift resulting in a premature stop codon in exon 7 (Fig. 1E, red asterisk). ES cell clones that carried a properly targeted 4.1B floxed allele were identified by Southern analysis (Fig. 1F). After germ line transmission, mice were genotyped using a combination of primers (Fig. 1E), which revealed various genotypes (Fig. 1G). Homozygous 4.1BFlox mice were born with expected Mendelian frequency and did not reveal any obvious behavioral or development deficits (data not shown). After Cre-mediated recombination using the ubiquitous β-Actin-Cre (Act-Cre), littermate mice with wild-type (+/+) or β-Act-Cre;4.1BFlox genotype were analyzed by immunoblotting. As shown in Figure 1H, spinal cord lysates from β-Act-Cre;4.1BFlox show complete loss of 145 and 125 kDa 4.1B protein isoforms, which were present in the wild-type lysates, indicating that 4.1B was ablated in β-Act-Cre;4.1BFlox mice. To ensure that 4.1B was also absent in the PNS myelinated axons, we performed immunostaining against 4.1B, paranodal Caspr, and nodal Ankyrin G (ANK) in teased sciatic nerves from littermate wild-type (+/+) and β-Act-Cre;4.1BFlox mice (Fig. 1I,J). As shown in Figure 1J, 4.1B immunoreactivity is completely absent in β-Act-Cre;4.1BFlox fibers (Fig. 1Ja, red; compare with Ia,d). Interestingly, the paranodal localization of Caspr in β-Act-Cre;4.1BFlox sciatic nerve fibers was altered, displaying punctate and stretched localization (Fig. 1Jb,d, green; compare with Ib,d). The nodal ANK was not affected (Fig. 1Jc,d, arrows; compare with Ic,d). To further confirm that this altered Caspr localization reflected a disorganization of other paranodal proteins, we immunostained β-Act-Cre;4.1BFlox sciatic nerve fibers against Neurofascin (NFAS) (which recognizes both NF155 and NF186), Caspr, and ANK. The wild-type fibers revealed a typical paranodal localization of NF155 (Fig. 1Ka, NFAS, green arrowheads) and Caspr (Fig. 1Kb, red arrowheads) at the paranodes and ANK (Fig. 1Kc,d, blue, arrow) at the nodes and all in the merged image (Fig. 1Kd). In β-Act-Cre;4.1BFlox sciatic nerve fibers, both Caspr and NF155 localization at the paranodes was disrupted, and their distribution stretched away from the paranodes (Fig. 1La,b,d, asterisks). The ANK localization was normal at the nodes (Fig. 1Lc,d, blue, arrow). Together, these results demonstrate that we have generated 4.1B-null mutants and that loss of 4.1B in β-Act-Cre;4.1BFlox mice affects the localization of paranodal proteins in myelinated axons. These results are in contrast to those of Horresh et al. (2010), who reported that paranodal proteins are not altered in 4.1B mutants (see below).
4.1B is required for the organization and stabilization of the paranodal and juxtaparanodal regions in myelinated axons
To determine the consequences of loss of 4.1B on axonal domain organization, we decided to analyze both 4.1B mutants [i.e., β-Act-Cre;4.1BFlox (this study) and 4.1B−/− generated previously (Yi et al., 2005)]. To date, no detailed phenotypic characterization of myelinated axons in any 4.1B mutants has been performed. In this study, we examined the localization of well characterized paranodal and juxtaparanodal proteins in teased sciatic nerve fibers and spinal cord sections (Figs. 2, 3). In wild-type (+/+) sciatic nerve fibers, 4.1B is enriched at the paranodes and juxtaparanodes and also localizes along the length of the internode (Fig. 2Aa, red). Caspr served as a marker for the paranodes (Fig. 2Ab, green), and the cytoskeletal adaptor protein AnkyrinG (AnkG) (Fig. 2Ac, blue) was used as a nodal marker. A merged image of all the markers is shown in Figure 2Ad (arrow points at the node). In 1-month-old β-Act-Cre;4.1BFlox sciatic nerves, 4.1B is absent (Fig. 2Ba), whereas AnkG (Fig. 2Bc) localization remains intact at the nodes. The Caspr localization is elongated and disrupted at the paranodes in β-Act-Cre;4.1BFlox sciatic nerves (Fig. 2Bb,d, asterisks). At 4 months, in β-Act-Cre;4.1BFlox sciatic nerves, 4.1B is absent, as expected (Fig. 2Ca), and AnkG (Fig. 2Cc) remains at the nodes. The Caspr localization at the paranodes in 4-month-old β-Act-Cre;4.1BFlox sciatic nerves is broken and occasionally stretches toward the juxtaparanodes. Additional examination of 1- and 4-month-old β-Act-Cre;4.1BFlox sciatic nerves revealed that all juxtaparanodal markers Kv1.1 (Fig. 2Ea,d;Fa,d), Kv1.2 (data not shown), PSD95 (data not shown), and Caspr2 (data not shown) were diffuse and failed to localize properly at the juxtaparanodes, compared with wild-type fibers (Fig. 2Da,d) (data not shown). In 1-month-old β-Act-Cre;4.1BFlox sciatic nerves, paranodal Caspr (Fig. 1Lb,d; 2Bb,d;Eb,d, asterisks) and NF155 (Fig. 1La,d, asterisks) localization were altered and showed diffuse distribution at the paranodes, often stretching into the juxtaparanodal areas. Nodal markers NF186 (Fig. 2Ec,d) and ANK (Figs. 1Lc,d; 2Bc,d) were not affected. In addition, Caspr (Fig. 2Cb,d;Fb,d;Hb,d, asterisks) and NF155 (Fig. 2Ha,d, arrowhead) localization at the paranodes in 4-month-old β-Act-Cre;4.1BFlox sciatic nerves were relatively more compact than at 1 month, but still contained broken and disrupted areas (Hd, arrowhead). These results indicate that loss of 4.1B affects juxtaparanodal organization and proper localization of axoglial junctional components at the paranodes (Table 1).
Figure 2.
