

Abstract
Glycerophospholipids represent a common class of lipids critically important for integrity of cellular membranes. Oxidation of esterified unsaturated fatty acids dramatically changes biological activities of phospholipids. Apart from impairment of their structural function, oxidation makes oxidized phospholipids (OxPLs) markers of “modified-self” type that are recognized by soluble and cell-associated receptors of innate immunity, including scavenger receptors, natural (germ line-encoded) antibodies, and C-reactive protein, thus directing removal of senescent and apoptotic cells or oxidized lipoproteins. In addition, OxPLs acquire novel biological activities not characteristic of their unoxidized precursors, including the ability to regulate innate and adaptive immune responses. Effects of OxPLs described in vitro and in vivo suggest their potential relevance in different pathologies, including atherosclerosis, acute inflammation, lung injury, and many other conditions. This review summarizes current knowledge on the mechanisms of formation, structures, and biological activities of OxPLs. Furthermore, potential applications of OxPLs as disease biomarkers, as well as experimental therapies targeting OxPLs, are described, providing a broad overview of an emerging class of lipid mediators. Antioxid. Redox Signal. 12, 1009–1059.
-
III. Accumulation and Potential Role of OxPLs in Pathology
-
IV. OxPLs as Biomarkers of Disease and Targets for Therapy
I. Mechanisms of Phospholipid Oxidation
A wealth of data points to oxidized lipids as markers and pathogenic factors in a variety of disease states. A prototype example demonstrating the importance of lipid oxidation in pathology is oxidation of low density lipoproteins (LDL), a process well studied in vitro and thought to play a key role during initiating stages of atherogenesis (66, 258, 277, 323). Studies performed in the 1990s identified oxidized phospholipids (OxPLs) as the major active principle of minimally modified (oxidized) LDL (MM-LDL) responsible for their ability to initiate monocytic inflammation characteristic of atherosclerosis (21, 368). Later work showed potential relevance of OxPLs to a number of other pathologies. This review provides an update of this rapidly growing field and describes emerging topics that were not covered by previous reviews (11, 19, 75, 134), for example, the role of OxPLs in adaptive immunity and cellular stress reactions. The review is focused on biological activities of OxPLs, but also describes major types of OxPLs and mechanisms of their generation and catabolism.
A. Oxidation of PLs: General mechanisms and biologically active products
Glycerophospholipids comprise an abundant class of lipids consisting of a glycerol backbone, phosphate-containing polar head group and two fatty acid residues. PL-bound polyunsaturated fatty acids (PUFAs) represent the major target for nonenzymatic or enzymatic oxidation that is not linked to the generation of metabolic energy. Although oxidative modifications of polar head groups have also been reported (316, 386), the biological importance of these reactions in insufficiently studied and will not be discussed here.
Oxidative fragmentation of a PL molecule generates several biologically active products, including small chemically reactive fragments of PUFAs, such as unesterified oxidized fatty acids (e.g., hydroperoxides and isoprostanes) and lyso-phospholipids. These products demonstrate multiple biological activities but will not be discussed here since the review specifically focuses on oxidation products having complete PL structure: glycerol backbone linked to polar head groups and two fatty acid residues, one or both of which are oxidized.
Available evidence suggests that nonenzymatic oxidation of PL-PUFAs proceeds according to the same basic mechanisms as oxidation of free (unesterified) PUFAs. This assumption is supported by identification of similar classes of molecular species generated by oxidation of free and PL-bound PUFAs that are described below. In contrast to the nonenzymatic oxidation, oxidation of PL-PUFAs by enzymes significantly differs from oxidation of unesterified PUFAs. While free PUFAs can be oxidized by multiple enzymes belonging to different protein families and introducing various oxidized groups, only one group of lipoxygenases (12/15 lipoxygenases) accepts PL-PUFAs as substrates producing PL-hydroperoxides (376). Further oxidation and rearrangements continue without participation of enzymes, and therefore oxidation initiated by enzymatic and nonenzymatic mechanisms produces many similar advanced PL oxidation products. Below follows a brief overview of pathways generating OxPLs. The description is focused on PUFA oxidation products that were shown to exist in PL-esterified form. Therefore, some original publications describing oxidation of unesterified PUFAs are not cited.
B. Initiation of oxidation
1. Nonenzymatic oxidation of PL-PUFAs
Nonenzymatic oxidation of PLs containing mono- and polyunsaturated fatty acids can be initiated by free radicals or nonradical reactive oxygen species (ROS) (Fig. 1). Free radical-mediated chain reaction initiated by formation of carbon-centered radicals and/or hydroperoxides of PUFAs (peroxidation of PUFAs) is one of the best understood and biologically relevant oxidative processes. The mechanisms described below were characterized for free PUFAs, but they are likely to be similar for PL-bound PUFAs.
FIG. 1.

Mechanisms initiating peroxidation of PUFAs esterified in phospholipids. Peroxidation of phospholipids containing PUFAs is initiated via both enzymatic and nonenzymatic mechanisms. Lipoxygenases from the 12/15 family accept PL-esterified PUFAs as substrate and insert dioxygen, producing hydroperoxides. PUFA peroxidation can also be induced by nonradical ROS (singlet oxygen), or by free radicals, which either penetrate from the environment, or are produced endogenously by enzymes, such as NADPH oxidase (NOX), myeloperoxidase (MPO), nitric oxide synthase (NOS), xanthine oxidase (XO), or respiratory chain in mitochondria. Reactions induced by different free radicals followed by addition of oxygen produce the same type of primary oxidation products (i.e., peroxyl radicals), which in turn transform into hydroperoxides after reacting with other PUFA molecules. *Cyt c is selective for cardiolipin and phosphatidylserine as compared to PC (164).
PUFAs are more susceptible to oxidation as compared to saturated FAs due to the presence of methylene groups located between double bonds (bisallylic methylene groups) and as a result characterized by weakened hydrogen-carbon bonds. As a consequence, free radicals easily abstract hydrogen from bisallylic methylene leading to the formation of carbon-centered radicals within PUFAs. Carbon-centered radicals rapidly react with molecular oxygen, producing peroxyl radicals. This reaction represents the initiation step of lipid peroxidation. Peroxyl radicals react with bisallylic methylene groups in other PUFA molecules, leading to the transformation of peroxyl radicals to hydroperoxides and generation of new carbon-centered radicals. Thus, additional cycles of peroxidation are initiated. PUFA hydroperoxides in turn produce reactive alkoxyl and hydroxyl radicals via iron- or copper-catalyzed Fenton-like reactions, further propagating the chain reaction.
2. Sources of free radicals inducing oxidation of PLs
Oxidation of PUFAs can be induced by organic or inorganic free radicals. In addition to exogenous physical and chemical sources, such as air pollution, smoking, UV-light, or ionizing radiation, also multiple endogenous sources of free radicals exist. Several enzymes (i.e., NADPH oxidase, xanthine oxidase, uncoupled NO synthase) and electron transport system in mitochondria (Fig. 1) produce superoxide anion radical that interacts with other radical and nonradical oxidant species to produce several biologically important oxidant molecules, the most important of which are presented in Fig. 1. Myeloperoxidase produces highly reactive radicals (e.g., NO2 radical) from ROS characterized by lower reactivity, such as H2O2.
Enzymes inducing oxidation of PLs due to their ability to generate free radicals should be distinguished from enzymes directly oxidizing PUFAs (i.e., lipoxygenases and cytochrome c) (Fig. 1).
3. Oxidation of PUFAs by nonradical ROS
PL-hydroperoxides can be formed as a result of oxidation by unstable, energy-rich singlet molecular oxygen characterized by significantly higher reactivity as compared to ground state (triplet) oxygen (117). Singlet oxygen is generated by recombination of other ROS (224), or produced from ground-state triplet oxygen after irradiation with UV-light in the presence of endogenous photosensitizers such as all-trans retinal, NAD(P)H, FAD, etc. (12, 377). UV-light was shown to stimulate oxidation of PUFA-PLs in vitro and in vivo (125, 128, 331).
Ozone, which is produced in the atmosphere by photochemical reactions and also generated by industry, represents an important oxidizing air pollutant attacking epithelial cells and surfactant of lungs. Ozone readily reacts with double bonds in mono- and polyunsaturated fatty acids yielding fragmented species, such as 1-palmitoyl-2-(9-oxononanoyl)-PC that was found in lung surfactant extracts treated with ozone (349). Ozonolysis is used for generation of oxidatively fragmented saturated species of OxPLs (294).
4. Nitration and halogenation of PLs
In addition to peroxidation mechanism resulting in insertion of oxygen into PUFA residues, PLs can be oxidized by halogen- and nitrogen-containing compounds leading to the formation of PL-PUFAs containing Cl or Br atoms, or NO2 group.
NO2 is a highly reactive radical molecule produced by myeloperoxidase or by nonenzymatic reactions (e.g., breakdown of peroxinitrite) (274). NO2 can induce peroxidation of PUFAs (51); however, in addition to that, reaction of NO2 with mono- and polyunsaturated PL-esterified FAs can generate nitrated forms (246). The balance between oxidation and nitration of PUFA is determined by several factors including NO2 concentration and oxygen tension (103). Presence of esterified (including PL-esterified) nitrated FAs has been documented in normal human plasma and red blood cells (13). Biological activities of nitrated PLs were not studied.
MPO and eosinophil peroxidase produce HClO and HBrO, which are able to modify PLs via two types of reactions (i.e., PL peroxidation or halogenation). Recombination of HClO with oxygen superoxide anion, Fe2+, or hydrogen peroxide generates highly reactive hydroxyl radical or singlet oxygen, both of which are potent inducers of lipid peroxidation (205). Direct oxidation of PL-esterified FAs by HClO and HBrO results in insertion of halogenide and hydroxy groups into double bonds leading to formation of halohydrins (53, 158, 374). Alternatively, HClO reacts with amino groups of PLs to form N-chloramines of PE and PS (172). N-chloramines of PE readily break down with formation of N-centered radicals, which can further initiate PL peroxidation (172). Both peroxidation and chlorination of PLs were detected in HClO-treated lipoproteins and cells (158, 317). Chlorohydrins of PL-esterified oleic acid were shown to induce proinflammatory effects in vascular wall (79).
5. Enzymatic oxidation of PUFA-PLs
Lipoxygenases (LOXs) recognize 1,4-pentadiene motifs within unsaturated fatty acids and introduce molecular oxygen with high regio- and stereoselectivity producing hydroperoxides. LOXs are classified according to the position of oxygen insertion in the arachidonate substrate. The majority of LOXs (e.g., physiologically important 5-LOX enzymes) oxidize only unesterified PUFAs. Among known LOXs, only one group (12/15-LOX) is capable of oxidizing PL-esterified fatty acids. This class of enzymes is present in different biological species and includes mouse, rat, rabbit, bovine, and porcine leukocyte-type 12-LOX, rabbit and human reticulocyte-type 15-LOX, and soybean LOX (146, 376). These enzymes can oxidize PL-PUFA substrates in micellar form and in organized structures such as cellular membranes (183) or lipoprotein particles (16).
Available evidence suggests that 12/15-LOXs may play a role in pathological lipid oxidation. Oxidation of PLs by mammalian 15-LOX selectively generates (S)-isomers of esterified hydroperoxides. In contrast, nonenzymatic lipid oxidation produces a racemic mixture of (R)- and (S)-hydroperoxides. The analysis of 15-LOX products by chiral chromatography showed accumulation of (S)-isomers at early stages of atherosclerotic lesion formation, thus pointing to the important role of this enzyme as initiator of PL oxidation in vivo (184). However, in advanced lesions enantioselectivity of 15-LOX products was lost, suggesting that nonenzymatic mechanisms also play important role in oxidation of lipids in arterial wall. Which factors determine the balance between enzymatic and nonenzymatic oxidation of PLs at initiating and advanced stages of atherosclerosis and other pathologies, requires further investigation.
