

Abstract
Elucidating the sources of genetic variation within microsatellite alleles has important implications for understanding the etiology of human diseases. Mismatch repair is a well described pathway for the suppression of microsatellite instability. However, the cellular polymerases responsible for generating microsatellite errors have not been fully described. We address this gap in knowledge by measuring the fidelity of recombinant yeast polymerase δ (Pol δ) and ε (Pol ε) holoenzymes during synthesis of a [GT/CA] microsatellite. The in vitro HSV-tk forward assay was used to measure DNA polymerase errors generated during gap-filling of complementary GT10 and CA10 -containing substrates and ∼90 nucleotides of HSV-tk coding sequence surrounding the microsatellites. The observed mutant frequencies within the microsatellites were four to 30-fold higher than the observed mutant frequencies within the coding sequence. More specifically, the rate of Pol δ and Pol ε misalignment-based insertion/deletion errors within the microsatellites was ∼1000-fold higher than the rate of insertion/deletion errors within the HSV-tk gene. Although the most common microsatellite error was the deletion of a single repeat unit, ∼ 20% of errors were deletions of two or more units for both polymerases. The differences in fidelity for wild type enzymes and their exonuclease-deficient derivatives were ∼two-fold for unit-based microsatellite insertion/deletion errors. Interestingly, the exonucleases preferentially removed potentially stabilizing interruption errors within the microsatellites. Since Pol δ and Pol ε perform not only the bulk of DNA replication in eukaryotic cells but also are implicated in performing DNA synthesis associated with repair and recombination, these results indicate that microsatellite errors may be introduced into the genome during multiple DNA metabolic pathways.
Keywords: microsatellite instability, indel mutation, DNA replication fidelity, polymerase proofreading, HSV-tk gene
1. Introduction
Microsatellite sequences are repetitive sequences of one to six base pairs per repeat unit that are non-randomly distributed throughout all eukaryotic genomes. Dinucleotide microsatellites are highly abundant; specifically, GT/CA dinucleotides comprise 19% of all microsatellites in the human genome [1]. A defining attribute of microsatellites is their high frequency of both expansion and deletion mutation, which results in allele length variation and a high degree of genetic polymorphism among individuals in populations [2]. The transition from low mutability, characteristic of short tandem repeat sequences, to high mutability, characteristic of microsatellites, occurs once a threshold number of repeat units in the allele has been reached [3]. Allele length polymorphisms at common mono- and dinucleotide microsatellites are implicated as genetic risk factors in several human diseases [4], including cystic fibrosis (CFTR gene) [5,6] and breast cancer (EGFR gene) [7,8]. Individuals with compromised postreplication mismatch repair (MMR), a pathway that repairs insertion/deletion (indel) mutations exceptionally well, are predisposed to the development of cancer (for recent review, see [9]). Tumors arising in these patients display widespread mononucleotide and dinucleotide microsatellite instability, and mutations within microsatellites associated with critical target genes are believed to play a causative role in the evolution of MMR-defective tumors [10,11]. Undoubtedly, elucidating the sources of genetic variation within common microsatellite alleles has important implications for understanding the etiology of human diseases.
Classically, indel mutations are proposed to arise during DNA replication by a slippage mechanism [12,13]. The slippage event creates a misaligned intermediate containing one or more extrahelical nucleotides, and depending on the stability of the misaligned stretch of DNA, the unpaired bases will be either inserted into, or deleted from, the DNA strand during the following round of replication [12,14]. Strong structural evidence supporting the slippage hypothesis emerged in 2006 when DNA polymerase λ was crystallized with a single-base deletion intermediate containing an unpaired nucleotide in the template strand [15,16]. The structure displayed the extrahelical nucleotide, the correct base pair at the primer terminus, and the geometry of a polymerase active site that was compatible with catalysis [15,16]. Experimental data obtained using purified DNA polymerases, bacteriophage, bacteria, yeast and human cell model systems are consistent with strand slippage models, in that all show mutation rates within tandemly repeated sequences or microsatellites that increase with an increase in the length of the repeat [3,17-23]. Consequently, interruptions in a repeated array dramatically reduce the mutation frequency [24,25].
DNA polymerases δ and ε are the only nuclear DNA polymerases in eukaryotic cells with an intrinsic 3′→ 5′ exonuclease (proofreading) activity. Thus, these DNA polymerases can be considered the “front-line” DNA repair mechanism for maintaining genome stability [26]. The critical importance of polymerase proofreading in the maintenance of genome stability and avoidance of disease has been demonstrated elegantly in exonuclease-deficient mouse models [27,28]. In yeast model systems, the 3′→ 5′ exonuclease activity was shown to contribute to the removal of indel mutations within short repeated sequences [29,30]. However, the contribution of polymerase proofreading activity in suppressing indel mutations diminished with increasing length of a mononucleotide tandem repeat [30]. Similarly, a previous in vitro study using the T7 DNA polymerase demonstrated that proofreading efficiency is diminished with an increase in the repeat tract length [31].
The fidelity of replicative eukaryotic DNA polymerases within microsatellites has not been investigated previously, despite the prevalence and potential of such sequences to modify disease risk. Here, we examined mutagenesis associated with in vitro DNA synthesis by the holoenzyme forms of yeast polymerase δ (Pol δ) and polymerase ε (Polε), using the established HSV-tk microsatellite assay [32]. The Pol δ holoenzyme is comprised of three subunits: Pol3, Pol31, and Pol32. Pol3 is the catalytic subunit containing both the polymerase and exonuclease active sites. The Pol ε holoenzyme is comprised of four subunits: Pol2, Dpb2, Dpb3, and Dpb4. Pol2 is the catalytic subunit, and similar to Pol3 of Pol δ, contains both the polymerase and exonuclease activities of the enzyme. We compared the frequency of indel errors created by each polymerase within a [GT/CA]10 microsatellite, and compared this to the frequency of indel errors within the HSV-tk gene coding region. To quantitatively assess the contribution of proofreading to microsatellite stability, we also conducted synthesis reactions using holoenzyme preparations that are exonuclease deficient. The results of this study emphasize the vital role played by cellular MMR in yeast for the suppression of DNA sequence variation within genomic microsatellites.
2. Material and Methods
2.1. Overexpression and purification of Pol δ and Pol ε holoenyzmes
Overexpression of proteins was performed in Saccharomyces cerevisiae strain BJ2168 (MATa,ura3-52,trp1-289,leu2-3,112,prb1-1122,prc1-407,pep4-3). For Polδ holoenzyme overexpression, BJ2168 was transformed with pBL341 and either pBL335 or pBL335-DV and growth and induction was as described previously [33]. Cells (60g of packed cells resuspended in 8ml of dH2O) frozen in liquid nitrogen in popcorn form were ground using a Spex Sample Prep 6870 freezer mill, which lysed the cells by magnetic motion. Purification continued as previously described in [33]. For the Pol ε holoenzyme, pJL1 or pJL1-exo and pJL6 were transformed, grown, and expressed as previously reported [34]. The four subunit Pol ε was purified in the same manner as previously described [34]. Activity of the purified enzyme was determined by a specific activity assay using labeled activated calf thymus DNA. In order to establish the contribution of exonucleolytic proofreading to microsatellite sequence stability, we also purified holoenzyme forms with catalytic subunits that are deficient in proofreading activity. The mutations introduced for the exonuclease deficient polymerase forms (pol δ = D520V in domain ExoIII; pol ε = double mutation of D289A and E291A) have been described previously, and have been shown to completely abolish in vitro proofreading activity [35-37].