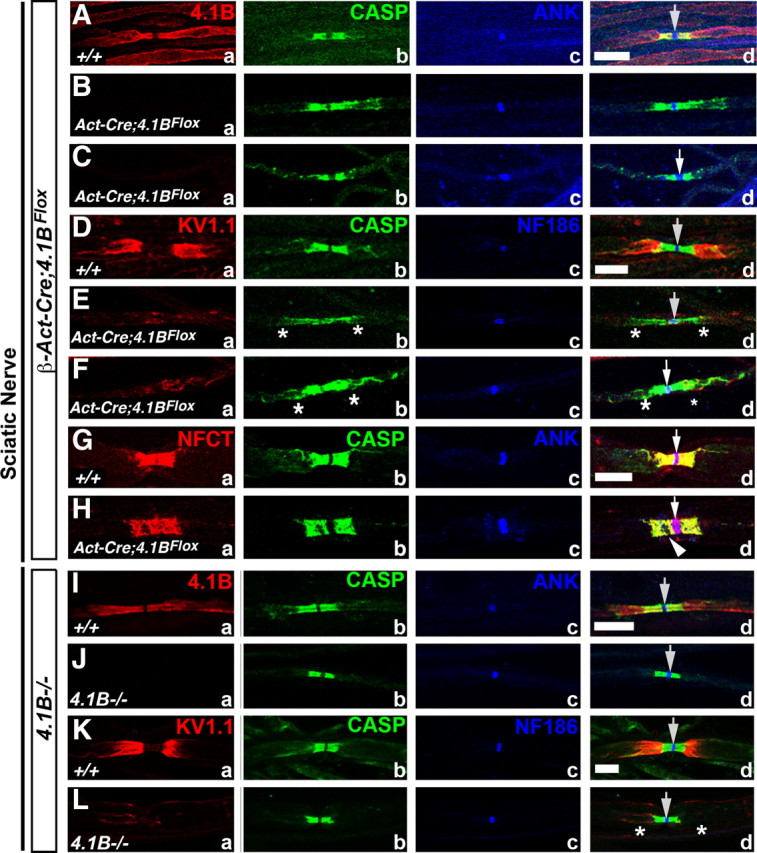
Loss of 4.1B disrupts paranodal and juxtaparanodal organization in the PNS myelinated axons. A–H, P30 wild-type (+/+) (Aa–Ad, Da–Dd, Ga–Gd), P30 β-Act-Cre;4.1BFlox (Ba–Bd, Ea–Ed), and 4-month-old β-Act-Cre;4.1BFlox (Ca–Cd, Fa–Fd, Ha–Hd) sciatic nerve fibers immunostained against 4.1B (Aa, Ba, Ca, red), Kv1.1 (Da, Ea, Fa, red), Caspr (Ab–Hb, green), pan-NFAS (Ga, Ha), AnkG (Ac–Cc, blue), NF186 (Dc–Hc, blue), and merged images (Ad–Hd). Note that 4.1B is absent from the mutant fibers (Ba, Ca) and Kv1.1 (Ea, Ed; Fa, Fd) is either undetectable at the juxtaparanodes or severely mislocalized. In all 4.1B mutant panels, Caspr (Bb, Bd; Cb, Cd; Eb, Ed; Fb, Fd; Hb, Hd, asterisks) and paranodal NF155 (NFAS) (Ha, Hd, arrowhead) fail to localize properly at the paranodes. Nodal AnkG or NF186 are not affected (Ac,d–Hc,d, arrows). I–L, P90 wild-type (+/+) (Ia–Id, Ka–Kd) and 4.1B−/− (Yi et al., 2005) (Ja–Jd, La–Ld) sciatic nerve fibers immunostained against 4.1B (Ia, Ja, red), Kv1.1 (Ka, La, red), Caspr (Ib–Lb, green), AnkG (Ic, Jc, blue), NF186 (Kc, Lc, blue), and merged images (Id–Ld). 4.1B is absent from the mutant fibers, as expected (Ja, Jd), and Kv1.1 is either undetectable at the juxtaparanodes or severely mislocalized (La, Ld, asterisks). However, Caspr localizes properly at the paranodes (Jb, Jd; Lb, Ld, asterisks). Nodal AnkG or NF186 are not affected (Jc, Jd; Kc, Kd; Lc, Ld, arrows). Scale bars, 10 μm (all).
Figure 3.
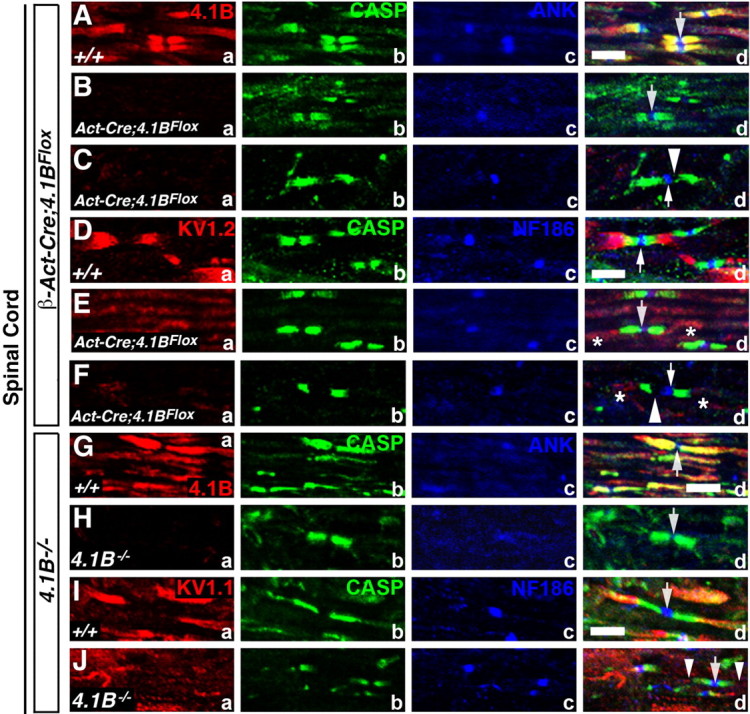
Mislocalization of juxtaparanodal components and destabilization of the paranodes in the CNS myelinated axons of 4.1B mutants. A–F, P30 wild-type (+/+) (Aa–Ad, Da–Dd), P30 β-Act-Cre;4.1BFlox (Ba–Bd, Ea–Ed), and 4-month-old β-Act-Cre;4.1BFlox (Ca–Cd, Fa–Fd) spinal cord sections immunostained against 4.1B (Aa, Ba, Ca, red), Kv1.2 (Da, Ea, Fa, red), Caspr (Ab–Fb, green), AnkG (Ac, Bc, Cc, blue), NF186 (Dc, Ec, Fc, blue), and merged images (Ad–Fd). Note that 4.1B is absent from the mutant fibers (Ba, Ca), and Kv1.1 (Ea, Ed; Fa, Fd) is either undetectable at the juxtaparanodes or severely mislocalized (Ed, Fd, asterisks). In P30 4.1BFlox mutants, Caspr localization in the CNS paranodes is not severely affected. In 4-month-old 4.1BFlox mutants, gaps form between the node and Caspr localization (Cd, Fd, arrowheads). G–J, P90 wild-type (+/+) (Ga–Gd, Ia–Id) and 4.1B−/− (Ha–Hd, Ja–Jd) spinal cord sections immunostained against 4.1B (Ga, Ha, red), Kv1.1 (Ia, Ja, red), Caspr (Gb–Jb, green), AnkG (Gc, Hc, blue), NF186 (Ic, Jc, blue), and merged images (Gd–Jd). Note that 4.1B is absent as expected (Ha) and Kv1.1 (Ja, Jd) is either undetectable at the juxtaparanodes or severely mislocalized (Jd, arrowheads). Caspr localization in the CNS paranodes in 4.1B−/− is not severely affected. Scale bars, 5 μm (all).
Table 1.
Phenotypic differences between two independently generated 4.1B mutant alleles
| Phenotype | β-Act-Cre;4.1BFloxa | 4.1B−/−b |
|---|---|---|
| Sciatic nerve | ||
| Paranodal proteins | Abnormal localizationc | Normal localizationd |
| Paranodal AGSJs | Missing transverse septa | No missing transverse septa |
| Destabilized AGSJs | Normal AGSJs | |
| Juxtaparanodal proteins | Abnormal localization | Abnormal localization |
| Spinal cord | ||
| Paranodal proteins | Abnormal localizatione | Normal localizationf |
| Paranodal AGSJs | Missing transverse septa | Missing transverse septa |
| Destabilized AGSJs | Destabilized AGSJs | |
| Juxtaparanodal proteins | Abnormal localization | Abnormal localization |
| 4.1R localization | Not detected at the paranodes | Not detected at the paranodes |
| 4.1R mRNA levels in DRGs | No change | No change |
a4.1B mutants generated in this study.
b4.1B mutants generated by Yi et al. (2005).
cParanodal proteins Caspr and NF155 show abnormal localization with stretched morphology and gaps.
dParanodal proteins Caspr and NF155 show normal localization and morphology.
eParanodal protein Caspr shows normal localization at P30, but at P130 the paranodal areas show gaps and abnormal morphology.
fParanodal protein Caspr shows relatively normal localization and morphology.