It has been suggested that cytochrome c (cyt c) may switch its activity from electron transport in mitochondria to peroxidase activity (166). The transformation begins from binding of cyt c to negatively charged cardiolipin (CL), leading to conformational changes with consequent release of PL-protein complex from mitochondria into cytosol. The complex of cyt c with CL upon activation by traces of PUFA-OOH or H2O2 acquires the ability to oxidize CL, PS, or PI, with formation of PL-OOH (164). CL-containing linoleic acid residues were preferentially oxidized by cyt c in vitro (149, 345) and in vivo (15, 347). Other PLs like PC or PE were not peroxidized by cyt c in spite of much higher unsaturation grade (164). The PL-peroxidation caused by cyt c may be essential for cell apoptosis (149, 346).
An alternative mechanism generating OxPLs is re-esterification of free oxidized PUFAs into lyso-PLs. Several types of OxPLs were shown to be formed via this mechanism in vitro and in vivo, including PLs containing monohydroxylated PUFAs (6, 27, 324), PUFA epoxides (86), esterified hepoxillins, and other oxidized residues (5). 5- and 12-LOXs and enzymes from 2C and 2J classes of cytochrome P-450 are examples of enzymes oxidizing free PUFAs whose products can be re-esterified into PLs (5, 42, 69, 169, 315). Further studies are required in order to clarify the impact of re-esterification as compared to other enzymatic and nonenzymatic pathways producing OxPLs under normal and pathological conditions.
C. Evolution of OxPLs
Oxidation of PUFAs can be initiated by a wide spectrum of reactions including enzymatic and nonenzymatic, free-radical, and radical-free processes. However, the vast majority of these reactions produce identical primary oxidation products (i.e., peroxyl radicals and hydroperoxides). Following the initiating step, subsequent oxidation of OxPLs is an enzyme-independent stochastic process producing a wide spectrum of OxPLs (Fig. 2).
FIG. 2.

Oxidation of esterified PUFA generates a variety of nonfragmented and fragmented OxPLs. Top panel presents mass spectrum of synthetic palmitoyl-arachidonoyl-phosphatidylcholine (PAPC). Bottom panel shows multiple products generated from PAPC upon prolonged exposure of pure dry lipid to air. Note formation of multiple fragmented (m/z < 782) and nonfragmented (m/z > 782) OxPAPC species that were generated by nonenzymatic oxidation of just one precursor molecule.
Generation of advanced peroxidation products proceeds according to several major mechanisms finally generating either full-length residues incorporating several oxygen atoms, or shortened fatty acid residues. These mechanisms include further oxidation of PUFA residue, cyclization of peroxyl radical, or oxidative fragmentation of esterified PUFAs (Figs. 3 and 4).
FIG. 3.

Evolution of phospholipid oxidation products. Peroxidation of PL-esterified PUFAs is initiated by formation of hydroperoxides or peroxyl radicals. Further evolution of primary PL oxidation products proceeds without participation of enzymes via three major pathways. First, additional oxidation within the same PUFA generates OxPLs with various combinations of functional groups such as hydroperoxides, hydroxides, keto- and epoxy-groups. Second pathway involves intramolecular cyclization, rearrangement, and further oxidation. If bicyclic endoperoxide is formed as an intermediate product, three groups of products are generated, including isoprostanes, isolevuglandins, and isothromboxanes, while cyclization leading to formation of monocyclic peroxide finally produces isofurans. Third group of transformations results from several chemical reactions all leading to fragmentation of PUFAs and generation of short residues having various combinations of hydroxide and carbonyl groups, or terminal furan.
FIG. 4.

Major forms of oxidatively modified residues that were identified in OxPLs. The figure presents example structures from all groups of PUFA oxidation products that were shown to exist in PL-esterified form. R is 1-acyl-2-lyso-sn-glycero-3-X; X, phosphocholine, phosphoserine, phosphoethanolamine, phosphoinositol, phosphoglycerol, phosphate; R' is an alkyl residiue.
1. Formation of polyoxygenated PLs
Introduction of additional oxygen atoms into PUFAs is a common mechanism increasing complexity of OxPL mixtures. Since monohydroperoxides of PUFAs contain double bonds, further peroxidation is possible, yielding advanced oxidation products with variable combinations of hydroxy-, hydroperoxy-, keto-, and epoxy groups. Some of these complex OxPLs, including dihydroxy-PC and epoxy-monohydroxy-PC, were detected in vivo (81). Biological activities of polyoxygenated PLs are not characterized.
2. Cyclization of peroxyl radical/generation of nonfragmented OxPLs
Cyclization of peroxyl radical produces cyclic peroxide, which undergoes either rearrangements yielding bicyclic endoperoxide, or oxidation introducing additional noncyclic or cyclic peroxide group. Cyclization of peroxyl radical is only possible for FAs having three or more double bonds (291). Bicyclic endoperoxide is a precursor of isoprostanes, isothromboxanes, and isolevuglandins, whereas rearrangement and further oxidation of molecules containing one cyclic and one noncyclic peroxide groups produces isofurans (282).
a. Esterified isoprostanes
The name “isoprostanes” originates from prostanoic acid, a 20-carbon carboxylic acid containing cyclopentane ring (also called prostane ring) within the aliphatic chain. Isoprostanes (isoPs) are prostaglandin-like compounds that differ from prostaglandins by stereo isomeric position of side chains relatively to cyclopentane ring, as well as stereo position of substituents within the prostanoic acid residue. The side chains in isoPs are located predominantly in cis- orientation in contrast to trans-position in prostaglandins (282). Moreover, naturally produced isoPs consist of several regio isomers, while prostaglandins are regio-specific. Prostaglandins are produced from unesterified arachidonic acid via enzymatic conversion by cyclooxygenases followed by PG-synthases (313). In contrast, isoPs are produced in vivo mainly from PL-esterified arachidonic acid via a free radical-induced oxidation (227). Bicyclic endoperoxide isoPG is formed as a random mixture of regio- and stereoisomers. As a result, rearrangement of bicyclic endoperoxides generates all possible stereoisomers of four regio isomers of isoPs, including stereospecific prostaglandins (104). Several classes of isoPs differing by type of substitutions and their location within the cyclopentane ring are formed, including F, E, and D series of isoPs (222). IsoPs of F-series are stable and therefore are widely used as markers of oxidative stress, both as PL-esterified and free forms (225), whereas isoPs E and D undergo spontaneous dehydration within the cyclopentane ring, yielding cyclopentenone-isoPs A and J, respectively (62). Formation of cyclopentenone-isoPs at least partially occurs in PL-esterified form since cyclopentenone-isoP-PLs were found in vivo (e.g., in liver and brain) (62, 231). Cyclopentenone-isoPs are highly reactive and readily form covalent complexes with reduced glutathione and cysteine residues of proteins (223).
An alternative way of transformation of bicyclic endoperoxide-containing PLs is formation of epoxyisoprostane-PLs such as PEIPC (Fig. 4) (370). Similarly to other isoPs, different regio- and stereo isomers of epoxyisoprostane-PC are formed (329). Dehydration of the cyclic isoprostane ring of PEIPC produces PECPC containing highly reactive cyclopentenone group (329). Elevated levels of PEIPC and PECPC were detected in ECs upon stimulation with inflammatory cytokines (329). Different positional isomers of PEIPC demonstrate similar ability to induce IL-8 expression and stimulate monocyte binding by ECs (329). In addition, epoxyisoprostane-PC demonstrates anti-inflammatory (360), angiogenic (40), and endothelial barrier-protective properties (28), and also induces antioxidant genes (125, 161).
Isoprostane-like compounds analogous to isoPs F, A, and J can be formed from PUFAs having three or more double bonds, including linolenic acid, eicosapentaenoic acid, adrenic acid, and docosahexaenoic acids (50, 154, 356, 383). PL-esterified forms of isoP-like compounds were detected in vivo in normal human samples (50, 280, 356, 383).
b. Esterified isothromboxanes
Apart from formation of (epoxy)isoprostanes, bicyclic endoperoxide spontaneously transforms into isothromboxane (isoTx) A2 (Fig. 4), which undergoes further transformation into isoTxB2 (283). PL-esterified isoTxs were found in rat liver at concentrations comparable with isoPs, and were strongly elevated during oxidative stress induced by CCl4 (228). Biological activities of PL-esterified isoTx A2 and B2 were not studied.
c. Esterified isolevuglandins
Isolevuglandins (also called isoketals, Fig. 4) represent an additional product of bicyclic endoperoxide rearrangement (292). Similarly to isoPs, isolevuglandins (isoLGs), are produced mainly in PL-esterfied form (290). Due to high reactivity of aldehyde groups, isoLG-PLs rapidly form covalent complexes with proteins. Following phospholipase cleavage, isoLG-protein adducts are formed that were found in oxidized LDL and inflamed tissues, and due to their long half-life in circulation were suggested as integral markers of oxidative stress (267, 293).
d. Esterified isofurans
Isofurans (Fig. 4) are PUFA oxidation products with a substituted tetrahydrofuran ring within the aliphatic chain (94). Isofurans can be formed either from cyclic peroxide or from monohydroperoxide according to different mechanisms (281). Formation of isofurans from cyclic peroxides is preferred over isoPs at enhanced oxygen tension (281). Isofuran-PLs are stable oxidation products and therefore can be used as conventional biomarkers of lipid oxidation. Isofurans formed from docosahexaenoic acid are called neurofurans (314). Elevated levels of PL-isofurans were found in hyperoxia-injured lungs (94), brain tissue from patients with Parkinson disease (93), and in brain samples of mouse model of Alzheimer disease (314).
3. Oxidative cleavage/formation of fragmented OxPL species
Fragmentation of hydroperoxides represents third important pathway whereby peroxides/peroxyls are transformed into advanced oxidation products.
γ-Hydroxy (or oxo) α,β-unsaturated PLs with terminal aldehyde groups (Fig. 4) are produced from hydroperoxides via oxidation/fragmentation or polymerization/cleavage. Oxidative fragmentation of hydroperoxides occurs via several mechanisms including β-scission, Hock rearrangement, or cyclization of alkoxy radical produced from hydroperoxide (128). On the other hand, peroxyl radical can cross-react with double bonds present in hydroperoxides yielding peroxydimers; these are unstable products and spontaneously break down forming either new radicals or α,β-unsaturated aldehydes (297).
a. Oxidatively truncated unsaturated OxPLs
γ-Hydroxy (or oxo)-α,β-unsaturated OxPLs containing terminal aldehyde (also called core aldehyde) or carboxyl groups (Fig. 4) demonstrate several biological activities, including interaction with scavenger receptor CD36 (264), induction of proinflammatory effects in ECs (327), and inhibition of toll-like receptor 4 (TLR4) (327). These OxPLs were found in vivo, and their levels are known to be elevated in plasma of hyperlipidemic mice, atherosclerotic plaques of mice and rabbits, and human damaged retina (263, 264, 331).
γ-Hydroxy (or oxo)-α,β-unsaturated aldehyde PLs are highly reactive compounds, able to covalently link to amino groups of proteins, as well as thiol groups of biomolecules (137). Alternatively, they can be further oxidized either to carboxylic α,β-unsaturated fragmented PLs or to saturated fragmented species.
OxPLs with terminal furan groups are produced from γ-hydroxy-α,β-unsaturated aldehyde PLs via spontaneous intramolecular reaction between aldehyde and hydroxyl groups (105) (Fig. 4). These are stable oxidation products generated in physiological oxidant systems in vitro and in vivo (105). Due to the loss of aldehyde groups, these compounds cannot react with proteins and are not toxic. In contrast to the parent γ-hydroxy-α,β-unsaturated aldehyde PLs, terminal-furan-PLs are not recognized by CD36 (105).
b. Oxidatively truncated saturated OxPLs
In addition to the products described above, oxidative fragmentation of PUFA-PLs also directly produces saturated fragmented species containing terminal carbonyl groups. Most common species are oxononanoate and azelaoate formed from linoleic acid, oxovaleroate, and glutaroate generated from arachidonic acid (Fig. 4), or oxobutyrate and succinate produced from docosahexaenoic acid (126, 265). In addition to direct formation from hydroperoxides, saturated fragmented OxPLs can be formed by further oxidation of γ-hydroxy (or oxo)-α, β-unsaturated PLs (265). Due to the lack of double bonds, saturated fragmented OxPLs are resistant to further oxidation. The absence of substituents and double bonds within fragmented chains results in diminished reactivity of aldehyde-containing saturated OxPLs as compared to α,β-unsaturated fragmented OxPLs. Thus, saturated fragmented OxPLs are chemically stable end products found inter alia in plasma and vessels of hypercholesterolemic patients and apoe−/− mice (263, 264, 368). These compounds demonstrate a variety of activities in vitro, including proinflammatory (368), angiogenic (40), anti-LPS effects (360), induction of antioxidant genes (161), and modulation of adaptive immune reactions (302).