2.2. In vitro HSV-tk gap-filling assay
An in vitro assay for the quantitation of DNA polymerase errors within microsatellite sequences has been described previously [32]. In this assay, an artificial microsatellite sequence, [GT/CA]9, was inserted in-frame between bases 111 and 112 of the HSV-tk target, in the sequence context [GT (insert) TCTC] on the sense strand. Bases flanking the insert are considered part of the microsatellite region; therefore, the entire microsatellite motif is [GT/CA]10. For the current study, a StuI restriction site at HSV-tk position 180 was created and subcloned into the aforementioned vectors as described [38], allowing our coding region mutational target to be shortened from ∼200bp to ∼90 bp. Linear DNA fragments bearing a functional cat gene and ssDNA bearing a nonfunctional cat gene were prepared, and used to construct gapped duplex (GD) molecules as described [32,38]. The pSStu2 GD substrate contains the [GT]10 microsatellite and surrounding HSV-tk coding sequence within the single-stranded gap, which serves as the template for DNA polymerase reaction. The pSAStu2 GD substrate contains the complementary [CA]10 microsatellite and HSV-tk sequences within the template sequence (Figure 1).
Figure 1. Determination of complete gap-filling by replicative polymerases.
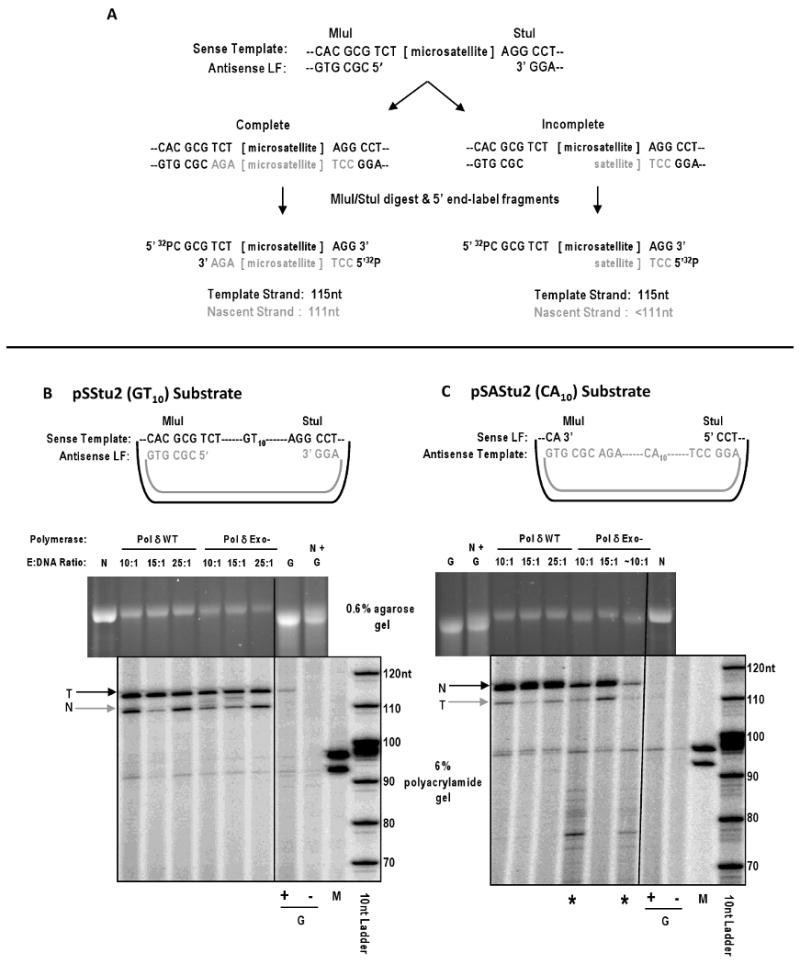
(A) Schematic representation of a polyacrylamide gel-based assay for detection of complete gap-filling. Cartoon depicts the pSStu2 gapped substrate created by hybridizing a sense ssDNA template to an antisense large fragment. The ∼100 nt MluI to StuI region of the gapped heteroduplex is used as a template for in vitro DNA synthesis. Potential products of the polymerase reaction can be completely (left) or incompletely (right) filled heteroduplexes. To determine the extent of the polymerase reaction, the reaction products are digested with MluI and StuI and the fragments are 5′ end-labeled through an exchange reaction with [γ-32P] ATP and T4 kinase. Complete reactions will give two products of 111nt (nascent strand) and 115nt (template strand), whereas incomplete reactions will result in products less than 111nt. Complete reactions performed using the complementary gapped substrate (pSAStu2) with produce the opposite pattern: a 111nt product from the template strand and a 115nt product from nascent strand. (B) Analyses of the extent of gap-filling by Pol δ forms using the pSStu2 (GT10) gapped substrate. Top panel: Cartoon depicting the gapped heteroduplex substrate. Middle panel: Agarose gel (0.6%) analysis of pol δ WT and Pol δ Exo- reaction products at varying enzyme:DNA molar ratios. Complete gap-filling reaction products will migrate at the position of the nicked substrate marker (N); G, unfilled gapped heteroduplex (starting substrate). Bottom panel: Polyacrylamide gel (6%) analysis of the same Pol δ reaction products after restriction enzyme digestion and [γ-32P] end-labelling of DNA products. Arrow labeled T indicates the DNA band corresponding to the template strand. Arrow labeled N indicates the DNA band corresponding to the nascent strand. An identical amount of unfilled gapped substrate was either digested with MluI and StuI (+) or untreated (-), and analyzed as controls. M, 93nt and 97nt markers; 10nt ladder, 5′ end-labeled ladder. (C) Analyses similar to those shown in (B) of the extent of gap-filling by Pol δ forms using the pSAStu2 (CA10) gapped substrate. Examples of incomplete reactions as shown by both the agarose and radioactive assays are shown in lanes indicated with asterisks (10:1 and ∼10:1 pol δ Exo- reactions). Fragments as short as 10 nt can be observed in the incomplete reactions with shorter electrophoresis times (data not shown).