Next, we examined localization of paranodal Caspr, juxtaparanodal KV1.1, and nodal ANK in the sciatic nerves of 3 month 4.1B−/− mice (Yi et al., 2005). As expected, 4.1B is absent at the paranodes and juxtaparanodes in 4.1B−/− mutant fibers (Fig. 2Ja, red). Interestingly, as observed by Horresh et al. (2010), Caspr (Fig. 2Jb,d;Lb,d, green) localization at the paranodes was not affected in 4.1B−/− mutant fibers. KV1.1 localization at the juxtaparanodes is compromised in 4.1B−/− mutants (Fig. 2La,d, red, asterisks), as is also seen in β-Act-Cre;4.1BFlox mutants (Fig. 2Ea,d;Fa,d, red). In addition, Kv1.2, Caspr2, and PSD95 were similarly mislocalized from the juxtaparanodes (data not shown). These data indicate that, in 4.1B−/− mutants, localization of the juxtaparanodal proteins is compromised, whereas the paranodal components are not affected (Table 1).
We also analyzed the localization of paranodal and juxtaparanodal proteins in the spinal cords of β-Act-Cre;4.1BFlox and 4.1B−/− mutants. In wild-type (+/+) spinal cord sections, 4.1B is enriched at the paranodes and the juxtaparanodes (Fig. 3Aa, red). In addition, Caspr (Fig. 3Ab,Db, green), AnkG (Fig. 3Ac, blue), and NF186 (Fig. 3Dc, blue) are localized at the paranodes and nodes, respectively (Fig. 3Ad,Dd). As in the PNS, 4.1B is absent in 1- and 4-month-old β-Act-Cre;4.1BFlox spinal cord myelinated axons (Fig. 3Ba,Ca, red). However, the paranodal localization of Caspr (Fig. 3Bb,d;Eb,d, green) was not disrupted in 1-month-old β-Act-Cre;4.1BFlox spinal cord axons. Interestingly, in 4-month-old β-Act-Cre;4.1BFlox spinal cord myelinated axons, Caspr (Fig. 3Cb,d;Fb,d, green) localization was not normal. There were gaps between paranodal Caspr and the nodal proteins AnkG and NF186 (Fig. 3Cc,d;Fc,d, arrowheads). Again, the juxtaparanodal Kv1.2 was mislocalized in both 1- and 4-month-old β-Act-Cre;4.1BFlox mutant spinal cord axons (Fig. 3Ea,d;Fa,d, red, asterisks). AnkG and NF186 were both maintained at the nodes in all β-Act-Cre;4.1BFlox mutants (Fig. 3Bc,d;Cc,d;Ec,d;Fc,d, respectively). Analysis of 4.1B−/− mutant spinal cord axons also revealed absence of 4.1B in 4.1B−/− mutants (Fig. 3Ha, red). Paranodal Caspr localization remained relatively unaffected (Fig. 3Hb,d;Jb,d, green). The juxtaparanodal Kv1.1 was mislocalized (Fig. 3Ja,d, red, arrowheads). Nodal AnkG (Fig. 3Hc,d, blue) and NF186 (Fig. 3Jc,d, blue) were not affected. The juxtaparanodal proteins Kv1.2, PSD95, and Caspr2 were similarly mislocalized in the spinal cords of both 4.1B mutants (data not shown). Together, the immunohistochemical results indicate that 4.1B is critical for proper organization of the juxtaparanodal region in both the PNS and the CNS, and that 4.1B is required for the stabilization of the paranodes in the PNS, and progressively in the CNS (Table 1).
Molecular interactions between paranodal Caspr and complex formation between juxtaparanodal Caspr2, Kv1.2, and 4.1B
To further investigate the striking phenotypic differences between the PNS and CNS myelinated fibers, we immunoblotted sciatic nerve and spinal cord lysates from wild type (+/+), 4.1B−/−, and β-Act-Cre;4.1BFlox mutants (Fig. 4A,B). Surprisingly, we discovered that the wild-type sciatic nerves express close to eight isoforms of 4.1B, some with molecular weights exceeding 180 kDa, in contrast to the CNS, where only two major isoforms (145 and 125 kDa) are expressed (Fig. 4, compare Aa, Ba). All of these 4.1B isoforms were absent in both 4.1B mutants (Fig. 4Aa,Ba). Immunoblotting against Caspr did not reveal any significant changes in the relative Caspr levels between wild-type and 4.1B mutants (Fig. 4Ab,Bb). In addition, immunoblotting against juxtaparanodal proteins Kv1.2 and Caspr2 did not reveal any significant differences in their levels between the wild-type and 4.1B mutant sciatic nerves (Fig. 4Ac,d). For protein loading control, membranes were immunoblotted against Tub (Fig. 4Ae,Bc). We also wanted to determine whether disruption of the paranodal axoglial junctions in Caspr (Bhat et al., 2001) and Cgt (Dupree et al., 1999) mutants affected 4.1B protein levels in the spinal cords. As shown in Figure 4Ca, 4.1B protein levels are relatively unchanged between wild-type and the paranodal mutants. Similarly, Caspr levels are not affected in 4.1B and Cgt mutant spinal cords (Fig. 4Cb). For protein loading control, membranes were immunoblotted against α-Tubulin (Fig. 4Cc). These data show that sciatic nerves express multiple forms of 4.1B and that loss of 4.1B or Caspr only affects the subcellular localization of Caspr and 4.1B, respectively, and that stable expression of each of these proteins is not dependent on the other paranodal proteins.
Figure 4.

Multiple 4.1B isoforms in sciatic nerves and molecular interactions between 4.1B, Caspr, and Caspr2. A, Immunoblot analysis of wild-type (+/+), 4.1B−/−, and β-Act-Cre;4.1BFlox (4.1BAct-Flox) sciatic nerve lysates against 4.1B (Aa), Caspr (Ab), Caspr2 (Ac), Kv1.2 (Ad), and Tub (Ae) reveal expression of approximately eight isoforms of 4.1B in the sciatic nerves, which are absent in both 4.1B mutants. Caspr levels are unchanged in 4.1B mutants. B, Immunoblot analysis of wild-type (+/+), 4.1B−/−, and β-Act-Cre;4.1BFlox (4.1BAct-Flox) spinal cord lysates against 4.1B (Ba), Caspr (Bb), and Tub (Bc) reveal expression of two major isoforms in the CNS, which are absent in both 4.1B mutants. Note that Caspr levels are not affected in 4.1B mutants. C, Immunoblot analysis of wild-type (+/+), 4.1B−/−, Caspr−/−, and Cgt−/− spinal cord lysates against 4.1B (Ca), Caspr (Cb), and Tub (Cc) showed that the levels of 4.1B and Caspr are not affected in Caspr and 4.1B mutants, respectively. Cgt mutants also did not show any changes in 4.1B expression. D, IP analysis using anti-Caspr antibodies (Da) with wild-type (+/+) spinal cord lysates co-IPs 4.1B (Db). E, IP analysis using anti-Caspr2 antibodies with wild-type (+/+) (Ea, Ea′), β-Act-Cre;4.1BFlox (4.1BAct-Flox) (Eb, Eb′), and 4.1B−/− (Ec, Ec′) spinal cord lysates co-IPs Kv1.2 in all the genotypes, suggesting that Caspr2 interactions with Kv1.2 are independent of 4.1B.