D. Termination of PL-oxidation and detoxification of reactive OxPLs
Several processes contribute to the elimination and detoxification of OxPLs, including the mechanisms terminating peroxidation chain reaction and inactivating toxic chemically reactive groups produced by oxidation (Fig. 5). While the reactions of PL-esterified carbon-centered and peroxyl radicals with low molecular weight antioxidants are likely to be similar to those of free fatty acids, enzymatic pathways are different since only a limited number of enzymes accept OxPLs as substrates. In addition, enzymatic cleavage of OxPLs generating lyso-PLs and free oxidized fatty acids represents another level of regulation of activity and biodistribution of OxPLs.
FIG. 5.

Termination of phospholipid oxidation. Several processes play a role in termination of peroxidation chain reaction and detoxification of reactive groups in PL-esterified PUFAs. In addition to scavenging of radicals by antioxidants, reactive peroxide groups are reduced by specific form of glutathione peroxidase (GPx4) capable of reducing PL-esterified residues, as well as peroxiredoxin VI and glutathione transferase (GST). Reactive carbonyl groups in PL residues are reduced by aldo-keto-reductases from AKR1A and B families. Furthermore, several phospholipases A selectively cleave oxidized residues, leading to formation of lyso-PLs and free oxidized fatty acids. Similar activity is demonstrated by LCAT. Finally, electrophilic PLs can form covalent complexes with amino acids, which may inactivate reactive groups on PLs but on the other hand can damage sensitive proteins.
1. Enzymatic reduction of hydroperoxides
As compared to hydroperoxides, hydroxides are characterized by significantly lower chemical reactivity and therefore are considered to be stable and relatively non-toxic oxidation products (319). The most important type of enzyme catalyzing reduction of hydroperoxides to hydroxides is glutathione peroxidase (GPx). Lipid hydroperoxides are reduced in a reaction involving selenocysteine residue of GPx and glutathione. As a result, lipid hydroxide and oxidized glutathione are generated. Several species of GPx are known that are characterized by different tissue, cell and organelle distribution as well as variable substrate specificity (68). Among GPx enzymes, PL glutathione peroxidase (GPx4) has the highest activity in respect to membrane-bound hydroperoxides of PL-esterified PUFAs (295). GPx4 is expressed in many cell types where it is located in cytosol, mitochondria, and nucleus (295). This enzyme has been shown to suppress peroxidation of PLs induced by lipoxygenases (298) or UVA light (372).
Other enzymes such as peroxiredoxin VI (208) and glutathione transferase (379) can also reduce PL-hydroperoxides to corresponding hydroxides. In analogy to GPx, peroxiredoxin VI and glutathione transferase also use glutathione to recover their catalytic cysteines.
2. Reduction of carbonyls in OxPLs by aldo-keto reductases
Upon oxidation of OxPLs, a variety of products containing aldehyde and keto groups are formed. A class of enzymes called aldo-keto reductases transforms these functional groups to respective hydroxyl groups. Apart from their physiological role in metabolism of sugar aldehydes, aldo-keto reductases play a role in detoxification of toxic (phospho)lipid aldehydes (160). Aldo-keto reductases of the families AKR1A and B recognize as substrate aldehyde-containing OxPLs including diacyl-phosphatidylcholines, -ethanolamines, -glycerols, phosphatidic acids, as well as alkyl-acyl PCs (318, 320).
3. OxPL cleavage
Platelet activating factor acetylhydrolase (PAF-AH), also known as lipoprotein-associated phospholipase A2 (Lp-PLA2), is well recognized for its ability to cleave and thus inactivate PAF (217). This enzyme is very selective in respect to short sn-2 residues and does not cleave unmodified long-chain residues. However, upon oxidation of residues, this selectivity is lost and OxPLs become substrates for Lp-PLA2 even when they have nonfragmented oxidized sn-2 residues. The enzyme was shown to hydrolyze fragmented saturated OxPLs (326), as well as long-chain OxPLs, including esterified F2-isoprostanes, PC-hydroperoxides and PEIPC (73, 181, 321). In addition to circulating Lp-PLA2, also intracellular PAF-AH type II is known to hydrolyze fragmented OxPL species (133), and, with lower efficiency, isoP-PCs (321). Mice genetically deficient in PAF-AH type II demonstrated enhanced accumulation of esterified isoPs in response to CCl4-induced oxidative stress, and were more prone to oxidative injury (180). These data show that PAF-AH type II enzyme is important for degradation of PL-esterified isoPs and protection from oxidative stress in vivo. In addition to Lp-PLA2 and PAF-AH type II, also other isoforms of PLA2 can cleave OxPLs. Biolological importance of these reactions requires further investigation (241). Furthermore, it was hypothesized that paraoxonase-1 (PON-1), an HDL-associated organophosphonate triesterase, can cleave oxidized residues in OxPLs; however, more detailed study suggested that PON-1 has no PLA2 activity and that the phospholipase activity observed in previous experiments was explained by contamination of PON-1 preparations with Lp-PLA2 (212).
Lecithin:cholesterol acyltransferase (LCAT) catalyzes formation of cholesterol esters from free cholesterol present in lipoproteins using PCs and PEs as donors of acyl chains. As a result of the reaction catalyzed by LCAT, lyso-PLs and cholesterol esters are formed. The enzyme uses as substrates not only native but also oxidized PLs (233). In addition, LCAT was shown to hydrolyze OxPCs generating free oxidized residues rather than cholesterol esters (121). In contrast to PAF-AH, LCAT has preference for long-chain oxidation products as compared to OxPLs containing truncated residues (330). LCAT was shown to prevent accumulation of OxPLs and inhibit oxidation of LDL and PL micelles (358).
E. Formation of adducts
Formation of covalent adducts of OxPLs with proteins represents an additional pathway of their inactivation but at the same time can damage sensitive proteins. OxPLs containing aldehyde or electrophilic α,β-unsaturated carbonyl groups react with nucleophilic groups such as thiol and amino groups (for example, cysteinyl and lysyl residues of proteins) forming covalent Michael adducts or Schiff bases (126, 334) (Fig. 6). Spontaneous secondary reactions of Schiff bases produce pyrroles, lactams, and hydroxylactams that are stable protein–lipid adducts, which can be detected in vivo (392).
FIG. 6.

Chemical reactions of OxPLs containing ϖ-terminal aldehyde groups with amino- and sulfhydryl groups in proteins. OxPL residues having reactive carbonyl groups can form Schiff bases or Michael adducts with NH2- and SH-groups of proteins. Formation of covalent complexes can modulate activity and half-life of OxPLs, initiate electrophilic stress response and inactivate sensitive proteins.
Formation of covalent adducts with OxPLs can inactivate important amino acid residues or induce polymerization of proteins resulting in protein dysfunction. Formation of PL–isolevuglandin–protein adducts was shown to impair function of cardiac K+ channel (47). Furthermore, γ-hydroxy-α,β-unsaturated aldehyde-PCs react with cysteinyl residues of cathepsin B, forming Michael adducts and as a consequence reducing its proteolytic activity (137).
It is known that formation of covalent adducts with oxidized lipids plays a key role in activation of redox-sensitive transcription factor NRF2. Covalent reaction of cyclopentenone isoPs to specific cysteines within NRF2 partner protein KEAP-1 allows NRF2 to escape degradation and translocate to the nucleus, and thus promotes transcription of NRF2 target genes (195). For PL-esterified isoPs such covalent reaction has not yet been shown. However, Gugiu et al. showed that OxPLs can penetrate into the cell and covalently modify intracellular proteins (129). These data raise a possibility that OxPLs can directly regulate activity of redox-sensitive transcription factors inside the cell.
Many protein–OxPL adducts are stable and can serve as biomarkers of oxidative stress. The majority of circulating OxPCs, which can be recognized by specific E06 antibodies, is present in plasma in association with Lp(a). More than 85% of E06 immunoreactivity in human plasma co-immunoprecipitated with apo(a) (17). The linkage between OxPLs and Lp(a) is ∼50% covalent and ∼50% noncovalent (extractable lipids) (17). Two specific lysines of the kringle V domain of apo(a) were identified to be covalently modified by OxPCs (82). Moreover, Lp(a) but not LDL is modified by isolevuglandins (291). Thus, Lp(a) may serve as a major carrier of OxPLs in circulation (343). It has been hypothesized that Lp(a)-bound OxPLs retain their proinflammatory properties (82) and may represent one of factors underlying the well-known association of Lp(a) levels with atherosclerosis (343).
II. (Patho)physiological Effects of OxPLs
A. OxPLs as markers of “modified self”
Peroxidation of PLs generates a wide spectrum of reactive molecules that can modify endogenous structures, including proteins and other lipids. This leads to the generation of altered lipids as well as oxidized lipid–protein adducts, yielding “modified-self molecules” or so-called “neo-self determinants” that are recognized by specific innate and adaptive immune responses (Fig. 7).
FIG. 7.

OxPLs are recognized by soluble and cell-associated pattern-recognition receptors of innate immunity. Fragmented oxidized residues change their orientation within the cell membrane or lipoprotein outer layer, and protrude into the water phase, thus enabling recognition by cellular receptors (e.g., CD36, natural (germ-line encoded) immunoglobulins or C-reactive protein). It is likely that in analogy with other, better characterized ligands of these proteins, interactions with OxPLs result in removal and degradation of oxidized lipoproteins, senescent and apoptotic cells. Furthermore, binding of OxPLs to CD36 promotes formation of foam cells characteristic of atherosclerosis.
1. OxPLs as antigens
It is well known that subtle modifications of autologous proteins can render these immunogenic (322). Similarly, modifications with products of PL oxidation can lead to the generation of immunogenic molecules. In general, these antigenic adducts are recognized in a hapten-specific manner, in which neo-epitopes are formed on different endogenous proteins and lipids (e.g., PLs like PE containing a free amino group). For example, when the abundant PL phosphatidylcholine (PC) present in LDL and cell membranes is oxidized, decomposition of the unsaturated sn-2 fatty acid generates a wide spectrum of reactive molecules, such as malondialdehyde (MDA) and 4-hydroxynonenal (84), as well as the “core aldehyde” of the residual OxPL backbone such as POVPC (20). All these products are highly reactive and can form neo-epitopes. Importantly, apoptotic cells have been found to display an enriched content of OxPLs in their membranes, which renders them pro-inflammatory and immunogenic (56).
Both IgG and IgM antibodies to OxLDL have been documented in animal models of atherosclerosis and in humans (25, 140), and some of these antibodies recognize specifically OxPL epitopes that are present in OxLDL. While many studies show direct relationship of anti-OxLDL antibody titers with surrogate markers of disease or clinical endpoints, others fail to do so (152, 339). In recent years, evidence has accumulated demonstrating a different role for IgG and IgM antibodies against OxLDL. These studies suggest that titers of IgM but not IgG anti-OxLDL antibodies show an inverse relationship with atherosclerosis (170, 339). Of particular interest is the finding that some antiphospholipid antibodies (aPLs) of patients with antiphospholipid syndrome (APS) recognize oxidation-specific epitopes in OxLDL (143, 350). The APS is characterized by the presence of circulating aPLs and clinical features such as venous or arterial thrombosis, fetal loss, and autoimmune thrombocytopenia. Horkko et al. showed that many aPLs bound to CL only after it had been oxidized (OxCL), but not to a reduced CL analogue that could not undergo oxidation (142). Importantly, cardiolipin is also present in LDL particles (74). Horkko et al. further identified the neoepitopes of some aPLs as adducts formed between OxPLs and associated proteins, such as β2 glycoprotein 1 (β2GP1) or apoB (142). Thus, many aPLs seem to be directed at neo-epitopes of OxPLs. Antibodies against OxCL are clearly associated with atherosclerosis. In a subsequent study, it was shown that the levels of autoantibodies to OxCL correlated with the levels of aortic isoP F2α-VI and with the severity of atherosclerosis in apoe−/−mice, indicating that antibodies to OxCL reflect lipid peroxidation in vivo (270). The importance of these antibodies is further demonstrated by the fact that aPLs in patients with systemic lupus erythematosus were found to be primarily directed against OxCL and that the same patients exhibited increased levels of apoB-associated OxPLs as determined by the mAb E06 (99). Thus, antibodies to OxPLs are found in autoimmune diseases that predispose to cardiovascular disease and are associated with increased oxidative stress. This is further underscored by the characterization of a natural IgM Ab derived from the spleen of atherosclerotic ldlr−/−mice. This IgM was selected for binding to OxCL and binds to OxLDL, apoptotic cells, and immunostains atherosclerotic lesions (344).