In vitro gap-filling reactions contained 0.125 pmol of GD substrate and 250 μM dNTPs in 100 μL final volume. The minimal amount of polymerase required to achieve complete gap-filling was determined empirically by titration for each polymerase and GD preparation. Pol δ reactions contained 40 mM Tris pH 7.8, 8 mM MgOAc, and 1.25 - 3.1 pmol of Pol δ WT or Pol δ Exo-. Reactions were incubated at 30°C for 1 hour. Pol ε reactions contained 50 mM Tris pH 7.5, 8 mM MgCl2, 2 mM DTT, 100 μg/mL BSA, 10% glycerol, and 0.125 - 1 pmol Pol ε WT or 0.625 - 1.9 pmol Pol ε Exo-. Reactions were incubated at 30°C for 30 min. All reactions were terminated with 15 mM EDTA and the buffers exchanged into TE buffer. Because the determination of complete gap-filling was crucial for accurate mutational analyses, two different, yet complementary, methods were used (Figure 1B and 1C). First, 50 fmol of product were analyzed by separation through an 0.6% agarose gel for ∼18 hours, along with GD and nicked (completely filled) standards. This standard analysis will detect the presence of starting (i.e., unfilled) GD substrate, as well as intermediate reaction products migrating between the standards. However, the migration patterns of complete gap-filling products and intermediate gap-filling products can be ambiguous due to the low resolution of the agarose gel. To more definitively differentiate complete gap-filling products from intermediate products, a denaturing polyacrylamide gel analysis was developed (Figure 1A). In this approach, 50 fmol of polymerase reaction products or 50 fmol of starting GD substrate were digested with 2 units of MluI and StuI at 37°C for 1 hour. Reaction products were exchanged into TE buffer and 5′-end labeled in an exchange reaction using 10 μCi of [γ-32P]ATP and 10 units of T4 kinase at 37°C for 10 min. Reactions were terminated with 15 mM EDTA, purified through a G25 Sephadex column, added to an equal volume of stop dye (99% v/v formamide, 5 mM EDTA, 0.1% (w/v) xylene cyanol, and 0.1% (w/v) bromophenol blue), and the products separated through a 6% denaturing polyacrylamide gel. Polymerase synthesis of ∼five nucleotides from the GD 3′OH will create one complete double-stranded restriction enzyme site, whereas the starting GD substrates encode only partial sites (Figure 1A). Thus, little to no MluI-StuI digestion products are expected from the unfilled GD substrate, whereas 111nt and 115 nt strands are expected from complete gap-filling products (Figure 1B,C). The method is strictly qualitative in design, because the efficiency of the T4 kinase reaction differs between strands (due to different sequences surrounding the 5′PO4) and because the recovery of radiolabelled DNA fragments from the G25 columns is variable. Never-the-less, the analysis can clearly identify incomplete gap-filling DNA products as bands less than 111 or 115 nt (Figure 1C).
To select for HSV-tk mutations, an aliquot of DNA from complete reactions was used to transform recA13, upp, tdk E. coli strain FT334 by electroporation. The background frequency of the gap-filling in vitro assay was determined by using the unfilled (starting) GD substrates for electroporation. In all cases, electroporated bacteria were plated on VBA selective media as described [32,39]. The presence of 50 μg/mL chloramphenicol (Cm) selects for progeny of the polymerase-synthesized strand and the presence of 40 μM 5-fluoro-2′-deoxyuridine (FUdR) selects for bacteria bearing HSV-tk mutant plasmids. The observed HSV-tk mutant frequency (MF) is defined as the number of FUdRRCmR colonies divided by the number of CmR colonies.
2.3. DNA sequence analyses and polymerase mutant frequency estimations
Independent FUdR-resistant mutants were isolated as described [32,39] from at least two separate polymerase reactions for each enzyme and template combination, as well as from unfilled GD DNA. The mutant plasmids were sequenced to identify the location of the mutation within the HSV-tk target. Sequencing reactions were performed using the PE-Applied Biosystems automated sequencing reagents and the TK309-MSI sequencing primer (5′CCGCCAGTAAGTCAT). Analysis was completed on a Perkin-Elmer ABI Model 3100 Sequencer. Seqman a sequence alignment program created by Lasergene (licensed to the National Institute of Environmental Health Sciences) was utilized to analyze the data. In some cases, 5′ strand displacement synthesis or 3′ exonuclease digestion occurred, resulting in polymerase errors outside of the Mlu I-Stu I gap target. Such mutations were excluded from the data analyses. Fisher's exact test (two-sided) was used to determine the statistical significance of differences in the proportions of specific classes of mutations, using the absolute numbers of observed mutants within each class (provided in Tables).
To quantitate polymerase errors, the observed HSV-tk MF was first corrected to reflect only those mutational events arising within the MluI–StuI target region. This proportion ranged from 90-95% for the various Pol δ reactions, and 59-92% for the Pol ε reactions. The resulting frequency is referred to as the “overall” HSV-tk MF in Table 2. For example, the observed HSV-tk mutant frequency for Pol δ WT using the pSStu2 substrate was 43 × 10-4 (Table 1). Of the 96 mutant plasmids sequenced, 86 events were within the MluI-StuI target; thus, the overall Pol δ WT HSV-tk MF is (86/96)(43 × 10-4), or 38 × 10-4 (Table 2). Next, the frequency of specific types of mutational events was calculated by multiplying the overall MF by the proportion of sequence changes observed. For the Pol δ WT example, 76 events were within the GT10 microsatellite target; thus, the microsatellite HSV-tk MF is (76/86)(38 × 10-4), or 34 × 10-4 (Table 2). Finally, the estimated polymerase mutant frequency (Pol MFest) for specific errors was calculated by subtracting the unfilled GD substrate MF (also referred to as the background MF) from the HSV-tk MF. For the Pol δ WT example, the Pol MFest for errors within the GT10 microsatellite is estimated to be (34 × 10-4) - (2.6 × 10-4) = 31 × 10-4 (Figure 2A).
Table 2. Replicative polymerase mutant frequencies at microsatellite and coding region target sequences.
| Polymerase | HSV-tk Mutant Frequency × 10-4 (Number of errors observed a) | |||||
|---|---|---|---|---|---|---|
| pSStu 2 substrate | pSAStu2 substrate | |||||
| Overall b | GT10 | Coding | Overall | CA10 | Coding | |
| Unfilled gap c | 3.8 (36) | 2.6 (28) | 0.75 (8) | 9.3 (31) | 8.4 (28) | 0.9 (3) |
| Pol δ | ||||||
| WT | 38 (86) | 34 (76) | 4.5 (10) | 20 (84) | 19 (79) | 1.2 (5) |
| Exo- | 65 (93) | 51 (73) | 14 (20) | 44 (89) | 35 (71) | 8.8 (18) |
| Pol ε | ||||||
| WT | 15 (71) | 12 (55) | 3.4 (16) | 28 (56) | 25 (49) | 3.6 (7) |
| Exo- | 39 (36) | 25 (23) | 14 (13) | 38 (55) | 30 (43) | 8.4 (12) |
Number of HSV-tk mutant plasmids containing errors within the indicated region.
The overall frequency of mutational events occurring within the MluI-StuI target region. Data were derived from the frequencies given in Table 1 after DNA sequence analyses of independent mutants. In some instances, mutations were detected outside of the MluI-StuI mutational target, caused by either polymerase 5′ strand displacement/ 3′ exonucleolytic degradation or pre-existing mutations in the GD substrate. Such mutations have been excluded from the table. The overall frequency is the sum of mutations arising within the indicated microsatellite plus those arising within the HSV-tk coding region.
No polymerase background mutant frequencies, derived from electroporation of bacteria with unfilled gapped substrate, followed by DNA sequence analyses of independent mutants.