Our immunohistochemical data showed that 4.1B is required for the localization of potassium channels Kv1.1 and Caspr2 to the juxtaparanodal region (Figs. 2, 3). Previous studies revealed that a possible mechanism for potassium channel retention at the juxtaparanodal region could be attributable to molecular interactions between Caspr2 and potassium channels (Poliak et al., 2003; Horresh et al., 2010). We therefore examined whether 4.1B is required for Caspr2 interactions with potassium channels. It is established that 4.1B interacts with Caspr (Denisenko-Nehrbass et al., 2003; Garcia-Fresco et al., 2006; Ogawa et al., 2006) and Caspr2 (Denisenko-Nehrbass et al., 2003; Horresh et al., 2010). As shown in Figure 4D, a and b, immunoprecipitation with anti-Caspr antibodies using spinal cord lysates coprecipitated 4.1B. Additionally, in the wild-type spinal cord lysates, IPs with anti-Caspr2 antibodies coprecipitated potassium channels Kv1.2 (Fig. 4Ea,a′). Caspr2 and Kv1.2 still formed a molecular complex in the absence of 4.1B in β-Act-Cre;4.1BFlox (Fig. 4Eb,b′) and 4.1B−/− (Fig. 4Ec,c′) spinal cord lysates. These results suggest that 4.1B is critical for the maintenance of the juxtaparanodal complex of Caspr2 and potassium channels and not the formation of this molecular complex. Together, our results demonstrate that 4.1B is a critical component of the paranodal and juxtaparanodal domains and that expression of multiple isoforms in the PNS versus the CNS may allow for differential function of 4.1B in axonal domain organization.
4.1B is critical for the stability of the paranodal axoglial septate junctions
Our immunofluorescence studies revealed striking juxtaparanodal disorganization in both the PNS and the CNS in β-Act-Cre;4.1BFlox and 4.1B−/− mutants (Figs. 2, 3). In addition, we found severe paranodal destabilization in the PNS, and progressive paranodal disruption in the CNS, of β-Act-Cre;4.1BFlox mice (Fig. 3). To further investigate the role of 4.1B in axonal domain organization, we used electron microscopy to specifically look at the ultrastructural architecture of the paranodal region in wild-type, β-Act-Cre;4.1BFlox, and 4.1B−/− mutant sciatic nerve and spinal cord myelinated axons (Figs. 5, 6). A low-magnification electron micrograph of P30 littermate wild-type (+/+) (Fig. 5A) and β-Act-Cre;4.1BFlox mutant (Fig. 5B) sciatic nerve myelinated axons, at the level of the nodal/paranodal region, shows a typical domain distinction between the nodal and the paranodal region. At this magnification, the wild-type and β-Act-Cre;4.1BFlox mutant fibers look essentially similar with respect to the presence of individual domains. At a higher magnification, the paranodal AGSJs show characteristic electron-dense septa between the myelin loops and the axolemma in wild-type fibers (Fig. 5C, arrowheads). In 1-month-old β-Act-Cre;4.1BFlox mutant fibers, a few electron-dense septa can be found (Fig. 5D, arrowhead), whereas most septa appear diffuse and ill defined (Fig. 5D, asterisks), suggesting that septa may be undergoing destabilization. In 3-month-old β-Act-Cre;4.1BFlox mutant fibers, the electron density of the septa is diffuse and fuzzy (Fig. 5E,F, asterisks). In comparison, ultrastructural analysis of previously generated 4.1B−/− mutants revealed no clear differences in the ultrastructural morphology of the paranodal axoglial septa at 3 months (Fig. 5H; compare with G, arrowheads). However, at 6 month and 1 year time points, septa were missing in the sciatic nerve paranodal regions (data not shown) in 4.1B−/− mutants. Quantification of the percentage of paranodes with a normal array of septa in the sciatic nerve of each genotype at 3 months revealed that, in the wild-type mice, 90% of paranodes contain a full complement of septa. In sciatic nerve fibers of 3-month-old β-Act-Cre;4.1BFlox mice, only 60% of the paranodes counted retained a full complement of septa, whereas 75% of paranodes in 3-month-old 4.1B−/− sciatic nerves contained a normal set of septa. In β-Act-Cre;4.1BFlox mice, paranodes often displayed severe morphological defects, which occluded visualization of missing septa suggesting that paranodal disorganization was more severe than were included in the quantification. Together, these results are consistent with our immunostaining results (Fig. 2), which showed disrupted localization of the paranodal proteins in β-Act-Cre;4.1BFlox mutant sciatic nerve fibers.
Figure 5.
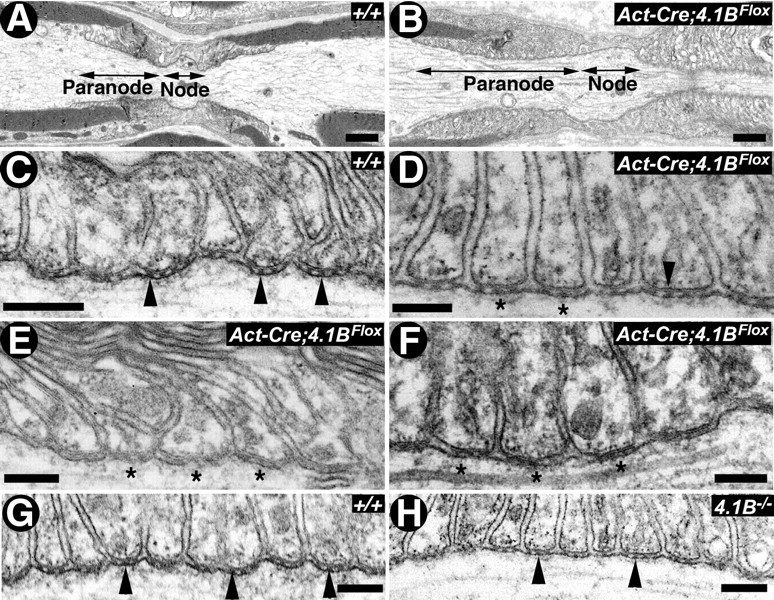
4.1B is required for the stability of paranodal axoglial septate junctions in sciatic nerve myelinated axons. A–D, Electron micrographs of longitudinal sections through the nodal/paranodal regions of sciatic nerves from P30 wild type (A, C) (+/+) and β-Act-Cre;4.1BFlox (B, D). At low magnification, the paranodal myelin loops in the β-Act-Cre;4.1BFlox mutant fibers are properly arrayed and show normal profiles, as in the wild type. At a higher magnification, the wild-type (+/+) paranodal loops are tightly apposed and indent the axon, with the characteristic transverse septa (C, black arrowheads). In β-Act-Cre;4.1BFlox mutant fibers, the paranodal septa display a fuzzy, ill defined appearance (D, asterisks). E, F, Electron micrographs of paranodal regions of P90 β-Act-Cre;4.1BFlox sciatic nerves. Septa are less electron-dense than wild-type septa, a sign of destabilization (asterisks). G, H, Electron micrographs of longitudinal sections through the paranodal regions of sciatic nerves from P90 wild type (J, +/+) and 4.1B−/− generated by Yi et al. (2005) (K). The paranodal septa in 4.1B−/− mutants display similar morphology and density as is seen in the wild-type (+/+) axons. Scale bars: A, B, 1 μm; C–H, 0.2 μm.
Figure 6.