Clearly, multiple OxPL epitopes exist, but the best described one is the phosphocholine group of oxidized phosphatidylcholine (OxPC). Indeed, phosphocholine-containing OxPLs have been identified as important antigens, in which the phosphocholine headgroup of oxidized but not of native PC is the epitope recognized by specific antibodies. A detailed understanding about the importance of phosphocholine evolved out of the detailed characterization of the prototypic monoclonal anti-OxLDL IgM Ab E06 that was derived from a panel of B-cell hybridomas from the spleens of atherosclerotic apoe−/−mice (251). E06, which was originally selected for the binding to copper-oxidized LDL, was subsequently found to specifically bind to OxPLs containing the phosphocholine headgroup but not to native unoxidized PCs (141). Moreover, in studies using synthetic OxPLs it was found that the specific OxPC epitope for E06 was the phosphocholine headgroup (98). Although the phosphocholine headgroup is present in native unoxidized PCs, only upon oxidation it becomes available for immune recognition by E06. Thus, it is hypothesized that structural changes as a result of PL oxidation lead to the generation of this neo-epitope. By the sequencing of the variable region of E06, Shaw et al. discovered that E06 is in fact a germline-encoded natural antibody identical to the previously characterized antibody T15 (303), which is known to bind the phosphocholine present in the capsular polysaccharide of S. pneumonia and many other microbes, and to protect mice from pneumococcal infections (48). Thus, molecular mimicry exists between the phosphocholine headgroup of OxPCs and the phosphocholine found in pneumococci and other microbes. E06 binds both the lipid and the protein moiety of OxLDL (containing free and protein-conjugated OxPCs) and inhibits its uptake by macrophages (141) as well as the uptake of apoptotic cells (55), suggesting that the same OxPL moiety is important for the recognition by macrophage scavenger receptors. Thus, phosphocholine is an important target of innate immunity recognized by a whole array of innate immune receptors (22).
2. OxPLs as ligands for scavenger receptors
Apart from serving as antigens for natural antibodies, OxPLs may also function as ligands for scavenger receptors. Scavenger receptors expressed on macrophages recognize both free OxPLs present in OxLDL, as well as OxPLs covalently linked to apoB-100 (26). Interactions of OxPLs with CD36, a member of class B scavenger receptor family (89), are by far the best characterized. Macrophage CD36 is involved in recognition and engulfment of apoptotic cells, and it also mediates the uptake of OxLDL and foam cell formation (89). The necessity of CD36 for the macrophage uptake of OxLDL and atherogenesis has also been demonstrated in vivo (90, 275), although a conflicting report exists (226). Podrez et al. have studied extensively the molecular characteristics of OxPLs interacting with CD36, and have demonstrated that a structurally conserved family of OxPLs with a truncated sn-2 acyl group incorporating a terminal γ-hydroxy(or oxo)-α,β-unsaturated carbonyl serves as high affinity ligand for CD36, and that these species are enriched in atherosclerotic lesions (264, 265). These oxidatively modified side chains protrude from lipid bilayers and lipoprotein particles to the aqueous compartment, thereby enabling their recognition by CD36 in neighboring cells (122). Recently, the structural basis of OxPC binding to CD36 has been studied in detail and amino acids 160–168 were identified as the core of the OxPC binding site, with two positively charged lysine residues 164 and 166 interacting with sn-2 acyl chains of OxPLs, thereby being indispensable for the binding (168). In addition to PC, also oxidized PS and phosphatidic acid were characterized as ligands for CD36 (122, 123) additionally suggesting that oxidized sn-2 residue but not polar head group is the major structural motif recognized by CD36. However, Boullier et al. presented conflicting data (46). They showed that binding of OxLDL to CD36 was inhibited by POVPC covalently bound via sn-2 group to a short peptide. This adduct does not contain γ-hydroxy(or oxo)-α,βunsaturated carbonyl motif and therefore is likely to interact with CD36 via different mechanisms possibly involving recognition of phosphocholine group (46).
In addition to CD36, OxPLs are recognized by another member of the class B scavenger receptors, SR-BI, and were hypothesized to serve as ligands for SR-BI-mediated internalization of OxLDL (116). Furthermore, OxPC species that are ligands for CD36 were shown to inhibit SR-BI-dependent HDL binding and reverse cholesterol transport (10). Thus, OxPLs may promote atherosclerosis not only by enhancing the uptake of modified lipoproteins by macrophages, but also by interfering with the reverse cholesterol transport by preventing SR-BI-mediated cholesterol uptake in hepatocytes.
3. OxPLs as ligands for CRP
The notion that OxPLs constitute a class of pathogen-associated molecular patterns (PAMPs) that are recognized by innate immune receptors is further supported by the finding that the ancient innate defense molecule C-reactive protein (CRP) binds OxPLs (57). CRP is an acute-phase protein that specifically binds to phosphocholine of the capsular polysaccharide of many microbes (359). Elevated levels of CRP are an independent risk factor for cardiovascular disease (199). In analogy to E06/T15 IgM, CRP binds to OxLDL and oxidized PC in a Ca2+-dependent manner, but does not bind to native LDL nor to unoxidized PC (57). CRP binding is mediated through the recognition of the phosphocholine moiety, as the binding to OxLDL was competed by KLH conjugated with phosphocholine and vice versa. Moreover, binding of CRP to phosphocholine-KLH could be competed by OxPC but not native unoxidized PC. Consistent with the presence of OxPLs in the membranes of apoptotic cells, CRP was also found to bind apoptotic cells via the phosphocholine moiety of OxPC. These data suggest that recognition of OxPCs in apoptotic and damaged cells may play a role in their clearance via CRP-dependent mechanisms (112).
B. Modulation of intracellular signaling by OxPLs
The data discussed in the previous chapter show that OxPLs are markers of disease of the ‘modified-self’ type that are recognized by natural antibodies and pattern-recognition receptors, and direct elimination of oxidized lipoproteins as well as senescent and apoptotic cells. However, increasing evidence shows that OxPLs are not only markers of disease but also ‘makers’ that are likely to play an active role in variable pathological states. This section describes intracellular signaling mechanisms activated by OxPLs.
1. Signal-transducing receptors initiating effects of OxPLs
OxPLs were shown or hypothesized to stimulate several types of signal-transducing receptors located on the cell surface or in the nucleus, including G protein-coupled receptors, receptor tyrosine kinases, Toll-like receptors, receptors coupled to endocytosis, and nuclear ligand-activated transcription factors such as PPARs.
a. PAF receptors
Probably the best understood molecular mechanism whereby OxPLs initiate biological effects is activation of receptor specific for PAF, which is an important lipid mediator of inflammation and platelet aggregation. This receptor specifically recognizes alkyl-acyl-phosphatidylcholines containing ether bond at the sn-1 position in combination with unusually short sn-2 acetyl residue. A proportion of PCs present in LDL particles contains sn-1 alkyl residues. Oxidative fragmentation of sn-2 PUFAs in alkyl-PCs generates products such as 1-alkyl-2-butenoyl and 1-alkyl-2-butanoyl (Fig. 4) that are recognized by PAF receptor (4, 210). These and similar ligands, usually referred to as PAF-like lipids, were found in atherosclerotic lesions and are known to form in vitro during oxidation of LDL (211). Although the affinity of PAF receptor for these ligands is 10-fold lower than for authentic PAF, they are likely to reach concentrations sufficient to activate the receptor (210). PAF-like lipids were shown to activate all major types of cells expressing PAF receptor (213). Whether similarly to alkyl-acyl PCs also fragmented diacyl-PCs can activate PAF receptor is still an open question. Similarly to PAF, POVPC was shown to stimulate adhesion of neutrophils to a gelatin matrix; the effect was inhibited by three different PAF receptor antagonists (312). In addition, POVPC competed with PAF for binding to macrophages and mimicked some (but not all) effects of PAF in this cell type (259). The role of the PAF receptor in the overall biological activity of OxPCs is not characterized. Many effects of OxPCs on ECs, for example, their proinflammatory and angiogenic action are neither mimicked by PAF, nor inhibited by PAF receptor antagonists (40, 191).
b. Prostaglandin receptors
OxPCs containing esterified isoPs (PEIPC, Fig. 4) activate receptors recognizing prostaglandins E2 and D (197) (EP2 and DP receptors, respectively). The EP2 receptor is expressed in all cell types relevant to atherosclerosis including endothelial cells (ECs), monocytes, macrophages, and vascular smooth muscle cells (VSMCs) (197). Activation of EP2 receptor on ECs results in activation of integrins and increased binding of monocytes similar to that induced by OxPAPC, while EP2 receptor antagonists inhibit action of OxPAPC. It has to be established whether prostaglandin receptors are activated by esterified isoPs directly or after release of isoPs from the glycerol backbone.
c. Scavenger receptors
Scavenger receptors were described in Section II.A.2 in the context of removal of oxidized lipoproteins and apoptotic cells containing OxPLs. In addition to that, scavenger receptor CD36, which is known to recognize fragmented unsaturated OxPLs (265), is likely to initiate some signaling effects of OxPLs. CD36 was shown to mediate activation of platelets by OxLDL acting by recruiting SRC family kinases FYN and LYN followed by activation of MKK4 and JNK2 protein kinases (60). It was hypothesized that CD36-dependent signaling stimulated by unsaturated fragmented OxPLs may explain hyperreactivity of platelets in patients with dyslipidemia (263).
d. VEGF receptors
Some effects of OxPLs may be mediated by VEGF receptors. Zimman et al. demonstrated enhanced phosphorylation (activation) of VEGFR2 within the first minutes of incubation with OxPAPC (396). Blocking antibodies to VEGF-A inhibited activation of VEGFR2 by exogenously added VEGF-A but did not influence the effects of OxPAPC, suggesting that the activation of VEGFR2 was ligand independent. It was hypothesized that trans-activation of VEGFR2 in OxPAPC-treated cells was mediated by c-SRC (396). Inactivation of VEGFR2 by siRNA and pharmacological inhibitors showed that this receptor plays a role in various effects of OxPAPC on ECs, including activation of signaling pathways (SREBP, ERK1/2) and expression of IL-8, tissue factor, and LDL receptor (396). In addition, OxPAPC, OxPAPG, OxPAPA, and OxPAPS were shown to induce VEGF-A production by ECs, peripheral blood mononuclear cells, monocyte-derived macrophages, fibroblasts, keratinocytes, lung epithelial cells, and epithelial tumor cell lines of different tissue origin (40). Therefore, in addition to transactivation of VEGFR2, VEGF-A can stimulate OxPL-treated cells acting as an autocrine and paracrine mediator.
e. Sphingosine-1-phosphate (S1P) receptor 1
Another example of receptor transactivation induced by OxPLs is S1P1 receptor (310). It was shown that OxPAPC stimulates recruitment of S1P1 to caveolin-enriched membrane microdomains, and induces its phosphorylation (activation) by AKT. These processes were important for OxPAPC-induced activation of RAC-1, leading to cytoskeleton reorganization necessary for enhancement of endothelial barrier. These results suggest that transactivation of S1P1 by OxPAPC plays a role in barrier-protective function of OxPLs (Section III.A.8).