Table 1. Observed HSV-tk mutant frequencies resulting from in vitro replicative polymerase DNA synthesis.
| Polymerase | HSV-tk mutant frequency ×10-4 (± std. dev) a | |||
|---|---|---|---|---|
| pSStu2 | pSAStu2 | |||
| [GT]10 substrate | N b | [CA]10 substrate | N | |
| None | ||||
| ssDNAc | 3.8 | 1.5 | ||
| Unfilled Gapd | 4.6 ± 0.6 (4) | 44 | 12.0 ± 3 (4) | 40 |
| Polδ | ||||
| WT | 43.0 ± 13 (5) | 96 | 21.0 ± 6 (5) | 90 |
| Exo- | 69.0 ± 18 (5) | 99 | 46.0 ± 23 (3) | 94 |
| Polε | ||||
| WT | 20 ± 3 (4) | 95 | 31.0 ± 6 (3) | 61 |
| Exo- | 42 (1) | 39 | 65 (2) | 93 |
Number of independent determinations is given in parentheses
Number of independent mutants sequenced
Electroporation of ssDNA used to create the gapped substrates
Electroporation of unfilled gapped DNA substrate
Figure 2. The high fidelity of WT replicative polymerases is not maintained in dinucleotide microsatellite sequences.
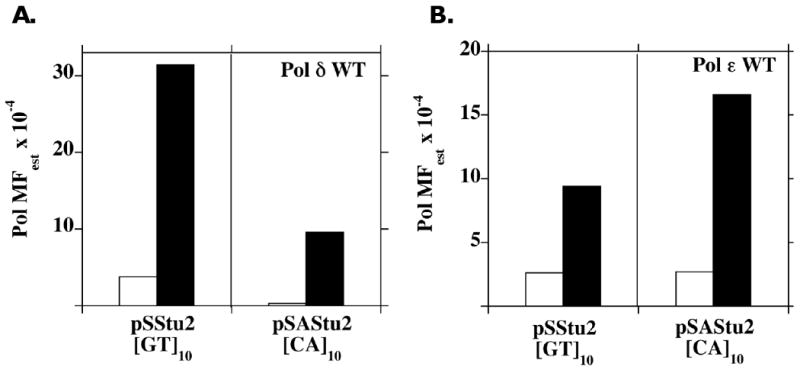
Gap-filling DNA synthesis reactions were performed using either (A). Polδ WT, or (B). Polε WT and the pSStu2/pSAStu2 complementary DNA substrates. The Pol MFest was calculated from the HSV-tk mutant frequency, using the data presented in Table 2: Pol MFest = [(HSV-tk MF of the indicated polymerase reaction) – (unfilled gap MF)] for each region. Graphs compare the Pol MFest within the HSV-tk coding sequence (Open bars) to that within the [GT]10 or [CA]10 microsatellite sequences (solid bars) for each substrate.
3. Results
DNA polymerases δ and ε are widely believed to perform the bulk of DNA replication elongation in eukaryotic cells, with one current model suggesting that Pol δ replicates primarily the lagging-strand template [40] and Pol ε replicates primarily the leading-strand template [41]. In addition, these polymerases have been implicated in the DNA synthesis associated with several DNA repair pathways [42,43]. Both Pol δ and Polε are highly accurate during in vitro synthesis of the lacZ target sequence, and introduce limited base substitution and indel errors [37,44]. This high fidelity is due, in part, to the intrinsic proofreading exonuclease activities [26]. Although previous in vitro studies have shown that the 3′→ 5′ exonuclease activity of replicative polymerases can remove indel errors within short (≤ 5 bases) mononucleotide repeats, the efficiency of proofreading microsatellites with larger repeat sizes has not been determined directly.
3.1. Wild-type replicative polymerase mutant frequencies
Gap-filling DNA polymerization reactions were performed using complementary DNA substrates, containing either an in-frame [GT]10 (pSStu2) or [CA]10 (pSAStu2) microsatellite sequence surrounded by HSV-tk gene coding sequence. The gapped duplex (GD) molecules contain a single-stranded gap of 91 - 95bp of HSV-tk sequence and 20bp of microsatellite sequence. The HSV-tk coding region sequence serves as a monitor for polymerase indel fidelity, as it contains 23 repeated mono- and dinucleotide sequences of two to three units each. (The DNA sequences of the target regions are provided in Supplemental Figure 1.) In these constructs, the target sequence contains few detectable sites of base substitution mutations, and is biased towards the detection of strand misalignment-based errors. Complementary agarose gel and denaturing polyacrylamide gel electrophoresis analyses were used to ensure that all enzymes completely filled both GD substrates under the stated reaction conditions (Figure 1). The Pol δ WT reactions generated DNA products with mutant frequencies that were two- to nine-fold higher than the corresponding unfilled GD background reactions (i.e., “no polymerase” control), while the Pol ε WT mutant frequencies were increased three- to four-fold over unfilled GD background (Table 1). As expected, the HSV-tk mutant frequencies measured for the exonuclease-deficient Pol δ and Pol ε reactions were higher than those measured for the WT holoenzymes (Table 1).
The HSV-tk forward mutation assay detects any mutation that inactivates the protein, both within the artificial microsatellite and within the surrounding protein-coding sequences present in the target. Therefore, independent mutant colonies were collected and sequenced to identify the location and type of mutation. A complete listing of the errors observed within the mutational target by all four enzymes and two substrates can be provided upon request. The majority of both Pol δ and Pol ε errors produced during the gap-filling reactions were within the [GT]10 or [CA]10 microsatellite sequences (Table 2).
The HSV-tk MF measured for the pSStu2 and pSAStu2 unfilled GD substrates differ by ∼3-fold (Table 1). Therefore, in order to directly compare polymerase mediated errors generated on the complementary DNA strands, we estimated the polymerase mutant frequency (Pol MFest) within each region by subtracting the unfilled GD background frequency from the corresponding HSV-tk frequencies for each polymerase reaction. The resulting Pol MFest for Pol δ WT within the [GT]10 and [CA]10 microsatellites is eight to 30-fold higher than the corresponding Pol MFest within the HSV-tk coding region (Figure 2A). The Pol MFest for Pol ε within the microsatellite sequences is four to six-fold higher than the corresponding coding region (Figure 2B). In addition, a strand bias for Pol δ WT microsatellite errors was observed, in that the Pol MFest within the [GT]10 allele is three-fold higher than the Pol MFest within the complementary [CA]10 allele (Figure 2A). A smaller but opposite trend was observed for Pol ε WT, in that the Pol MFest within the CA allele is 1.8-fold higher than that for the GT allele (Figure 2B). Mutational strand biases have been observed previously for polymerases α and β within microsatellite sequences [22].
3.2. Specificity of wild-type polymerase errors within microsatellite sequences
We observed previously that DNA polymerases in vitro produce two classes of mutations within a microsatellite sequence: unit-based indel errors and interruption errors [32,38,45]. Although Pol δ WT and Pol ε WT also produced both classes of errors within the [GT]10 and [CA]10 microsatellite sequences, the majority of errors (82-100%) were unit-based indel mutations (Table 3). Both polymerases are highly biased towards the production of deletion errors, compared to insertion errors, within the microsatellite sequences. Moreover, approximately 20% of the indel deletions were of two or more repeat units (Table 3). The largest Pol δ WT deletion observed was the removal of four repeat units (eight nucleotides) in the [CA]10 substrate, while the largest Pol ε WT deletion was five repeat units within the [GT]10 substrate (data not shown).