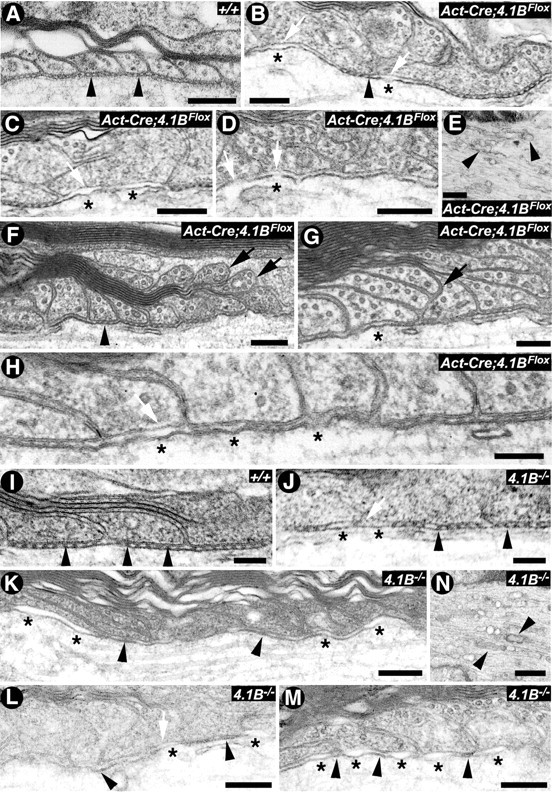
4.1B is required for the stability of paranodal axoglial septate junctions in the spinal cord myelinated axons. A–D, Electron micrographs of the paranodal regions of P30 spinal cord myelinated fibers from the wild type (+/+) show paranodal loops tightly apposed to and indent the axon, with the characteristic septa (A, black arrowheads). In β-Act-Cre;4.1BFlox mutant spinal cords, the paranodal region displays regions with normal septa (B, arrowheads) and regions in which septa are essentially absent (B–D, asterisks). The white arrows point to detached axolemma from the paranodal loops (B–D) or breaks in the axolemma (D). E, β-Act-Cre;4.1BFlox mutant myelinated axons often display accumulation of SER and mitochondria at the nodal/paranodal region, as has been previously observed in Caspr−/− mutants. F–H, Electron micrographs of the paranodal regions of spinal cord myelinated fibers from P90 β-Act-Cre;4.1BFlox mutants show lifting off of paranodal loops (F, G, arrows), stretches of missing septa (G, H), and detachment of the axolemma (G, H, asterisks), whereas some septa also remain at this time point (F, arrowhead). I–M, Electron micrographs of the paranodal regions of spinal cord myelinated fibers from P90 wild type (I) (+/+), P90 4.1B−/− mutants (J), P180 4.1B−/− mutants (K), and P365 4.1B−/− mutants (L, M). The spinal cords of P90 4.1B−/− mutants show paranodal regions with normal septa (arrowheads) and regions with abnormal or missing septa (J, K, asterisks). In P365 4.1B−/− spinal cords, majority of the paranodes did not display septa with normal morphology, but displayed diffuse electron densities (L, arrowheads). Large regions of the paranodes had essentially lost all the septa (M, asterisks) and displayed thinner axolemma. N, 4.1B−/− myelinated axons from 1-year-old mutants also displayed accumulation of SER and mitochondria at the nodal/paranodal regions (arrowheads), as observed in P30 β-Act-Cre;4.1BFlox mutants (see E). Scale bars: A–D, 0.2 μm; E, N, 0.5 μm; F–H, 0.1 μm; I–M, 0.2 μm.
Next, we analyzed the spinal cord myelinated axons in 1- and 3-month-old wild type and β-Act-Cre;4.1BFlox mutants. As shown in Figure 6A, wild-type spinal cord myelinated axons display a uniform arrangement of septa between the myelin loops and the axolemma (Fig. 6A, arrowheads). The fibers from 1-month-old β-Act-Cre;4.1BFlox mutant spinal cords revealed the presence of axoglial septa in some regions (Fig. 6B, arrowhead) and complete loss of septa in other regions at the same paranodes (Fig. 6B–D, asterisks). In the areas in which septa are missing, the axolemma showed clear detachments from the myelin loops and loss of electron densities (Fig. 6B–D, white arrows). At 3 months, some electron densities are still found in β-Act-Cre;4.1BFlox mutant spinal cords (Fig. 6F, arrowhead). However, many paranodal loops were detached from the axonal membrane (Fig. 6F,G, arrows). In addition, larger areas were found with missing septa (Fig. 6H, white arrow), and detached axolemma (Fig. 6G,H, asterisks). Detachment of paranodal loops abutting the node agrees with immunohistochemical data revealing gaps between the nodal proteins and Caspr in 4-month-old β-Act-Cre;4.1BFlox mutant spinal cords (Fig. 3Cd,Fd, arrowheads). The β-Act-Cre;4.1BFlox mutant fibers also revealed accumulation of smooth endoplasmic reticulum (SER) at the nodal/paranodal regions (Fig. 6E), suggesting a disruption of the local axonal cytoskeleton, as has been previously observed in Caspr, Cgt (Einheber et al., 2006; Garcia-Fresco et al., 2006), and NfascNF155 (Pillai et al., 2009) mutants. These data suggest that 4.1B is not required for the initial organization of the paranodal axoglial septa, but for their stabilization.
To determine the paranodal defects in the spinal cord myelinated axons in 4.1B−/− mutants, we analyzed them at P90, 6 month, and 1 year time points. As shown in Figure 6I, P90 wild-type spinal cord myelinated axons showed proper densities and arrangement of the septa (arrowheads). In contrast, P90 4.1B−/− mutants showed myelinated axons with normal septa (Fig. 6J, arrowheads) and regions with abnormal or missing septa (Fig. 6J, asterisks, white arrow). By 6 months, 4.1B−/− mutant myelinated axons revealed a more prominent loss of septa with increased spacing between the paranodal lateral loops and the axolemma, indicating a progressive deterioration of the paranode with age (Fig. 6K, asterisks). In these fibers, destabilization of septa becomes obvious as the densities of these fibers looked diffuse (Fig. 6K, arrowheads). Analysis of 1-year-old 4.1B−/− mutants spinal cord myelinated axons revealed additional destabilization of the paranodal regions, with essentially all septa lost and the axolemma detaching from the myelin loops (Fig. 6L,M, asterisks). The 1-year-old 4.1B−/− mutant spinal cord myelinated axons also revealed accumulation of organelles, including mitochondria and SER at the nodal/paranodal regions, suggesting a disruption of the axonal cytoskeleton, as observed in the sciatic nerves (Fig. 6N, arrowheads) (data not shown). To directly compare the phenotypes in the two 4.1B-null mice, the number of paranodes with a complete set of septa at 3 months were quantified. In wild-type spinal cords, 94% of paranodes contain a full set of septa. In β-Act-Cre;4.1BFlox spinal cords, only 36% of the paranodes retain a complete set of septa, whereas 50% of the paranodes in 4.1B−/− spinal cords maintain a complete set of septa. Interestingly, in the spinal cord of β-Act-Cre;4.1BFlox mice, loss of septa resulted in destabilization of paranodal loops, especially in the region abutting the node (Fig. 6F). Quantification revealed that 72% of paranodes in wild-type spinal cords contain a complete set of loops contacting the paranodal axolemma. In the β-Act-Cre;4.1BFlox spinal cords, only 33% of paranodes maintained loops in contact with the axolemma. In comparison, in 4.1B−/− spinal cords, 71% of paranodes retained a complete set of loops contacting the axolemma. These data suggest that loss of 4.1B in β-Act-Cre;4.1BFlox spinal cords results in destabilization of the septa and lifting of the paranodal loops from the axolemma. Together, the ultrastructural analyses in β-Act-Cre;4.1BFlox and 4.1B−/− mutants demonstrate that 4.1B is critical for the stabilization of paranodal AGSJs, and that the long-term stability of the AGSJs is dependent on 4.1B, as it provides a scaffolding link with the paranodal axonal cytoskeleton (Table 1).