f. Toll-like receptor 4
Several publications suggest that OxPLs may activate toll-like receptor 4 (TLR4). In particular, a role for TLR4 in OxPAPC-mediated induction of IL-8 in HeLa cells was suggested (366). More recently, OxPAPC was shown to induce lung injury and IL-6 production by mouse lung macrophages via the TLR4–TRIF–TRAF6 pathway (153). The conclusion about the involvement of TLR4 was based on the results of TLR4 knockout or knockdown experiments demonstrating attenuated effects of OxPLs. Whether OxPLs act as direct ligands of TLR4 is questionable, and it is more likely that a whole complex of receptors is involved in the recognition of OxPLs. Accordingly, several groups have shown that OxPLs do not induce many genes that are upregulated by LPS via TLR4. It has been shown that unlike LPS, various classes of OxPLs do not influence basal levels of E-selectin, ICAM-1, VCAM-1, TNFα, IL-6, IL-1α, IL-1β, and COX-2 in whole blood or individual cell types, including human umbilical vein ECs, blood monocytes, macrophage cell line, or fibroblasts (38, 83, 360). In contrast to LPS, OxPLs did not activate NF-κB-driven luciferase reporter in HEK cells stably transfected with TLR4 and MD-2 (360). These data show that OxPLs are not canonical TLR4 ligands inducing the same inflammatory effects as LPS or agonistic lipid A species. However, this conclusion does not exclude a role for TLR4 in OxPL-induced inflammation. TLR4 was shown to play an important role in aseptic inflammation induced by ischemia-reperfusion or mechanical ventilation of lungs (8, 355). It was hypothesized that in such pathologies TLR4 is stimulated by endogenous ligands (8). Similar mechanism might be activated by OxPLs. Furthermore, it is possible that OxPLs stimulate TLR4 only in combination with specific co-receptors and/or intracellular signaling adaptor proteins. Thus, agonistic effects of OxPLs on TLR4 may be selective for certain cell types and cell differentiation/activation state. Interestingly, in several in vitro and in vivo models OxPLs were shown to counteract acute inflammation induced by LPS. The hypothesized mechanisms of the anti-endotoxin activity of OxPLs are described in Section III.A.7.
g. PPARα and PPARγ
Peroxisome proliferator-activated receptors (PPARs) are intracellular ligand-activated transcription factors. Diacyl-OxPLs stimulated a PPAR response element-driven reporter construct in transfected HAECs (189). The effect of OxPAPC, POVPC, and PGPC was mediated by PPARα as indicated by activation of the ligand-binding domain of PPARα, but not PPARγ or PPARδ (189). Long-chain diacyl-OxPCs, such as PEIPC and PECPC, also activated PPARα in transfected HeLa cells (329). In addition to diacyl-OxPLs, also alkyl-PC hexadecyl-azelaoyl-PC was identified as a ligand and agonist of PPARγ in monocytes, where it stimulated expression of CD36 and COX-2 (72, 268), as well as in epidermal cells where it stimulated expression of COX-2 (391). Since oxidized FAs such as 9- and 13-HODE were shown to activate PPARs (234), it has to be established to which extent activation of PPARs by OxPLs depends on their cleavage catalyzed by PLA2 producing unesterified oxidized fatty acids, or on activation of LOX enzyme producing oxidized fatty acids intracellularly (77, 130).
h. Nonreceptor mechanisms
It is likely that certain cellular effects of OxPLs are initiated through nonreceptor mechanisms. The experimental support for this hypothesis is provided by the data showing that OxPLs induce depletion of cellular cholesterol leading to disruption of caveolae, which in turn results in activation of lipid-sensitive transcription factor SREBP and enhanced expression of IL-8 (380). Furthermore, OxPLs were shown to activate signaling events characteristic of electrophilic and unfolded protein stress responses. These cellular reactions, which are initiated independently of classical signal-transducing receptors, are described in detail in Section II.B.5.
2. Second messengers upregulated by OxPLs
a. Elevation of Ca2+i
Many hormones, growth factors, and lipid mediators induce biological effects by increasing cytosolic levels of calcium ions (Ca2+i) and stimulating Ca2+-sensitive intracellular effector pathways. MM-LDL was shown to induce elevation of Ca2+i in endothelial cells (139). In addition, OxPAPC was shown to induce rapid and reversible Ca2+-responses in ECs (39). One of the downstream effectors of OxPAPC in ECs was identified as Ca2+-sensitive phosphatase calcineurin that regulates nuclear translocation of transcription factor NFAT, which in turn mediates effects of OxPAPC such as induction of tissue factor (39).
b. Elevation of cAMP
MM-LDL causes a saturable dose-dependent increase in cAMP levels in aortic ECs (255, 261) that may result from stimulation of Gs and inhibition of Gi heterotrimeric G-protein complexes (255). Likewise, OxPAPC and its components POVPC and PEIPC increase intracellular cAMP levels (67). The amount of PEIPC required to cause cAMP-dependent cellular events was ∼10-fold lower than that of POVPC, suggesting that the PEIPC component of OxPAPC is a principal cAMP-elevating PL (67). Subsequent studies identified Gs-coupled prostaglandin E2 and D receptors (EP2 and DP) as mediators of cAMP elevation induced by OxPAPC and PEIPC, but not POVPC (197). Another study presented evidence for the existence of an adenylate cyclase-coupled membrane receptor that is activated by POVPC but not PGPC (192).
Elevation of cAMP induced by OxPAPC causes cellular effects relevant to inflammation. Intracellular cAMP elevation in ECs caused by OxPAPC, POVPC, or PEIPC activates small GTPase R-RAS, which stimulates apical expression of connecting segment-1-containing fibronectin and thus promotes the entry of monocytes into sites of chronic inflammation (67) (see Section III.A.1). On the other hand, cAMP-dependent PKA plays a role in OxPAPC-induced phosphorylation of transcription factor CREB leading to enhanced expression of heme oxygenase-1, an enzyme with prominent antioxidant and anti-inflammatory properties (182). In addition, cAMP was shown to inhibit activation of the major pro-inflammatory transcription factor, NF-κB (257). Furthermore, cAMP and PKA play mechanistic roles in barrier-protective effects of OxPLs in pulmonary vascular endothelium (30) (see Section III.A.8).
3. Intracellular signaling pathways activated by OxPLs
a. Protein kinases and phosphatases activated by OxPLs
OxPLs were shown to activate a number of protein kinases and phosphatases, which characterizes OxPLs as pleiotropic lipid mediators (Table 1). More detailed description of these enzymes as mediators of bilological effects of OxPLs is given in corresponding chapters.
Table 1.
Signaling Protein Kinases and Phosphatases Activated by Oxidized Phospholipids
| Activating OxPL | Cell response | References | |
|---|---|---|---|
| Signaling kinase | |||
| PKA | OxPAPC | Antioxidative response | 182 |
| PEIPC | Endothelial barrier protective response | 30 | |
| PECPC | |||
| PI3-kinase | OxPAPC | Endothelial cell/monocyte interactions | 67 |
| PEIPC | Endothelial barrier protective response | 310 | |
| c-SRC | OxPAPC | Transactivation of VEGF-A receptor | 396 |
| IL-8 production | 113, 381 | ||
| Focal adhesion remodeling | 30 | ||
| JAK | OxPAPC | IL-8 production | 113, 381 |
| PEIPC | |||
| ERK1/2 | OxPAPC | Expression of EGR-1 and tissue factor | 39, 396 |
| Antioxidative response | 182 | ||
| IL-8 production | 396 | ||
| JNK | OxPAPC | ATF-2 dependent gene expression | 29 |
| PKC | OxPAPC | Induction of tissue factor | 39 |
| Endothelial barrier protective response | 29, 30 | ||
| AKT | OxPAPC | S1P1 receptor transactivation | 310 |
| Endothelial barrier protective response | |||
| Signaling phosphatase | |||
| Calcineurin | OxPAPC | NFAT-mediated gene expression | 39 |
| Tissue factor expression | |||
| MKP-1 | OxPAPC | Monocyte chemotactic activity to endothelium | 278 |
b. Small GTPases regulated by OxPLs
Small GTPases control various cellular functions including cell proliferation, gene expression, cell motility, regulation of monolayer integrity, and barrier properties. The most extensively characterized members are RHO, RAC, and CDC42, which have distinct effects on actin cytoskeleton, cell adhesions, and cell motility (354), whereas RAS family GTPases mainly control MAP kinase signaling and gene expression (35). Different components of OxPLs activate small GTPases RAC, CDC42 (28), and R-RAS (67) in endothelial cells.
R-RAS
OxPAPC was shown to induce selective activation of R-RAS, without affecting H-RAS activity (67). OxPAPC components POVPC and PEIPC also activated R-RAS, leading to enhanced α5β1-integrin-dependent monocyte binding to aortic ECs. Notably, activation of R-RAS by OxPAPC, POVPC, and PEIPC occurred via a cAMP-dependent mechanism (67).
RAC and CDC42
RAC and CDC42 play a crucial role in enhancement of peripheral endothelial cytoskeleton and EC barrier properties via CDC42-mediated formation of filopodia (85, 335) and RAC-dependent lamellopodia extension, formation of new cell adhesions, and enhancement of peripheral endothelial cytoskeleton (220, 332, 335). Stimulation of pulmonary ECs with barrier-protective concentrations of OxPAPC or OxPAPS strongly activates RAC and CDC42 activities (28, 30, 31). HPLC-MS analysis revealed that PECPC co-eluted with barrier-protective activity (28). Another OxPAPC component PEIPC also induced barrier-protective effects in pulmonary and aortic endothelial monolayers (K. Birukov, J. Berliner, unpublished). Consistent with their barrier-protective effects, OxPLs containing full-length oxidized sn-2-residues induced activation of RAC and CDC42 (but not RHO) small GTPases (28). In contrast, barrier-disrupting fragmented products of PAPC oxidation (POVPC, PGPC, see Section III.8) did not activate RAC or CDC42. These findings demonstrate a key role of RAC- and CDC42-dependent mechanisms in EC barrier protection induced by OxPAPC.
Mechanisms of RAC and CDC42 activation by OxPAPC
Guanine nucleotide exchange factors (GEFs) facilitate exchange of GDP for GTP in the nucleotide-binding site of small GTPases (35, 41, 393). Recent study by Birukova et al. (32) showed involvement of RAC/CDC42-specific GEFs TIAM1 and βPIX in the OxPAPC-induced pulmonary EC barrier regulation. PAK-interacting exchange factor (βPIX) is localized within focal adhesions (209), whereas activated TIAM1 becomes recruited to caveolin-enriched cell membrane microdomains (311) or to adherens junctions in EC monolayers (207). Such localization of RAC-specific GEFs induced by OxPAPC appears to be important for local RAC/CDC42 activation, leading to active cytoskeleton remodeling and enhancement of cell–cell junctions critical for increased endothelial barrier function (7, 375). Protein depletion of TIAM1 and βPIX using siRNA approach abolished OxPAPC-induced RAC activation, cytoskeletal remodeling, and EC barrier protective response (32). Thus, OxPAPC via yet to be identified mechanisms, which may include PKC, PKA and tyrosine kinases (30, 32, 54, 301), activates TIAM1 and βPIX, which in turn stimulate RAC/CDC42 GTPases and induce cortical actin polymerization and EC barrier enhancement.
RAC–RHO crosstalk
Low concentrations of OxPAPC or PEIPC do not activate RHO (28, 32). OxPAPS and OxPAPC induce robust stimulation of RAC, and at the same time significantly attenuate RHO signaling activated by thrombin (31). Both OxPAPS and OxPAPC markedly attenuated RHO activation and endothelial barrier disruption in EC monolayers exposed to pathologic mechanical stress (244). Exact mechanisms of OxPAPC-induced downregulation of RHO activity remain to be clarified and may involve direct PKA-mediated RHO phosphorylation (63), or indirect suppression of RHO activation via PKA-catalyzed phosphorylation of RHO GDP dissociation inhibitor (271). Other mechanisms may involve modulation of RHO-specific GEFs by PKA, PKC, and SRC known to be activated by OxPAPC (29, 393). Activated RAC1 may downregulate RHO via p190–RHOGAP (RHO-specific GTPase activating protein)-dependent mechanism (135).