Table 3. Replicative polymerase error specificity within the [GT/CA]10 microsatellite.
| Microsatellite/ Mutational Events |
Proportion of Events (number observed) | |||
|---|---|---|---|---|
| Polδ | Polε | |||
| WT | Exo- | WT | Exo- | |
| [GT]10 | ||||
| Unit-based InDel | 0.82 (62) | 0.73 (53) | 0.96 (53) | 0.52 (12) |
| Deletion | 0.82 (62) | 0.73 (53) | 0.93 (51) | 0.52 (12) |
| 1 unit | 0.65 (49) | 0.49 (36) | 0.73 (40) | 0.30 (7) |
| > 1 unit | 0.17 (13) | 0.23 (17) | 0.20 (11) | 0.22 (5) |
| Insertion | 0 | 0 | 0.04 (2) a | 0 |
| Interruption | 0.18 (14) | 0.26 (19) | 0.04 (2) | 0.48 (11) |
| [CA]10 | ||||
| Unit-based InDel | 1.0 (79) | 0.92 (65) | 1.0 (49) | 0.93 (40) |
| Deletion | 0.92 (73) | 0.92 (65) | 0.84 (41) | 0.84 (36) |
| 1 unit | 0.61 (48) | 0.66 (47) | 0.65 (32) | 0.74 (32) |
| > 1 unit | 0.32 (25) | 0.25 (18) | 0.18 (9) | 0.09 (4) |
| Insertion | 0.08 (6) | 0 | 0.16 (8) | 0.09 (4) |
| 1 unit | 0.05 (4) | 0.10 (5) | 0.05 (2) | |
| > 1 unit | 0.03 (2) | 0.06 (3) | 0.05 (2) | |
| Interruption | 0 | 0.08 (6) | 0 | 0.07 (3) |
All mutants were 1 unit changes
Both unit-based microsatellite indels and traditional frameshift indel errors arise by a strand misaligment mechanism. To directly compare the likelihood of the two types of polymerase misaligment errors, we calculated the Pol MFest per site for indel errors within the 23 repeated sequence DNA motifs contained in our HSV-tk target sequence (Supplemental Figure 1). The replicative holoenzymes produce microsatellite misalignment errors at a rate that is nearly three orders of magnitude (∼900-fold) greater than indel errors within the HSV-tk sequence (Table 4).
Table 4. Comparison of wild-type polymerase fidelity for different types of misalignment-based errors.
| Substrate/ Mutational event |
Pol MFesta × 10-4 (number observed) | |
|---|---|---|
| Pol δ WT | Pol ε WT | |
| pSStu2 [GT]10 | ||
| Microsatellite Indelsb | 25 (62) | 9.0 (53) |
| Coding Indelc | 1.1 (3) | 0.16 (1) |
| Coding Indel/sited | 0.048 | 0.007 |
| Fold differencee | 500 | 1300 |
| pSAStu2 [CA]10 | ||
| Microsatellite Indels | 11 (79) | 17 (49) |
| Coding Indel | ≤ 0.06 (0) | ≤ 0.39 (0) |
| Coding Indel/site | ≤ 0.003 | ≤0.02 |
| Fold difference | ≥ 3600 | ≥ 800 |
Pol MFest values were calculated for each error from the data presented in Tables 2 and 3. Pol MFest = [HSV-tk MF for the indicated reaction – unfilled gap MF] × Proportion of total
Frequency of unit-based indel errors within the indicated microsatellite
Frequency of indel errors at 2-3 unit repeated sequences within the indicated HSV-tk target sequence.
Frequency of indel errors/ site. There are 23 sites of 2-3 unit repeated sequences within the MluI-StuI mutational target (Figure S1).
Microsatellite indel frequency / Coding indel frequency per site
Microsatellite interruptions have been observed in previous polymerase studies [32,38]. Both Pol δ WT and Pol ε WT produce more interruptions on the [GT]10 template compared to the [CA]10 template (Table 3). This difference is statistically significant for Polδ (p ≤ 0.0001, Fisher's exact test). In addition, Pol δ WT produces more interruptions (18% of errors) than does Pol ε WT (2% of errors) within the [GT]10 sequence (p=0.014, Fisher's exact test) (Table 3).
3.3. Polymerase fidelity in the absence of proofreading
To investigate the extent to which the exonuclease activities of Pol δ and Polε contribute to microsatellite sequence fidelity, we determined the polymerase mutant frequencies of exonuclease-deficient forms. The HSV-tk mutant frequencies within the GT10 and CA10 microsatellite regions observed for the exonuclease-deficient enzymes generally differed from those observed for WT enzymes by less than two-fold (Table 2). When examining specifically the frequency of indel errors within the microsatellites, we observed Pol MFest differences of 1.2-fold between the WT and exonuclease-deficient Pol ε forms (Figure 3). A larger two- to four-fold difference in the coding region Pol MFest was observed between WT and exonuclease-deficient Pol ε forms (Table 2). For the Pol δ forms, the differences between WT and exonuclease-deficient forms were 1.4-fold and 2.6-fold for the [GT]10 and [CA]10 templates, respectively (Figure 3). A three- to seven-fold difference in HSV-tk mutant frequencies within the coding regions was observed between the WT and exonuclease-deficient forms of Pol δ during synthesis of the two templates (Table 2). The differences in proofreading efficiency on the complementary pSStu2 and pSAStu2 templates likely reflect the influence of DNA sequence context on polymerase error production and/or exonuclease activity (i.e., hotspot differences on the complementary strands; see Figure S1). These magnitudes of Pol δ and Pol ε exonuclease enhancement of fidelity at the HSV-tk coding sequence are consistent with previous studies using the same enzymes and a lacZ indel reversion target sequence [44]. The data in Figure 3 demonstrate that the exonuclease activity does not contribute strongly to the fidelity of the holoenzymes during dinucleotide microsatellite DNA synthesis.
Figure 3. The 3′ to 5′ exonuclease activity contributes little to the correction of replicative polymerase indel errors within microsatellite sequences.
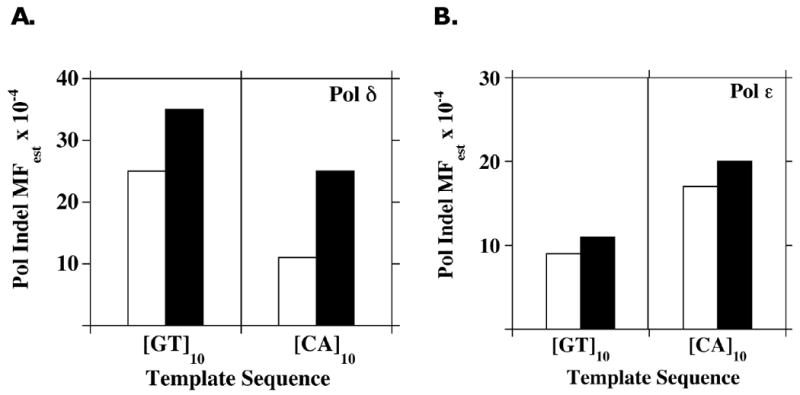
Gap-filling DNA synthesis reactions were performed using exonuclease-deficient and proficient forms of (A). Pol δ, or (B). Pol ε and pSStu2/pSAStu2 DNA substrates. The Pol MFest values specifically for unit-based Indel errors within each microsatellite sequence were calculated using the frequency data in Table 2 and the proportion data in Table 3. Graphs compare the Pol MFest for wild-type (open bars) and exonuclease deficient (solid bars) enzymes.