4.1R is not expressed at the PNS paranodes or enriched in 4.1B−/− mutants
One of the underlying reasons suggested to explain a lack of phenotype at the PNS paranodes in 4.1B−/− mutants was that 4.1R was expressed at the mutant paranodes and not at the wild-type paranodes (Horresh et al., 2010). We wanted to determine whether β-Act-Cre;4.1BFlox mutant sciatic nerves also had increased 4.1R expression at the paranodes, and as controls we used 4.1B−/− mutants. Immunostaining of wild-type (+/+), β-Act-Cre;4.1BFlox, and 4.1B−/− sciatic nerve fibers against 4.1R did not reveal expression of 4.1R at paranodes in any of the genotypes analyzed (Fig. 7Aa,c–Fa,c, red, arrowheads). Caspr localization was normal in the wild-type and 4.1B−/− mutant fibers and disrupted in β-Act-Cre;4.1BFlox mutant fibers (data not shown). These data show that 4.1R is not expressed at the paranodes and thus cannot possibly compensate for loss of 4.1B at the paranodes. To test the specificity of our 4.1R antibody, we performed immunoblot analysis of sciatic nerves from wild-type and 4.1R−/− mice and confirmed that 4.1R was missing in 4.1R−/− sciatic nerves (Fig. 7G) (Shi et al., 1999). We also performed immunoblot analysis of sciatic nerves and spinal cords from littermate wild type and β-Act-Cre;4.1BFlox and 4.1B−/− mutants. As shown in Figure 7, H and I, no significant differences, and especially no increases, were observed in 4.1R levels between wild type and both 4.1B mutants. Immunoblotting against Tub was used as a loading control. To confirm these results, we also completed RT-PCR with RNA extracted from dorsal root ganglia of littermate wild type and β-Act-Cre;4.1BFlox and 4.1B−/− mutants (Fig. 7J). Primers targeted to amplify DNA from exons 5 through 8 were used to show deletion of exon 6 in the β-Act-Cre;4.1BFlox mice (Fig. 7J). The genotype of 4.1B−/− mutants were confirmed using primers designed to exons 2 through 3, resulting in loss of the band in this mutant only (data not shown). Importantly, 4.1R levels were uniform in all genotypes (Fig. 7J). Actin was used as a housekeeping gene control. Together, the immunostaining, immunoblot, and RT-PCR analyses in two independent 4.1B mutants show that 4.1R is not expressed at the paranodes in the wild-type fibers and that 4.1R does not get enriched at 4.1B mutant paranodes (Table 1).
Figure 7.

4.1R is not enriched at sciatic nerve paranodes in 4.1B mutants. A–F, P30 wild-type (+/+) (Aa–Ac, Ca–Cc, Ea–Ec), β-Act-Cre;4.1BFlox (Act-Cre;4.1BFlox) (Ba–Bc, Da–Dc), and 4.1B−/− (Fa–Fc) sciatic nerves immunostained against 4.1R (Aa,c–Fa,c, red, arrowheads) and NF186 (Ab–Fb, green) and in merged images (Ac–Fc). Note that 4.1R is not expressed or enriched at mutant paranodes in any of the genotypes. G, Immunoblot analysis of wild-type (+/+) and 4.1R−/− sciatic nerve lysates against 4.1R (Ga) and Tub (Gb) reveal the specificity of the 4.1R antibody with the 80 kDa 4.1R present in wild-type lysate, but not 4.1R−/− lysate (Ga). H, Immunoblot analysis of wild-type (+/+), β-Act-Cre;4.1BFlox (4.1BAct-Flox), and 4.1B−/− sciatic nerve lysates against 4.1R (Da) and Tub (Db) reveal no change in 4.1R levels in either of the 4.1B mutants, compared with wild type. I, Immunoblot analysis of wild-type (+/+), β-Act-Cre;4.1BFlox (4.1BAct-Flox), and 4.1B−/− spinal cord lysates against 4.1R (Ia) and Tub (Ib) reveal no change in 4.1R expression in both 4.1B mutants (asterisks), compared with wild type. J, RT-PCR analyses of wild-type (+/+), β-Act-Cre;4.1BFlox (Act-Cre;4.1BFlox), and 4.1B−/− dorsal root ganglia using primers specific to 4.1B, 4.1R, and Actin (Act) reveal no change in the levels of 4.1R mRNA in either of the 4.1B mutants, compared with wild type. Scale bars, 10 μm.
Discussion
The organization of specialized membrane environments that create cellular diversification depends on linkage of cellular membranes with the underlying cytoskeleton (Jekely, 2007). The 4.1 protein family functions to link the plasma membrane with the actin cytoskeleton (Cohen and Foley, 1982; Conboy, 1993; Hoover and Bryant, 2000; Sun et al., 2002; Chen et al., 2005). In the nervous system, the cell adhesion molecules Caspr and Caspr2 contain 4.1-binding sites, which are required for their localization to the paranodes and juxtaparanodes, respectively (Poliak et al., 2003; Traka et al., 2003; Horresh et al., 2010). When Caspr expression is genetically ablated, AGSJs fail to form, leading to loss of axonal domain segregation and disrupted paranodal axonal cytoskeleton (Bhat et al., 2001; Garcia-Fresco et al., 2006), suggesting that paranodal AGSJs are linked to axonal cytoskeleton. In this study, phenotypic analyses of 4.1B mutants showed that 4.1B is required for the stability of paranodal AGSJs in myelinated axons.
Isoform-specific functions of 4.1B in PNS and CNS myelinated axons
Phenotypic analysis of β-Act-Cre;4.1BFlox mutant sciatic nerves showed paranodal defects as early as P30. The CNS paranodal defects at P30 were relatively mild, but at P90 transverse septa were often lost and paranodal loops were coming off the axolemma, indicating an important role for 4.1B in AGSJ stabilization. Phenotypic analysis of the previously reported 4.1B mutants (Yi et al., 2005) did not show disorganized Caspr localization or loss of paranodal septa in the PNS at P90, but revealed progressive destabilization of CNS paranodal septa at later stages of development. These variable phenotypes between 4.1B mutants could be attributed to the nature of 4.1B mutations, their genetic backgrounds, or the multiple splice isoforms of 4.1B. Strain genetic backgrounds could be a contributing factor to phenotypic discrepancies between the two 4.1B mutants. The 4.1B−/− mice are on a C57BL/6 background, whereas the β-Act-Cre;4.1BFlox mice are on a mixed background of C57BL/6 and 129SvEv. β-Act-Cre;4.1BFlox mice show a consistent phenotype throughout all mixed lines. We also examined the localization of 4.1B, Caspr, AnkG, Kv1.2, and NF186 in each background strain and did not observe any disruptions in the localization of these proteins or the morphology of the paranodes (data not shown). These results suggest that the paranodal phenotypes observed in β-Act-Cre;4.1BFlox mice are attributable to loss of 4.1B and not attributable to genetic background of the strains used.
In agreement with previous studies that showed two 4.1B isoforms in brain lysates (145 and 125 kDa) (Denisenko-Nehrbass et al., 2003), we also observed the same isoforms in the spinal cord. Differential alternative splicing in the PNS versus CNS may allow 4.1B to function in a tissue-specific manner. The expression of close to eight different isoforms in the PNS may explain the variation in phenotype seen between PNS and CNS, as well as the differences between β-Act-Cre;4.1BFlox and 4.1B−/− mutants. The phenotypic differences may lie in the nature of the 4.1B mutations, as specific isoforms may play different roles in domain organization and stability in the CNS and the PNS. In 4.1B−/− mutants, exon 3 was disrupted by insertion of a puromycin resistance cassette (Yi et al., 2005). It is possible that some compensatory isoforms or splice variants remain in 4.1B−/− mutants, which may not be detected by our antibodies directed to the U3 region of 4.1B, or by antibodies directed to the U2 region of 4.1B (Kang et al., 2009) (data not shown). We believe that our targeted deletion of a common exon 6, which encodes the middle portion of the FERM domain, generates a 4.1B-null mutation. Additional evidence for a 4.1B-null mutation lies in the paranodal disorganization phenotype displayed by β-Act-Cre;4.1BFlox compared with that observed in 4.1B−/− mutants, even at 1 year of age. In addition, our immunohistochemical, immunoblot, and RT-PCR analyses did not reveal compensatory expression of 4.1R at the paranodes, or an increase in 4.1R in the sciatic nerves of 4.1B mutants, as was reported by Horresh et al. (2010). Together, our data demonstrate that 4.1B is critically required for the stabilization of the paranodal AGSJs and the juxtaparanodal axoglial scaffolds in myelinated axons.