4. Transcription factors mediating effects of OxPLs
OxPLs have profound influence on gene expression. In human aortic ECs, OxPAPC modulates expression of ∼1,000 genes, including up- and downregulated mRNAs (107, 108). OxPLs regulate genes related to inflammation, lipid metabolism, cellular stress, proliferation, and differentiation. Transcription factors characterized as mediators of the effects of OxPLs are described in Table 2. The mechanisms of gene regulation by OxPLs often differ from those activated by well-characterized inducers of corresponding genes. For example, OxPLs induce VEGF-A in endothelial cells independently of its major transcriptional regulator HIF-1 but through activation of ATF4-dependent transcription (248). Another example is IL-8 that is regulated by inflammatory agonists predominantly through the NF-κB pathway. In contrast, the induction of IL-8 by OxPLs does not require NF-κB (382), but may involve SREBP, STAT3- and PPARα-dependent mechanisms (380, 381). Furthermore, expression of tissue factor is typically induced via NF-κB-dependent transcription, while OxPLs were shown to induce expression of this blood clotting initiator acting via transcription factors EGR-1 and NFAT (39). These examples illustrate that in some cases induction of gene expression by OxPLs involves specific combinations of transcription factors. As a result, kinetics of gene induction may be different such as significantly more prolonged elevation of IL-8 induced in ECs by OxPAPC as compared to the action of TNFα (382).
Table 2.
Transcription Factors Activated by OxPLs
| Transcription factor | Oxidized phospholipids | Cell types | Target genes | Upstream signaling or co-factors | References |
|---|---|---|---|---|---|
| STAT3 | OxPAPC, PEIPC | HAECs | IL-8 | c-SRC, JAK2 | 113, 381 |
| EGR-1 | OxPAPC | HUVECs, mouse carotid artery | Tissue factor | ERK1/2, PKC | 39, 102, 163 |
| CREB | OxPAPC | HUVECs | Heme oxygenase-1 | PKA, PKC, p38, ERK | 182 |
| NFAT | OxPAPC | HUVECs | Tissue factor | Calcineurin | 39 |
| SREBP | OxPAPC | HAECs, HeLa, HMECs | IL-8, LDL receptor | Cholesterol depletion, PI3K/Akt, eNOS, superoxide anion, VEGF-A receptor 2 | 114, 380, 396 |
| PPARs | UVB-irradiated alkyl-hexadecyl-arachidonoyl-PC, alkyl-azelaoyl-PC, PEIPC, PECPC, OxPAPC, PGPC, POVPC | Epidermal cells, human monocytes, HeLa, HAECs | COX-2, CD36, IL-8, MCP-1 | – | 72, 189, 268, 329, 391 |
| NRF2 | OxPAPC, OxPAPG, OxPAPA, OxPAPE, OxPAPS, OxPLPC, PEIPC, PAPC-OOH | HAECs, HUVECs | OKL38, GCLM, NQO1, HO-1 | NADPH oxidase, superoxide anion | 161, 196 |
| ATF4 | OxPAPC, OxPAPG, OxPAPA, OxPAPS, POVPC, PGPC, PEIPC, PAPC-OOH PAPC-OH | HAECs, HUVECs | VEGF-A, IL-8, IL-6, MCP-1, ATF3, TRB3, MGC4504 | Phospho-eIF2α | 107, 108, 248 |
| XBP1 | OxPAPC | HAECs | IL-8, IL-6 | – | 107 |
| ATF6 | OxPAPC | HAECs | – | – | 107 |
| KLF4 | OxPAPC, POVPC, PGPC | VSMCs, rat carotid artery | Downregulation of smooth muscle alpha-actin, smooth muscle myosin heavy chain, myocardin; Induction of type VIII collagen alpha1 chain | ERK1/2, ELK-1, histone deacetylases | 65, 260, 384 |
| ELK-1 | POVPC | VSMCs | Downregulation of smooth muscle alpha-actin, smooth muscle myosin heavy chain | ERK1/2, KLF4, histone deacetylases | 384 |
5. Cellular stress pathways activated by OxPLs
a. Electrophilic stress response
Exposure to electrophiles, including electrophilic α,β-unsaturatred aldehydes derived from oxidation of unsaturated FAs evokes a concerted response aiming at limiting their toxic effect in cells. This occurs through transcriptional induction of genes having a cis-acting element called antioxidant response element (ARE), or electrophile response element (EpRE), in their promoters (240). This sequence binds the transcription factor nuclear factor-E2-related factor 2 (NRF2), which forms heterodimers with other basic leucine zipper (bZip) transcription factors, particularly small MAF proteins, recruiting transcriptional coactivators and facilitating the expression of target genes (173). Under basal conditions, NRF2-dependent transcription is repressed by its negative regulator KEAP1, which functions as an adaptor for CUL3-based E3 ligase leading to proteasomal degradation of NRF2. When cells are exposed to electrophiles, NRF2 escapes KEAP1-mediated repression, translocates to the nucleus, and binds to the ARE (Fig. 8) (173). KEAP1 is a thiol-rich protein, the mouse Keap1 having a total of 25 and the human protein 27 cysteine residues. Many of these are prone to alkylation by electrophiles (80). Electrophilic lipid oxidation products such as s4-hydroxynonenal and 15-deoxy-A12,14-prostaglandin J2 are known to react to KEAP1 protein (144; 195). Although a number of cysteine residues in recombinant KEAP1 have been shown to be reactive towards various electrophiles (138), the functionality of Cys151, Cys273, and Cys288 has been demonstrated by testing the abilities of specific KEAP1 cysteine mutants to inhibit NRF2 activity in transient transfection assays (144, 195, 363, 389) as well as in vivo (378). The model proposed by Yamamoto et al. (378) suggests that Cys151 is essential for NRF2 activation in response to electrophiles while Cys273 and Cys288 are important for KEAP1 repression of NRF2 activity under unstressed conditions (378). In addition to direct sensing of electrophiles by KEAP1, a number of protein kinase pathways have been suggested to play a role in regulating NRF2 activity (173).
FIG. 8.

Activation of electrophilic stress response by OxPLs. Induction of antioxidant genes is initiated by reactive electrophilic compounds via covalent modification of thiol groups in KEAP1 leading to derepression of transcription factor NRF2. OxPLs were shown to induce nuclear accumulation of NRF2, stimulate its binding to promoters of target genes, and elevate mRNA and protein levels of major antioxidant genes. It has to be established whether OxPLs similarly to classical electrophiles form covalent complexes with thiol groups of KEAP1.
One of the earliest reports regarding the regulation of antioxidant genes by OxPLs was the observation that heme oxygenase-1 (HO-1) was induced by OxPAPC (157). The HO-1 gene is a classical ARE-regulated gene, and both human and mouse HO-1 promoters have two enhancer areas located approximately −4 and −10 kb relative to the transcriptional start site. Both enhancer areas contain multiple ARE-elements (287). Later on, a microarray analysis of human aortic ECs revealed that both modifier and catalytic subunits of glutamate-cysteine ligase (GCLM and GCLC, respectively), the rate-limiting enzyme of glutathione synthesis, were upregulated by OxPAPC (108). GCLM and GCLC also have well-characterized ARE sequences in their 5′-flanking regions. Later studies confirmed that OxPLs evoke a general NRF2-mediated response and induce antioxidant genes in ECs in vitro and mouse carotid arteries in vivo. Indeed, NRF2 was activated in OxPAPC-treated cells as assessed by its nuclear accumulation, while silencing of NRF2 expression by siRNA reduced HO-1 expression as well as the expression of GCLM and NAD(P)H:quinone oxidoreductase-1 (NQO1), one of the classical ARE-regulated genes (161). Moreover, chromatin immunoprecipitation (ChIP) experiments revealed a direct interaction of NRF2 with NQO1 ARE-containing promoter region as well as the distal enhancer region of HO-1 in OxPAPC-exposed cells. However, unlike with GCLM and NQO1, silencing of NRF2 with NRF2-specific siRNA had only a partial effect on HO-1 expression, suggesting that other redundant pathways such as that mediated by cAMP-responsive element-binding protein (CREB) contribute to its induction by OxPLs (182). It was also demonstrated that OxPAPC-inducible expression of HO-1, GCLM, and NQO1 was lower in NRF2-null than wild-type mouse carotid arteries in vivo (161). In addition to HO-1, GCLM and NQO1, also the expression of human tumor suppressor gene OKL38 by OxPLs has been shown to be NRF2-dependent (196).
The nature of NRF2-inducing OxPLs was also examined (161). Thiol reactivity of OxPAPC is important in the induction of NRF2-dependent genes, as small molecular weight thiol antioxidants glutathione and N-acetylcysteine were able to inhibit induction. On the other hand, reduction of electrophilic groups in OxPAPC by sodium borohydride significantly inhibited induction of ARE genes. Of the different species present in OxPAPC, isoP-PC was by far the most potent in inducing NRF2-dependent genes (161). However, the isoP ring structure was not absolutely necessary for the activity, as oxidized palmitoyl-linoleoyl-phosphatidylcholine (OxPLPC), unable to form the prostanoid structure (242), was almost equally effective in its ability to induce NRF2-dependent genes. Also other species such as PC-hydroperoxides were able to increase the expression of NRF2 target genes.
Selective reduction of PC-hydroperoxides to the oxidatively inert hydroxides by triphenylphosphine resulted in attenuated induction of ARE genes (161). These data implicate that the NRF2 activating capacity was shared with structurally rather diverse family of OxPLs demonstrating electrophilic properties.
What is the mechanism of NRF2 activation by OxPLs? One possible mechanism is that electrophilic OxPLs could be directly sensed by reactive cysteine residues in KEAP1, as intact PLs can be taken up by the cell by transbilayer movement (250, 337), or by receptor-mediated mechanisms (72). Moreover, using fluorescent or biotin labeling, OxPLs have recently been shown to be internalized (129, 229), and to bind to intracellular proteins such as H-RAS (129, 229). Intriguingly, structural analogs of 15-deoxy-A12,14-prostaglandin J2, a well-defined NRF2 activator known to bind KEAP1 protein thiols, have been found esterified in the sn-2 position in some of the OxPAPC species (144, 179, 195, 370). It is therefore alluring to speculate that KEAP1 would be a direct sensor of electrophilic OxPLs.
An alternative mechanism by which OxPLs activate NRF2 has been proposed (196). OxPAPC has previously been shown to increase endothelial superoxide anion production via NADPH oxidase (286). Li et al. (196) inhibited NADPH oxidase by diphenyleneiodonium (DPI) and found that the induction of OKL38 by OxPAPC was NADPH oxidase-dependent. As OKL38 induction was also inhibited by siRNA specific to NRF2, the assumption was made that NRF2 activation by OxPAPC is NADPH oxidase-dependent. However, activation of NADPH oxidase appears not to be necessary for the induction of all NRF2 target genes, as neither DPI, NADPH oxidase inhibitor apocynin, nor siRNA knocking down NOX4, the major endothelial NADPH oxidase isoform responsive to OxPAPC (1, 286), were able to affect the expression of ARE target genes in HUVECs (161). Regarding the role of NADPH oxidase in NRF2 signaling, there are only a few reports in the literature showing that NRF2 activation by any stimuli requires NADPH oxidase-derived radical production (254, 300, 367). It is unfortunate that these reports used DPI to inhibit NADPH oxidase. This compound is not a specific NADPH oxidase inhibitor, but inhibits flavoenzymes in general, and thus firm conclusions cannot be drawn from these studies. Moreover, the question still remains how NADPH oxidase-derived superoxide radical would impact on NRF2 signaling. With respect to KEAP1, protein and mixed disulfide formation with glutathione has been detected using recombinant KEAP1 protein exposed to different GSH/GSSG redox ratios (138). However, whether physiologic ROS messengers such as H2O2 are able to modify KEAP1 in this manner, is currently unknown. In contrast, there is strong evidence that KEAP1 thiol residues are directly alkylated by different inducers (138). Should modification of KEAP1 be the trigger for the ARE response by OxPLs, direct modification of thiol residues by them or their fragmented products should be the most likely mechanism.
b. Unfolded protein response
Endoplasmic reticulum (ER) is the site of folding and maturation of most secreted and transmembrane proteins within eukaryotic cells. Its complex protein folding machinery is highly sensitive to external pertubations, which may cause accumulation of unfolded or misfolded proteins within ER, a condition referred to as ER stress. The unfolded protein response (UPR) is an adaptive signaling pathway triggered by ER stress. It is a combined transcriptional and translational response aiming at restoring ER protein folding and homeostasis. The UPR signaling is sensed by three transmembrane signaling proteins residing in ER, double-stranded RNA-dependent protein kinase (PKR)-like ER kinase (PERK), inositol requiring 1 (IRE1), and activating transcription factor-6 (ATF6), each triggering downstream signaling events ultimately leading to increased expression of UPR target genes (Fig. 9) (299).