Interestingly, the proofreading exonuclease activity of both polymerases preferentially removes interruption errors within the microsatellites (Table 3). Exonuclease deficient Pol ε produced significantly more interruptions during [GT]10 DNA synthesis, compared to the wild type enzyme (p<0.0001, Fisher's exact test). Similarly, exonuclease deficient Pol δ produced significantly more interruptions during [CA]10 DNA synthesis, compared to the wild type enzyme (p=0.01, Fisher's exact test). A strand bias for interruptions was also apparent, with more interruptions occurring within the [GT] than the [CA] template sequence for both exonuclease-deficient polymerases (p=0.0043, Pol δ; p=0.0002, Pol ε; Fisher's exact test). These mutational biases may reflect, in part, the inherent error specificites of both enzymes previously measured within the lacZ mutational target [37,44].
4. Discussion
This study is the first to investigate the in vitro fidelity of replicative, eukaryotic holoenzymes within a tandem repeat array that can be defined as a microsatellite allele [3]. Using the well-defined in vitro HSV-tk polymerase fidelity assay [3,32,38,45], high polymerase mutant frequencies (∼10-3) were measured during microsatellite DNA synthesis by the wild-type holoenzymes, which contrasts with the low mutant frequencies (∼10-4) measured during synthesis of a non-repetitive sequence (Figure 2). Error correction by the respective proofreading exonucleases contributed little to the overall fidelity of the polymerases within the microsatellite sequences (Figure 3). These data support the previous suggestion that microsatellites are “at-risk” sequences for variation within the eukaryotic genome [46].
We investigated whether the high fidelity of replicative polymerases previously measured for base substitution and indel errors within genes would be mirrored in longer, tandem repeats (e.g., microsatellites). As clearly shown here, high fidelity is not maintained for either Pol δ or Pol ε within the [GT/CA]10 microsatellite. Directly comparing misalignment-based errors, the fidelity of Pol δ WT and Pol ε WT for unit-based indel errors created during synthesis of the microsatellite alleles is ∼1000-fold lower than the fidelity for indel errors created during HSV-tk gene synthesis (Table 4). The results for Pol ε were unexpected, as this polymerase is among the most accurate of DNA polymerases in vitro for single base indel errors [42]. The addition of the replication accessory proteins RPA, RFC and PCNA to yeast Pol δ in vitro reactions was previously shown not to alter the error rate for one nucleotide deletions [33]. Likewise, the addition of RFC and PCNA to human Pol δ reactions does not alter the frequency of errors within the GT10 microsatellite1. [GT/CA] mutagenesis during genomic DNA replication will be the summation of polymerase errors on the [GT] strand plus errors on the [CA] strand. Assuming that Pol δ and Pol ε replicate complementary strands and that the polymerases display similar fidelity in vivo, we estimate conservatively that the combined Pol δ + Pol ε error frequency during [GT+CA] synthesis is 2.5-5 × 10-3 (Table 5). This frequency corresponds to an expected mutation rate of 1 mutant per 200 – 400 [GT/CA] alleles per round of DNA replication.
Table 5. Estimated microsatellite mutation frequencies due to wild-type replicative holoenzyme errors during DNA replication.
| Orientation | Strand | Sequence | Pol EFa |
|---|---|---|---|
| 1 | Leading (ypol ε) | GT | 0.86 × 10-3 |
| Lagging (ypol δ) | CA | 1.7 × 10-3 | |
| Summation | GT + CA | 2.5 × 10-3 | |
| 2 | Leading (ypol ε) | CA | 2.4 × 10-3 |
| Lagging (ypol δ) | GT | 3.1 × 10-3 | |
| Summation | GT + CA | 5.5 × 10-3 | |
The polymerase indel error frequency (EF) was estimated as previously described [32]. Pol EF = [Overall MF – ssDNA MF] × proportion of errors within the indicated microsatellite sequence (data shown in Tables 1-3). This calculation may be an understimate, as it assumes that 100% of the mutations pre-existing within the ssDNA template are within the microsatellite.
Previously, the error rates of Pol δ WT and Pol ε WT during synthesis of seven consecutive thymines were determined using an in vitro lacZ gap-filling assay, similar to the HSV-tk assay [44]. Using those data, we calculated the error rate per repeat unit for Pol δ and Pol ε at the mononucleotide T allele versus the GT dinucleotide allele (Table 6). This analysis suggests that the fidelity of Pol δ and Pol ε for mononucleotide microsatellite DNA synthesis may be even lower, as the polymerases created errors more often within the [T]7 allele than within the [GT]10 allele. However, the effects of repeat unit size (mono-, di- tetra) on replicative polymerase error rates must be tested directly in future studies using the same mutational assay.
Table 6. Comparison of wild-type polymerase error rates within mononucleotide and dinucleotide tandem repeats.
| [T]7 Mononucleotidea (×10-4) | [GT]10 Dinucleotide (×10-4) | Ratio | |||
|---|---|---|---|---|---|
| Error rate | Error rate/repeat |
Error rate | Error rate/repeat |
Mono/di | |
| Pol δ | 310 | 44 | 25 | 2.5 | 18 |
| Pol ε | 23 | 3.3 | 9.0 | 0.9 | 3.7 |
Data originally reported in Table IV of Fortune et. al. 2005 [44].
We also examined the importance of the intrinsic 3′ to 5′ exonuclease activity to microsatellite sequence variation. The frequency of unit-based microsatellite indel errors produced by exonuclease-deficient enzymes differed by less than 1.4-fold from that measured for the corresponding exonuclease-proficient enzymes, with the exception of Pol δ and the CA allele (Figure 3). Interestingly, the exonucleases tend to preferentially remove interruption errors within the microsatellite alleles (Table 3). Such interruptions, if maintained over several rounds of DNA replication, would be expected to stabilize genomic microsatellites by breaking an allele into two, shorter tandem arrays that mutate at lower frequencies than the parent allele. Thus, the normally protective proofreading function may act instead to promote genome instability within microsatellite sequences. These in vitro results are consistent with previous in vivo studies showing that the proofreading activities of both Pol δ and Pol ε are inefficient at recognizing and repairing indel mutations in [GT/CA] repeats [29]. The low proofreading efficiency within microsatellites may be due to stabilization of the misaligned intermediate over the entire length of the repeated array, resulting in minimal distortion of the DNA substrate. Structurally, the mere physical distance over which to incorporate a bulge of unpaired bases within a long repetitive sequence may result in the physical separation of the polymerase active site from the misalignment, such that the bulge is rendered invisible to the proofreading function.