Organization of axonal domains and localization of 4.1B
A key question that remains unresolved is whether 4.1B is required in the formation of paranodal and juxtaparanodal domains, or for their stability. In the absence of 4.1B, paranodal AGSJs are formed (Figs. 5, 6), suggesting that 4.1B is not required for the formation, but for the stability of AGSJs (Bhat et al., 2001; Denisenko-Nehrbass et al., 2003; Poliak et al., 2003; Horresh et al., 2010). Furthermore, disruption of paranodal AGSJs in Caspr (data not shown) and Contactin (Gollan et al., 2002) mutants results in diffuse localization of 4.1B along the axon, suggesting that 4.1B requires AGSJs to become enriched at these domains. Paranodal localization of 4.1B may also be aided through its interactions with αII and βII spectrin, which are enriched at the paranodal region (Garcia-Fresco et al., 2006; Ogawa et al., 2006). These observations indicate that paranodal and juxtaparanodal proteins initially cluster independently of 4.1B, and once clustered through axon–glial interaction mechanisms, 4.1B interactions ensure that these axoglial adhesion complexes are stabilized.
Role of axonal cytoskeleton in stabilizing axon–glial interactions and septate junctions
The molecular identities of the complexes responsible for stabilizing axonal domains over the life of the axon are not well understood. At the paranodes, AGSJs are organized by interactions between Caspr, Cont, and NfascNF155 (Bhat et al., 2001; Boyle et al., 2001; Pillai et al., 2009). Loss of Caspr leads to disorganization of the paranodal region and axonal cytoskeleton, suggesting that AGSJs are linked to the axonal cytoskeleton (Garcia-Fresco et al., 2006; Ogawa et al., 2006). Paranodal disorganization in β-Act-Cre;4.1BFlox mutants provides additional evidence that AGSJs are linked to axonal cytoskeleton and that 4.1B is required to ensure stability of the paranodal region. Support for the role of 4.1B as a cytoskeletal adaptor protein comes from transgenic and rescue studies that show Caspr lacking its 4.1-binding domain is able to cluster at paranodes but fails to establish a barrier to prevent juxtaparanodal components from entering paranodal space (Horresh et al., 2010). Furthermore, Caspr with a deleted C-terminus can reach, but fails to stabilize at paranodes, suggesting that the C-terminus is essential for Caspr stabilization (Gollan et al., 2002). Protein 4.1B likely stabilizes the paranodes through direct interactions with Caspr and other cytoskeletal proteins such as AnkB and αII- and βII-spectrin (Garcia-Fresco et al., 2006; Ogawa et al., 2006). Interestingly, domain disorganization in 4.1B mutants did not result in conduction velocity differences in P30 wild-type, β-Act-Cre;4.1BFlox, or 4.1B−/− sciatic nerve fibers (data not shown). In addition, neither 4.1B mutants display obvious behavioral deficits, as of 4 months of age. However, precise electrophysiological recordings of individual axons and behavioral tests for motor function could reveal deficits in older 4.1B mutants.
The juxtaparanode is established by interactions between Caspr2 and TAG-1 (Poliak et al., 2003; Traka et al., 2003). In this region, no axoglial junctions are formed, but a loose axoglial scaffold is established that moves toward the paranode in paranodal mutants (Dupree et al., 1999; Bhat et al., 2001; Boyle et al., 2001; Pillai et al., 2009). These findings suggest that the juxtaparanode is not anchored with axonal cytoskeleton even though Caspr2 also has 4.1B-binding sequences (Denisenko-Nehrbass et al., 2003; Horresh et al., 2010). The juxtaparanodal region clusters potassium channels, which are lost in Caspr2 and Tag-1 mutants (Poliak et al., 2003; Traka et al., 2003). Interestingly, both the β-Act-Cre;4.1BFlox and 4.1B−/− mutants revealed disorganization of juxtaparanodal proteins, with loss of potassium channels and Caspr2 localization (Figs. 2, 3) (data not shown). This suggests that formation and/or stabilization of the juxtaparanodal axoglial scaffold critically depends on 4.1B, thus highlighting the role of axonal cytoskeletal adaptor proteins in the stability of axonal domains.
Evolutionary role of cytoskeletal proteins in stabilizing axoglial septate junctions
The founding member and Drosophila ortholog of the vertebrate Caspr family, Neurexin IV (Nrx IV), contains a 4.1-binding sequence in its C-terminus (Baumgartner et al., 1996; Bellen et al., 1998; Bhat, 2003, 2004; Banerjee et al., 2006a). An ortholog of the vertebrate 4.1 family that colocalizes with Nrx IV at septate junctions is Coracle (Cora) (Fehon et al., 1994). Null mutants of nrx IV and cora show mislocalization of Cora and Nrx IV, respectively, and fail to establish septate junctions (Fehon et al., 1994; Baumgartner et al., 1996; Lamb et al., 1998; Ward et al., 1998; Banerjee et al., 2006b). Furthermore, mutations in the FERM domain of Cora disrupts its membrane localization, suggesting that the interaction between Cora and its transmembrane-binding partners is required for localization of Cora at the membrane. Together, our data provide convincing evidence that 4.1B is critically required for proper maintenance of axonal domains in myelinated axons by acting as a molecular adaptor between membrane components at the paranodal and juxtaparanodal regions and the underlying axonal cytoskeleton. These interactions are central to the long-term stability of axonal domains and thus saltatory nerve conduction.
Footnotes
This work was supported by NIH Grants GM063074 and NS050356, the National Multiple Sclerosis Society, and the State of North Carolina (M.A.B.). We thank Alan Fanning and members of the Bhat Laboratory for helpful discussions, and Victoria Madden of University of North Carolina Microscopy Facility for assistance with electron microscopy.
The authors declare no competing financial interests.