FIG. 9.

Activation of unfolded protein response (UPR) by OxPLs. Multiple stress conditions leading to impaired processing of proteins in endoplasmic reticulum activate adaptive reaction called UPR, which is mediated via three branches, all initiated by binding of unfolded proteins to BIP/GRP78, thus leading to dissociation from, and de-repression of, ATF6, IRE1, and PERK. Dissociation of BIP/GRP78 initiates processing of ATF6 protein, splicing of XBP-1 mRNA by IRE1, and phosphorylation of eIF2α, resulting in selective translation of ATF4. OxPAPC and other classes of OxPLs were shown to activate all three arms of the UPR, leading to formation of transcriptionally active ATF6, XBP1, and ATF4, as well as induction of their target genes. However, the mechanism of activation is currently unknown. In particular, it is not clear whether similarly to unfolded proteins OxPLs directly bind to BiP/GRP78 and reverse repression of ATF6, IRE1, and PERK.
A microarray analysis of human aortic ECs exposed to OxPAPC revealed that a number of genes induced by OxPL treatment were known UPR target genes (108). Gargalovic et al. demonstrated activation by OxPAPC of all branches of UPR, including cleavage and nuclear translocation of ATF6, splicing of XBP1 mediated by IRE1, and phosphorylation of eIF2α catalyzed by PERK (107). Using the siRNA approach, the authors demonstrated that both ATF4 and XBP1 are important for transcriptional induction of proinflammatory genes IL-8, CXCL3, IL-6, and MCP-1 (107). In human atherosclerotic lesions, immunostaining for ATF3 and ATF4 revealed that ECs in shoulder areas of atheroma stained positively for both proteins (107). Moreover, lesion areas containing foam cells also stained positively. Staining with the E06 antibody recognizing oxidatively modified PCs showed positivity at the areas of lipid deposits and foam cells and in close proximity of endothelial cell layer (107). These data combined with earlier findings reporting increased expression of UPR marker genes at every stage of atherogenesis in apoe−/− mice (394) and the well-demonstrated role of UPR in macrophage apoptosis (92) and cytokine production (198) suggests that UPR may play an important role in the atherogenic process.
In addition to the role of UPR in OxPL-induced inflammation, recent findings suggest that UPR is also important for proangiogenic effects of OxPLs (40, 248). Specifically, induction of the critical proangiogenic growth factor VEGF-A was shown to be dependent on transcription factor ATF4 (248). Moreover, the structural characteristics of UPR inducing OxPLs were explored in this study. The results show that several different OxPLs having varying oxidative modifications in their sn-2 position were able to induce both VEGF-A and ATF3 (248). However, on a molar basis, PEIPC is the most active species evoking the UPR pathway. Moreover, the presence of oxidized sn-2 residues is a prerequisite, as lyso-PC is dramatically less potent than OxPLs (248).
The mechanisms by which OxPLs evoke the UPR pathway remain unclear. The involvement of previously characterized OxPL receptors, including PAF-, prostaglandin E2 receptor, PPARs, and TLR4 is not likely (107, 248). One possible mechanism is the effect of PL oxidation on biophysical properties of ER membranes, as has been suggested to occur upon accumulation of free cholesterol in macrophages (92). PL oxidation products differ in structure and polarity from their parent molecules and therefore have profound effects on membrane physical properties. It has been shown that oxidation of unsaturated PLs within the membrane increases its rigidity (44), and that incorporation of OxPLs into PL monolayer causes monolayer expansion and discontinuity, with reorientation of oxidatively modified side chains into the aqueous phase (288). These modifications should impact on the structure and function of membrane-associated proteins, and it is therefore enticing to speculate that the transmembrane sensors of ER stress could be affected in this manner. Another, alternative explanation would be that OxPLs are recognized by a currently unknown receptor which transduces the UPR signal. One promising candidate for this is GRP78/BiP, which forms complexes with all three UPR signaling mediators, IRE1, PERK, and ATF6 (Fig. 9) (285). As GRP78/BiP has been shown to bind lipid peroxidation product 4-hydroxynonenal (357) as well as biotinylated oxPAPE (129), one may envision that covalent modification of GRP78/BiP disrupts the interaction with its binding partners eliciting the UPR response.
c. Membrane stress
In addition to activation of specific cell signaling cascades, oxidation of PLs can have profound effects on cellular functions via altering biophysical properties of cellular membranes. As referred to in the previous section, oxidation of unsaturated PLs increases membrane rigidity (44), and incorporation of OxPLs into DPPC monolayer causes monolayer expansion as well as reorientation of oxidatively modified side chains and subsequent partitioning of OxPLs into the aqueous phase (288). Oxidative modification of PL bilayers not only affects plasma membrane but mitochondrial membranes as well, drastically altering their biophysical properties (218, 219). This membrane disorganization influences the conformation and function of membrane-associated proteins (177), and also exposes functionally important oxidized fatty acyl chains for recognition by their receptors, such as CD36 (134). Interestingly, oxidized PS was hypothesized to act as a “nonenzymatic scramblase” promoting externalization of unoxidized PS characteristic of apoptotic cells (348). The PS externalization serves as a signal for clearance of apoptotic cells, as discussed below.
d. Apoptosis-related signaling
Apart from their ability to evoke stress signaling pathways aiming at restoring cellular homeostasis, OxPLs, when exceeding the cell's capacity for adaptation, can trigger apoptosis. POVPC and PGPC have been reported to activate apoptotic cell signaling cascades in vascular smooth muscle cells (201). Both species were able to activate acid sphingomyelinase, and inhibition of its activity suppressed caspase-3 activation (201). Moreover, azelaoyl-PC, abundant in oxidized LDL (336), can damage mitochondria and trigger apoptosis via the intrinsic apoptotic cascade (61). In addition to exogenous OxPLs, endogenous PL oxidation products, such as CL oxidized by cyt c in mitochondria, have been implicated to play a significant role in early execution of the intrinsic apoptotic program (166). Oxidized CL epitopes can also be recognized within apoptotic cells by a natural antibody (344). It has also been argued that externalization of PS, an early event in apoptosis involved in phagocyte recognition and engulfment of apoptotic cells by macrophages, requires PS oxidation (134, 165).
In addition to their role in triggering and execution of apoptosis, increased levels of biologically active OxPLs are found in apoptotic cells (56). OxPLs are highly enriched in membrane vesicles released from activated cells and blebs derived from cells undergoing apoptosis (151). Membrane vesicles, also called microparticles, are found in circulation, and the levels are increased in patients with increased atherothrombotic risk (45). Membrane vesicles, derived in part from apoptotic VSMCs, are also highly abundant in atherosclerotic lesions (193). OxPLs within membrane vesicles have been shown to have inflammatory effects via enhancing monocyte binding to ECs, an effect inhibited by OxPL-recognizing antibodies (56, 151).
III. Accumulation and Potential Role of OxPLs in Pathology
A. Major biological activities of OxPLs
1. Proinflammatory effects of OxPLs
Intensive study of OxPLs as biologically active compounds began after these lipids were identified as an active principle of MM-LDL capable of stimulating production of chemokines and adhesion of monocytes to ECs (21). Inflammatory effects of MM-LDL may be mediated by PAF-like (alkyl-acyl) OxPLs (309), as well as diacyl-OxPLs that were reproducibly shown to induce inflammatory reactions in cultured ECs and in animal vessels in vitro, in vivo, and ex vivo (79, 102, 368). Available data show that proinflammatory effects of diacyl-OxPLs differ from the action of classical inflammatory agonists. While LPS, TNFα, or IL-1 induce extravasation of both monocytes and granulocytes via ICAM-1, VCAM-1, and E-selectin-dependent mechanisms, OxPAPC and MM-LDL do not induce expression of these adhesion molecules, and stimulate ECs to bind monocytes but not neutrophils (102, 150, 176, 192). This difference is nicely illustrated in the model of air pouch. Injection of OxPAPC stimulated accumulation of monocytes/macrophages in pouch wall, but not in the lumen. In contrast, injection of LPS resulted in accumulation of monocytes and neutrophils both in the lumen and the wall of the pouch (162). Specific induction of mononuclear cell adhesion supports the notion that OxPLs represent initiating factors in atherogenesis (18), which is characterized by selective accumulation of mononuclear cells in arterial intima. The mechanisms of selective binding of monocytes by OxPL-treated ECs are not completely understood since diacyl-OxPLs are known to stimulate expression in ECs of granulocyte-specific chemokines and adhesion molecules capable of binding neutrophils such as P-selectin (see below). Following is a description of currently identified inflammatory mechanisms activated by OxPLs.
a. Cell adhesion molecules activated by OxPLs
Two mechanisms mediating adhesion of monocytes to ECs treated with MM-LDL or OxPLs were identified. First, MM-LDL was shown to activate β1-integrins on the ECs (306). Activated β1-integrins bind a splice variant of fibronectin (FN) containing a 25-amino acid sequence called connecting segment-1 (CS-1 FN). Thus, CS-1 FN is captured from plasma and deposited on the apical surface of the endothelium where it serves as a ligand for VLA-4, a major integrin mediating firm adhesion of monocytes (306). This mechanism is likely to operate in atherogenesis as illustrated by the presence of activated β1-integrins and accumulation of CS-1 FN in atherosclerotis lesions at the sites of mononuclear cell infiltration (306). Furthermore, chronic infusion of a short peptide mimicking the VLA-4-binding site of CS-1 FN inhibited formation of lesions in LDL receptor knockout mice (305). Similarly to MM-LDL, OxPAPC was shown to stimulate adhesion of monocytes to the ECs via CS-1 FN-dependent mechanism (192). The activation of β1-integrins by OxPLs is initiated by elevation of cAMP followed by the activation of GTPase R-RAS and PI-3-kinase pathway (67). IsoP-containing PC (PEIPC, Fig. 4) was identified as a potent component of OxPAPC activating this pathway via prostaglandin E2 receptor (197).
The second mechanism of leukocyte adhesion activated by OxPLs depends on P-selectin, known to bind both monocytes and neutrophils (216). MM-LDL and its component OxPLs upregulate P-selectin in HAECs (361). P-selectin was localized intracellularly in quiescent cells but was immediately translocated to the cell surface after treatment with histamine or LDL oxidation products. More recently, it was found that this effect can be reproduced by OxPAPC in isolated vessels. Blocking antibodies to P-selectin inhibited rolling of monocytic cell line in mouse carotid arteries preperfused for 4 h with OxPAPC but not its unoxidized precursor (102). OxPAPC did not significantly elevate the levels of P-selectin mRNA, suggesting that the effect most likely resulted from mobilization of Weibel–Palade bodies (102). Upregulation of P-selectin protein and P-selectin-dependent adhesion of leukocytes were also observed ex vivo in mouse aortic segments pretreated with PL-chlorohydrin generated by oxidation of esterified unsaturated fatty acid with hypochlorite known to be generated by myeloperoxidase (79). Podrez et al. described enhanced expression of P-selectin on the surface of blood platelets incubated with OxPLs, suggesting that in addition to monocyte-endothelial interaction, OxPLs can stimulate formation of platelet-monocyte aggregates thought to play a role in pathogenesis of vascular disease (263).
b. Chemokines
OxPLs stimulate expression in ECs and VSMCs of several chemokines (Table 3), some of which are specific for mononuclear leukocytes, while other attract both monocytes and granulocytes, or are selective for granulocytes. The induction of chemokines in ECs by OxPLs is regulated by genetic and/or epigenetic factors (108), as well as by shear stress (145).