Evolutionary models of microsatellite mutation have been developed for use in estimates of genetic distances between populations (reviewed in [2]). The widely used stepwise mutation models for microsatellite mutation assume that insertions or deletions of a single unit occur at fixed rates as a function of allele length. A full 20% of the indel mutations created by the wild-type replicative polymerases within the dinucleotide alleles were deletions of two or more repeat units, as measured in the HSV-tk assay (Table 3). These observations suggest that Pol δ and Pol ε may be able to accommodate large loops of extrahelical nucleotides during extension synthesis. Alternatively, the multi-unit deletions may result from the simultaneous formation of multiple, single unit bulges within the long [GT/CA]10 repeated tract. Further investigation is required to elucidate the mechanistic underpinnings of the multi-unit repeat deletions. Regardless of mechanism, the incidence of multi-unit deletions within the [GT/CA] microsatellite should be taken into account in future mathematical models of microsatellite evolution.
Finally, small strand biases in replicative polymerase fidelity during [GT]10 versus [CA]10 microsatellite synthesis were observed (Figure 2). This bias is intriguing, as a current model for genome replication is that Pol δ synthesizes the lagging strand template [40] and Pol ε synthesizes the leading strand template [41]. The in vitro data presented here predict a strand bias in microsatellite mutability may exist in vivo, depending on the position of the GT versus the CA dinucleotide sequence relative to the origin of DNA replication (Table 5). We plan to further investigate a microsatellite strand bias for DNA polymerase errors in vivo, using a yeast reporter cassette adjacent to a well-defined origin of replication in a yeast strain with specialized Pol δ and Pol ε mutator alleles with wild type catalytic activity and strong mutational specificity [40,41]. In yeast, differences in the specificity and efficiency of MMR correction have been observed among A10, T10, C10, and G10 alleles [47,48]. The results of this study support the hypothesis that these biases reflect, in part, the error specificities of the replicative polymerases that initiate the mutation.
5. Conclusions
The high replicative DNA polymerase fidelity associated with synthesis of gene target sequences is not maintained during microsatellite DNA synthesis. Both Pol δ and Pol ε holoenzymes produce a high frequency of indel errors within the [GT/CA] microsatellite sequence. While the majority of indel errors are deletion of one repeat unit, a significant proportion (∼20%) are of multiple units. The proofreading exonuclease activities of polymerases δ and ε contribute little to the repair of unit-based indel errors within the microsatellite. The B family DNA polymerases δ and ε are widely believed to be responsible for the bulk of DNA replication elongation synthesis within eukaryotic genomes, but also have been implicated in the DNA synthesis associated with several DNA repair pathways and recombination [43]. The high holoenzyme error rates within microsatellites, coupled with the low efficiency of proofreading correction of polymerase indel errors, is expected to place a high burden of premutational intermediates upon the mismatch repair system in vivo.
Supplementary Material
Acknowledgments
The DNA sequencing Research Support group at the NIEHS completed the sequencing reactions. We thank Ms. Noelle Strubczweski for determining the unfilled gapped substrate background mutant frequency. We also thank E. Johansson and E.B. Lundström for providing purified Pol ε exonuclease deficient enzyme for in vitro reactions. This work was supported by grant R01 CA100060 from the National Institutes of Health to K.A.E and by project Z01 ES065070 to T.A.K from the Department of Intramural Research of the National Institutes of Health. The funding sources had no role of any type in the study.
Abbreviations used
- EF
error frequency
- GD
gapped duplex
- Indel
insertion/deletion
- MF
mutant frequency
- MMR
mismatch repair
- Pol δ
yeast DNA polymerase delta holoenzyme
- Pol ε
yeast DNA polymerase epsilon holoenzyme
Footnotes
S.E. Hile, M. Y.W.T. Lee, and K.A. Eckert, manuscript in preparation
6. Conflict of Interest statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Subramanian S, Mishra RK, Singh L. Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions. Genome Biol. 2003;4:R13. doi: 10.1186/gb-2003-4-2-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435–445. doi: 10.1038/nrg1348. [DOI] [PubMed] [Google Scholar]
- 3.Kelkar YD, Strubczewski N, Hile SE, Chiaromonte F, Eckert KA, Makova KD. What Is a Microsatellite: A Computational and Experimental Definition Based upon Repeat Mutational Behavior at A/T and GT/AC Repeats. Genome Biol Evol. 2010;2:620–635. doi: 10.1093/gbe/evq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannan AJ. Tandem repeat polymorphisms: modulators of disease susceptibility and candidates for ‘missing heritability’. Trends Genet. 2010;26:59–65. doi: 10.1016/j.tig.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Chu CS, Trapnell BC, Curristin S, Cutting GR, Crystal RG. Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat Genet. 1993;3:151–156. doi: 10.1038/ng0293-151. [DOI] [PubMed] [Google Scholar]
- 6.Cuppens H, Lin W, Jaspers M, Costes B, Teng H, Vankeerberghen A, Jorissen M, Droogmans G, Reynaert I, Goossens M, Nilius B, Cassiman JJ. Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (Tg)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J Clin Invest. 1998;101:487–496. doi: 10.1172/JCI639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buerger H, Packeisen J, Boecker A, Tidow N, Kersting C, Bielawski K, Isola J, Yatabe Y, Nakachi K, Boecker W, Brandt B. Allelic length of a CA dinucleotide repeat in the egfr gene correlates with the frequency of amplifications of this sequence--first results of an inter-ethnic breast cancer study. J Pathol. 2004;203:545–550. doi: 10.1002/path.1542. [DOI] [PubMed] [Google Scholar]
- 8.Gebhardt F, Zanker KS, Brandt B. Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. J Biol Chem. 1999;274:13176–13180. doi: 10.1074/jbc.274.19.13176. [DOI] [PubMed] [Google Scholar]
- 9.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135:1079–1099. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imai K, Yamamoto H. Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis. 2008;29:673–680. doi: 10.1093/carcin/bgm228. [DOI] [PubMed] [Google Scholar]
- 12.Levinson G, Gutman GA. Slipped strand mispairing: a major mechanism for DNA sequence evolution. Molecular Biology Evolution. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- 13.Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, Inouye M. Frameshift mutations and the genetic code. This paper is dedicated to Professor Theodosius Dobzhansky on the occasion of his 66th birthday. Cold Spring Harb Symp Quant Biol. 1966;31:77–84. doi: 10.1101/sqb.1966.031.01.014. [DOI] [PubMed] [Google Scholar]