References
- Banerjee S, Sousa AD, Bhat MA. Organization and function of septate junctions: an evolutionary perspective. Cell Biochem Biophys. 2006a;46:65–77. doi: 10.1385/CBB:46:1:65. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Pillai AM, Paik R, Li J, Bhat MA. Axonal ensheathment and septate junction formation in the peripheral nervous system of Drosophila. J Neurosci. 2006b;26:3319–3329. doi: 10.1523/JNEUROSCI.5383-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner S, Littleton JT, Broadie K, Bhat MA, Harbecke R, Lengyel JA, Chiquet-Ehrismann R, Prokop A, Bellen HJ. A Drosophila neurexin is required for septate junction and blood-nerve barrier formation and function. Cell. 1996;87:1059–1068. doi: 10.1016/s0092-8674(00)81800-0. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Lu Y, Beckstead R, Bhat MA. Neurexin IV, caspr and paranodin—novel members of the neurexin family: encounters of axons and glia. Trends Neurosci. 1998;21:444–449. doi: 10.1016/s0166-2236(98)01267-3. [DOI] [PubMed] [Google Scholar]
- Bhat MA. Molecular organization of axo-glial junctions. Curr Opin Neurobiol. 2003;13:552–559. doi: 10.1016/j.conb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Bhat MA. The neurexin and NCP gene families. In: Lazarini RA, editor. Myelin biology and disorders. New York: Elsevier; 2004. pp. 579–597. [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–397. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- Chen K, Merino C, Sigrist SJ, Featherstone DE. The 4.1 protein coracle mediates subunit-selective anchoring of Drosophila glutamate receptors to the postsynaptic actin cytoskeleton. J Neurosci. 2005;25:6667–6675. doi: 10.1523/JNEUROSCI.1527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CM, Foley SF. The role of band 4.1 in the association of actin with erythrocyte membranes. Biochim Biophys Acta. 1982;688:691–701. doi: 10.1016/0005-2736(82)90281-4. [DOI] [PubMed] [Google Scholar]
- Conboy JG. Structure, function, and molecular genetics of erythroid membrane skeletal protein 4.1 in normal and abnormal red blood cells. Semin Hematol. 1993;30:58–73. [PubMed] [Google Scholar]
- Denisenko-Nehrbass N, Oguievetskaia K, Goutebroze L, Galvez T, Yamakawa H, Ohara O, Carnaud M, Girault JA. Protein 4.1B associates with both Caspr/paranodin and Caspr2 at paranodes and juxtaparanodes of myelinated fibres. Eur J Neurosci. 2003;17:411–416. doi: 10.1046/j.1460-9568.2003.02441.x. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Girault JA, Popko B. Axo-glial interactions regulate the localization of axonal paranodal proteins. J Cell Biol. 1999;147:1145–1152. doi: 10.1083/jcb.147.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S, Bhat MA, Salzer JL. Disrupted axo-glial junctions result in accumulation of abnormal mitochondria at nodes of Ranvier. Neuron Glia Biol. 2006;2:165–174. doi: 10.1017/S1740925X06000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehon RG, Dawson IA, Artavanis-Tsakonas S. A Drosophila homologue of membrane-skeleton protein 4.1 is associated with septate junctions and is encoded by the coracle gene. Development. 1994;120:545–557. doi: 10.1242/dev.120.3.545. [DOI] [PubMed] [Google Scholar]
- Garcia-Fresco GP, Sousa AD, Pillai AM, Moy SS, Crawley JN, Tessarollo L, Dupree JL, Bhat MA. Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc Natl Acad Sci U S A. 2006;103:5137–5142. doi: 10.1073/pnas.0601082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JA, Labesse G, Mornon JP, Callebaut I. Janus kinases and focal adhesion kinases play in the 4.1 band: a superfamily of band 4.1 domains important for cell structure and signal transduction. Mol Med. 1998;4:751–769. [PMC free article] [PubMed] [Google Scholar]
- Gollan L, Sabanay H, Poliak S, Berglund EO, Ranscht B, Peles E. Retention of a cell adhesion complex at the paranodal junction requires the cytoplasmic region of Caspr. J Cell Biol. 2002;157:1247–1256. doi: 10.1083/jcb.200203050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover KB, Bryant PJ. The genetics of the protein 4.1 family: organizers of the membrane and cytoskeleton. Curr Opin Cell Biol. 2000;12:229–234. doi: 10.1016/s0955-0674(99)00080-0. [DOI] [PubMed] [Google Scholar]
- Horresh I, Bar V, Kissil JL, Peles E. Organization of myelinated axons by Caspr and Caspr2 requires the cytoskeletal adapter protein 4.1B. J Neurosci. 2010;30:2480–2489. doi: 10.1523/JNEUROSCI.5225-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jekely G. Eukaryotic membranes and cytoskeleton. New York: Springer; 2007. [Google Scholar]
- Kang Q, Wang T, Zhang H, Mohandas N, An X. A Golgi-associated protein 4.1B variant is required for assimilation of proteins in the membrane. J Cell Sci. 2009;122:1091–1099. doi: 10.1242/jcs.039644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RS, Ward RE, Schweizer L, Fehon RG. Drosophila coracle, a member of the protein 4.1 superfamily, has essential structural functions in the septate junctions and developmental functions in embryonic and adult epithelial cells. Mol Biol Cell. 1998;9:3505–3519. doi: 10.1091/mbc.9.12.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Schafer DP, Horresh I, Bar V, Hales K, Yang Y, Susuki K, Peles E, Stankewich MC, Rasband MN. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J Neurosci. 2006;26:5230–5239. doi: 10.1523/JNEUROSCI.0425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara R, Yamakawa H, Nakayama M, Ohara O. Type II brain 4.1 (4.1B/KIAA0987), a member of the protein 4.1 family, is localized to neuronal paranodes. Brain Res Mol Brain Res. 2000;85:41–52. doi: 10.1016/s0169-328x(00)00233-3. [DOI] [PubMed] [Google Scholar]
- Parra M, Gascard P, Walensky LD, Gimm JA, Blackshaw S, Chan N, Takakuwa Y, Berger T, Lee G, Chasis JA, Snyder SH, Mohandas N, Conboy JG. Molecular and functional characterization of protein 4.1B, a novel member of the protein 4.1 family with high level, focal expression in brain. J Biol Chem. 2000;275:3247–3255. doi: 10.1074/jbc.275.5.3247. [DOI] [PubMed] [Google Scholar]
- Peters LL, Weier HU, Walensky LD, Snyder SH, Parra M, Mohandas N, Conboy JG. Four paralogous protein 4.1 genes map to distinct chromosomes in mouse and human. Genomics. 1998;54:348–350. doi: 10.1006/geno.1998.5537. [DOI] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res. 2009;87:1773–1793. doi: 10.1002/jnr.22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, Furley AJ, Peles E. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J. Role of glial cells in the differentiation and function of myelinated axons. Int J Dev Neurosci. 1988;6:3–24. doi: 10.1016/0736-5748(88)90025-1. [DOI] [PubMed] [Google Scholar]
- Salzer JL. Polarized domains of myelinated axons. Neuron. 2003;40:297–318. doi: 10.1016/s0896-6273(03)00628-7. [DOI] [PubMed] [Google Scholar]
- Shi ZT, Afzal V, Coller B, Patel D, Chasis JA, Parra M, Lee G, Paszty C, Stevens M, Walensky L, Peters LL, Mohandas N, Rubin E, Conboy JG. Protein 4.1R-deficient mice are viable but have erythroid membrane skeleton abnormalities. J Clin Invest. 1999;103:331–340. doi: 10.1172/JCI3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CX, Robb VA, Gutmann DH. Protein 4.1 tumor suppressors: getting a FERM grip on growth regulation. J Cell Sci. 2002;115:3991–4000. doi: 10.1242/jcs.00094. [DOI] [PubMed] [Google Scholar]
- Thaxton C, Bhat MA. Myelination and regional domain differentiation of the axon. Results Probl Cell Differ. 2009;48:1–28. doi: 10.1007/400_2009_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Pillai AM, Pribisko AL, Labasque M, Dupree JL, Faivre-Sarrailh C, Bhat MA. In vivo deletion of immunoglobulin domains 5 and 6 in neurofascin (Nfasc) reveals domain-specific requirements in myelinated axons. J Neurosci. 2010;30:4868–4876. doi: 10.1523/JNEUROSCI.5951-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Pillai AM, Pribisko AL, Dupree JL, Bhat MA. Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron. 2011;69:244–257. doi: 10.1016/j.neuron.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Goutebroze L, Denisenko N, Bessa M, Nifli A, Havaki S, Iwakura Y, Fukamauchi F, Watanabe K, Soliven B, Girault JA, Karagogeos D. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J Cell Biol. 2003;162:1161–1172. doi: 10.1083/jcb.200305078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RE, 4th, Lamb RS, Fehon RG. A conserved functional domain of Drosophila coracle is required for localization at the septate junction and has membrane-organizing activity. J Cell Biol. 1998;140:1463–1473. doi: 10.1083/jcb.140.6.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa H, Ohara R, Nakajima D, Nakayama M, Ohara O. Molecular characterization of a new member of the protein 4.1 family (brain 4.1) in rat brain. Brain Res Mol Brain Res. 1999;70:197–209. doi: 10.1016/s0169-328x(99)00139-4. [DOI] [PubMed] [Google Scholar]
- Yi C, McCarty JH, Troutman SA, Eckman MS, Bronson RT, Kissil JL. Loss of the putative tumor suppressor band 4.1B/Dal1 gene is dispensable for normal development and does not predispose to cancer. Mol Cell Biol. 2005;25:10052–10059. doi: 10.1128/MCB.25.22.10052-10059.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]