- 14.Streisinger G, Owen J. Mechanisms of spontaneous and induced frameshift mutation in bacteriophage T4. Genetics. 1985;109:633–659. doi: 10.1093/genetics/109.4.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Structural analysis of strand misalignment during DNA synthesis by a human DNA polymerase. Cell. 2006;124:331–342. doi: 10.1016/j.cell.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 16.Bebenek K, Garcia-Diaz M, Foley MC, Pedersen LC, Schlick T, Kunkel TA. Substrate-induced DNA strand misalignment during catalytic cycling by DNA polymerase lambda. EMBO Rep. 2008;9:459–464. doi: 10.1038/embor.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ripley LS, Clark A, deBoer JG. Spectrum of spontaneous frameshift mutations. Sequences of bacteriophage T4 rII gene frameshifts. J Mol Biol. 1986;191:601–613. doi: 10.1016/0022-2836(86)90448-1. [DOI] [PubMed] [Google Scholar]
- 18.Schaaper RM, Dunn RL. Spectra of spontaneous mutations in Escherichia coli strains defective in mismatch correction: the nature of in vivo DNA replication errors. Proc Natl Acad Sci U S A. 1987;84:6220–6224. doi: 10.1073/pnas.84.17.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaaper RM, Dunn RL. Spontaneous mutation in the Escherichia coli lacI gene. Genetics. 1991;129:317–326. doi: 10.1093/genetics/129.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunkel TA. Frameshift mutagenesis by eucaryotic DNA polymerases in vitro. J Biol Chem. 1986;261:13581–13587. [PubMed] [Google Scholar]
- 21.Kunkel TA, Patel SS, Johnson KA. Error-prone replication of repeated DNA sequences by T7 DNA polymerase in the absence of its processivity subunit. Proc Natl Acad Sci U S A. 1994;91:6830–6834. doi: 10.1073/pnas.91.15.6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckert KA, Hile SE. Every microsatellite is different: Intrinsic DNA features dictate mutagenesis of common microsatellites present in the human genome. Mol Carcinog. 2009;48:379–388. doi: 10.1002/mc.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weirdl M, Dominska M, Petes TD. Microsatellite instability in yeast: dependence on the length of the microsatellite. Genetics. 1997;146:769–779. doi: 10.1093/genetics/146.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petes TD, Greenwell PW, Dominska M. Stabilization of microsatellite sequences by variant repeats in the yeast Saccharomyces cerevisiae. Genetics. 1997;146:491–498. doi: 10.1093/genetics/146.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bebenek K, Kunkel TA. Streisinger revisited: DNA synthesis errors mediated by substrate misalignments. Cold Spring Harb Symp Quant Biol. 2000;65:81–91. doi: 10.1101/sqb.2000.65.81. [DOI] [PubMed] [Google Scholar]
- 26.Morrison A, Sugino A. The 3′-->5′ exonucleases of both DNA polymerases delta and epsilon participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol Gen Genet. 1994;242:289–296. doi: 10.1007/BF00280418. [DOI] [PubMed] [Google Scholar]
- 27.Albertson TM, Ogawa M, Bugni JM, Hays LE, Chen Y, Wang Y, Treuting PM, Heddle JA, Goldsby RE, Preston BD. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A. 2009;106:17101–17104. doi: 10.1073/pnas.0907147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldsby RE, Hays LE, Chen X, Olmsted EA, Slayton WB, Spangrude GJ, Preston BD. High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc Natl Acad Sci U S A. 2002;99:15560–15565. doi: 10.1073/pnas.232340999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strand M, Prolla TA, Liskay RM, Petes TD. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature. 1993;365:274–276. doi: 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- 30.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroutil LC, Register K, Bebenek K, Kunkel TA. Exonucleolytic proofreading during replication of repetitive DNA. Biochemistry. 1996;35:1046–1053. doi: 10.1021/bi952178h. [DOI] [PubMed] [Google Scholar]
- 32.Eckert KA, Mowery A, Hile SE. Misalignment-mediated DNA polymerase beta mutations: comparison of microsatellite and frame-shift error rates using a forward mutation assay. Biochemistry. 2002;41:10490–10498. doi: 10.1021/bi025918c. [DOI] [PubMed] [Google Scholar]
- 33.Fortune JM, Stith CM, Kissling GE, Burgers PM, Kunkel TA. RPA and PCNA suppress formation of large deletion errors by yeast DNA polymerase delta. Nucleic Acids Res. 2006;34:4335–4341. doi: 10.1093/nar/gkl403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chilkova O, Jonsson BH, Johansson E. The quaternary structure of DNA polymerase epsilon from Saccharomyces cerevisiae. J Biol Chem. 2003;278:14082–14086. doi: 10.1074/jbc.M211818200. [DOI] [PubMed] [Google Scholar]
- 35.Jin YH, Ayyagari R, Resnick MA, Gordenin DA, Burgers PM. Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3′-5′-exonuclease activities of Pol delta in the creation of a ligatable nick. J Biol Chem. 2003;278:1626–1633. doi: 10.1074/jbc.M209803200. [DOI] [PubMed] [Google Scholar]
- 36.Jin YH, Obert R, Burgers PM, Kunkel TA, Resnick MA, Gordenin DA. The 3′-->5′ exonuclease of DNA polymerase delta can substitute for the 5′ flap endonuclease Rad27/Fen1 in processing Okazaki fragments and preventing genome instability. Proc Natl Acad Sci U S A. 2001;98:5122–5127. doi: 10.1073/pnas.091095198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shcherbakova PV, Pavlov YI, Chilkova O, Rogozin IB, Johansson E, Kunkel TA. Unique error signature of the four-subunit yeast DNA polymerase epsilon. J Biol Chem. 2003;278:43770–43780. doi: 10.1074/jbc.M306893200. [DOI] [PubMed] [Google Scholar]
- 38.Hile SE, Eckert KA. DNA polymerase kappa produces interrupted mutations and displays polar pausing within mononucleotide microsatellite sequences. Nucleic Acids Res. 2008;36:688–696. doi: 10.1093/nar/gkm1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eckert KA, Hile SE, Vargo PL. Development and use of an in vitro HSV-tk forward mutation assay to study eukaryotic DNA polymerase processing of DNA alkyl lesions. Nucleic Acids Res. 1997;25:1450–1457. doi: 10.1093/nar/25.7.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pursell ZF, Kunkel TA. DNA polymerase epsilon: a polymerase of unusual size (and complexity) Prog Nucleic Acid Res Mol Biol. 2008;82:101–145. doi: 10.1016/S0079-6603(08)00004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bebenek K, Kunkel TA. Functions of DNA polymerases. Adv Protein Chem. 2004;69:137–165. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 44.Fortune JM, Pavlov YI, Welch CM, Johansson E, Burgers PM, Kunkel TA. Saccharomyces cerevisiae DNA polymerase delta: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J Biol Chem. 2005;280:29980–29987. doi: 10.1074/jbc.M505236200. [DOI] [PubMed] [Google Scholar]
- 45.Hile SE, Eckert KA. Positive correlation between DNA polymerase alpha-primase pausing and mutagenesis within polypyrimidine/polypurine microsatellite sequences. J Mol Biol. 2004;335:745–759. doi: 10.1016/j.jmb.2003.10.075. [DOI] [PubMed] [Google Scholar]
- 46.Gordenin DA, Resnick MA. Yeast ARMs (DNA at-risk motifs) can reveal sources of genome instability. Mutation Research. 1998;400:45–58. doi: 10.1016/s0027-5107(98)00047-5. [DOI] [PubMed] [Google Scholar]
- 47.Gragg H, Harfe BD, Jinks-Robertson S. Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol Cell Biol. 2002;22:8756–8762. doi: 10.1128/MCB.22.24.8756-8762.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harfe BD, Jinks-Robertson S. Sequence composition and context effects on the generation and repair of frameshift intermediates in mononucleotide runs in Saccharomyces cerevisiae. Genetics. 2000;156:571–578. doi: 10.1093/genetics/156.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.