Table 3.
Chemokines Regulated by OxPLs
| Chemokine | OxPLs | Cell or tissue type | Target leukocytes [major groups, (262)] | References |
|---|---|---|---|---|
| MCP-1 (CCL2) | OxPAPC, POVPC, PGPC, HOOA-PC, PEIPC, PECPC | HAECs, rat aortic SMCs, mouse carotid artery, skin air-pouch | Monocytes | 102, 189, 260, 327, 329 |
| MCP-3 (CCL7) | POVPC, OxPAPC | Rat aortic SMCs, mouse skin air-pouch | Monocytes | 260 |
| MCP-5 (CCL12) | OxPAPC | Mouse skin air-pouch | Monocytes | 162 |
| GROα (CXCL1) | OxPAPC | HAECs | Neutrophils | 107 |
| CXCL3 (GROγ, MIP2β) | OxPAPC | HAECs | Monocytes | 107 |
| IL-8 (CXCL8) | OxPAPC, POVPC, PGPC, HOOA-PC, PEIPC, PECPC | HAECs | Neutrophils, macrophages | 189, 327, 329 |
| KC | OxPAPC | Mouse carotid artery | Neutrophils | 102 |
| MIP-1α (CCL3) | OxPAPC | Mouse carotid artery | Granulocytes, monocytes, lymphocytes | 102 |
| MIP-1β (CCL4) | OxPAPC | Mouse carotid artery | Granulocytes, monocytes, lymphocytes | 102 |
| IP-10 (CXCL10) | OxPAPC | Mouse skin air-pouch | Monocytes, lymphocytes | 162 |
| RANTES (CCL5) | OxPAPC | Mouse skin air-pouch | Monocytes | 162 |
| BRAK (CXCL14) | OxPAPC | Mouse skin air-pouch | Monocytes | 162 |
VEGF-A produced in monocytes stimulated by OxPLs is another candidate cytokine that may direct migration of leukocytes. Enhanced secretion of VEGF-A in response to OxPAPC was shown in human blood monocytes and monocyte-derived macrophages (40). In addition to its angiogenic growth factor activity, VEGF-A is a potent chemoattractant for monocytes acting via FLT-1 receptor (14). Furthermore, overexpression of VEGF-A was shown to induce expression of VCAM-1 and PECAM-1 on endothelium and stimulate adhesion of monocytes leading to the generation of bigger and more inflamed atherosclerotic lesions in apoe−/− mice (202).
c. Direct effects of OxPLs on leukocytes
Experiments testing direct action of OxPLs on leukocytes produced controversial data describing both pro- and anti-inflammatory effects. Imai et al. showed that BSA-conjugated OxPAPC stimulated production of IL-6 in mouse lung tissue macrophages (153). The effects were inhibited by mAb E06 specific for OxPCs (see Section IV.A). Similar inhibitory effects of E06 were observed after stimulation of alveolar macrophages with bronchoalveolar lavage fluid derived from mice with acute lung injury, or in peritoneal macrophages treated with artificially oxidized lung surfactant (153). Because the lung injury was ameliorated in il-6−/− animals, the authors concluded that IL-6 induced by OxPLs in macrophages plays important role in the pathogenesis of acute lung injury (153). Smiley et al. showed that similarly to PAF, POVPC stimulated adhesion of neutrophils to a gelatin matrix (312), while Pegorie et al. observed POVPC-stimulated expression of IL-8, TNFα, and IL-1β in human monocyte-derived macrophages (259). Furthermore, treatment of monocytic cell line THP-1 with PC-hydroperoxide and PC-hydroxide, but not with unoxidized or oxidatively truncated PCs, stimulated LFA-1-dependent adhesion of cells to immobilized ICAM-1 (9).
The limited data presented above describe several proinflammatory effects of OxPLs in monocytes/macrophages, which however seem to be different from the action of well-recognized inducers of inflammation. To make the picture more complicated, OxPLs induce in leukocytes certain effects that are usually regarded as anti-inflammatory. For example, OxPAPC and PEIPC acting via the EP2 receptor were shown to inhibit basal production of TNFα and enhance production of IL-10 by human macrophages and macrophage cell line (197). Another publication demonstrated OxPL-induced inhibition of IL-12 synthesis with concomitant elevation of anti-inflammatory IL-10 expression in primary human monocytes stimulated by TLR2/1 ligand (70). Treatment of murine bone marrow macrophages with OxPAPC-treated LDL did not upregulate inflammatory genes but induced genes of protective antioxidant response (124).
In addition to regulating production of cytokines or adhesion, OxPLs can influence other functions of leukocytes directly related to inflammation and antibacterial defense. OxPAPC and other classes of OxPLs interefere with the ability of leukocytes to kill bacteria by inhibiting oxidative burst in human neutrophils (37), suppressing phagocytosis of bacteria by mouse peritoneal macrophages and polymorphonuclear cells (178), and blocking production of antibacterial peptide cathelicidin by human monocytes stimulated by TLR2/1 agonist (70). It is likely that the anti-inflammatory effects may compromise antibacterial defense and aggravate the course of infectious disease (178).
In summary, OxPLs induce both pro- and anti-inflammatory effects in monocytes and macrophages. The net effect likely depends on the type of inflammation and differentiation/activation state of leukocytes.
2. Effects of OxPLs on generation of ROS
There are indications that OxPAPC, but not its unoxidized precursor, may increase intracellular and extracellular levels of superoxide radical in human and bovine aortic ECs (78, 196, 286). Elevated activity of NADPH oxidase, partially resulting from increased expression of NOX4 mRNA, was suggested as one potential mechanism mediating enhanced production of superoxide radical in OxPAPC-treated cells (286). Another mechanism of NADPH oxidase activation depends on recruitment of RAC1 to the membrane, leading to activation of superoxide anion radical production (190). On the other hand, activation and uncoupling of endothelial NO synthase was hypothesized to be an additional source of superoxide radical in human aortic ECs treated with OxPAPC and PEIPC (114).
3. Effects of OxPLs on blood coagulation and activation of platelets
a. Modulation of blood coagulation
Since membrane PLs are key components of blood coagulation cascade, it is not surprising that oxidation of esterified PUFAs influences clot formation. The inclusion of OxPLs in liposomes resulted in significant enhancement of prothrombinase activity (371). On the other hand, oxidation of liposomes containing PUFA–PLs stimulated activites of anticoagulant proteins C and S (289). However, oxidized PS and PE were also found to stimulate the opposite reaction, namely inactivation of protein C by protein C inhibitor (206). Thus, OxPLs were reported to exert functionally opposite effects on different steps of coagulation cascade.
Additional procoagulant mechanisms activated by OxPLs regulate expression or activity of pro- and anticoagulant proteins on the surface of endothelium. Three effects of OxPLs enhancing thrombogenic potential of endothelium were described (Fig. 10). First, MM-LDL (91), OxPAPC, and PGPC (39) were shown to upregulate on ECs expression and activity of tissue factor, which is a master-switch initiating clotting cascade. OxPAPC upregulated mRNA, protein, and activity of tissue factor acting via mechanisms involving protein kinases ERK1/2, phosphatase calcineurin, and transcription factors NFAT and EGR-1 (39). In addition, OxLDL and OxPCs downregulated expression of the major anticoagulant protein located on ECs, thrombomodulin. The effect resulted from decreased nuclear accumulation and promoter binding of transcription factors mediating basal expression of thrombomodulin, including retinoic acid receptor β, retinoid X receptor α, Sp1, and Sp3 (156). Furthermore, peroxidation products of PC and PE containing esterified linoleic acid were shown to influence the initial steps in blood coagulation by inactivating tissue factor pathway inhibitor. The effect was explained by direct binding of OxPLs to the C-terminus of tissue factor pathway inhibitor leading to its inhibition (247).
FIG. 10.

OxPLs induce pro-coagulant shift in endothelium. OxPLs were shown to activate three pro-coagulant mechanisms in endothelium. First, OxPAPCs was shown to upregulate via EGR-1- and NFAT-dependent mechanisms expression and activity of the major inducer of coagulation, tissue factor (TF, mechanism 1). Furthermore, peroxidation products of PC and PE containing oxidized linoleic acid were shown to inactivate tissue factor pathway inhibitor (TFPI, mechanism 2) as a result of direct binding of OxPLs to the C-terminus of TFPI. Finally, OxPAPC was shown to inhibit expression of the major anticoagulant protein on ECs, thrombomodulin (TM, mechanism 3). The effect was explained by decreased activity of transcription factors mediating basal expression of TM (i.e., retinoic acid receptor β, retinoid X receptor α, Sp1 and Sp3).
In summary, OxPLs induce multiple effects on blood coagulation, and the majority of these effects are likely to induce procoagulant shift in endothelium and promote blood clotting (Fig. 10).
b. Activation of platelets
Oxidized lipoproteins and lipids extracted from atherosclerotic lesions are known to induce proaggregant effects in platelets, which may be partially mediated by the action of OxPLs. PAF-like (alkyl-acyl) OxPLs and LysoPA accumulate in MM-LDL and atheroma and act as potent inducers of platelet activation working in concert with other agonists (132, 213).
In contrast to the well-characterized receptors mediating action of PAF-like (alkyl-acyl) OxPLs and LysoPA, the mechanisms whereby platelets are activated by diacyl-OxPLs are not completely understood. Diacyl-OxPCs were shown to activate platelets independently of PAF-, LysoPA-, LysoPC-, or thromboxane A2 receptors (119). Diacyl-OxPLs are weak agonists since they induce platelet shape change and surface expression of P-selectin but do not induce platelet aggregation when added alone (119, 131). Although relatively weak, the effects of OxPLs may be of biological relevance, as suggested by the ability of unsaturated fragmented diacyl-OxPCs to stimulate platelets synergistically with agonists such as ADP (263). Experiments using CD36 knockout mice showed that the proaggregant action of OxLDL and unsaturated fragmented diacyl-OxPLs on platelets may be initiated by CD36 (263, 265). The role for CD36 is further supported by the inhibition of platelet activation by peptide mimicking the CD36 binding site (168). Recognition of OxLDL by CD36 is followed by recruitment and activation of SRC-family kinases, MKK4 and JNK2. It is likely that activation of this cascade underlies the enhanced reactivity of platelets in hyperlipidemic patients known to have elevated plasma levels of OxPLs (168, 263).
In summary, in addition to inducing thrombogenic shift in the endothelium, OxPLs are likely to increase the risk of thrombosis through direct activation of platelets mediated by CD36, PAF, LPA and probably other unidentified receptors.
4. Modulation of vascular smooth muscle cell phenotype
A unique feature of vascular smooth muscle cells (VSMC) is their phenotypic plasticity. During early atherogenesis, SMCs undergo phenotypic switching from contractile to synthetic phenotype characterized by decreased expression of SMC differentiation marker proteins, a high rate of cellular proliferation, and increased synthesis of extracellular matrix proteins (249). Accumulating evidence suggests that OxPLs may play a role in phenotypic modulation of VSMCs. OxPAPC and its oxidatively fragmented component POVPC profoundly reduced protein expression of the markers of differentiated SMC such as smooth muscle (SM) α-actin and smooth muscle myosin heavy chain (SM MHC), while simultaneously increasing expression of the proinflammatory genes MCP-1, MCP-3, and cytolysin (260). OxPL-induced inhibition of SM α-actin and SM MHC expression were mediated at least in part by Krüppel-like transcription factor 4 (KLF4), the potent repressor of SMC differentiation marker genes. POVPC-induced KLF4 activation suppressed expression of VSMC differentiation transcription factor myocardin, thus pointing to a potential mechanism of POVPC effects on VSMC phenotypic transition (260).
In addition to phenotypic effects on vascular SMC, fragmented product of PAPC oxidation, POVPC, alters extracellular matrix production and stimulates VSMC migration (65). POVPC increased expression of type VIII collagen alpha1 chain (Col8a1) mRNA in vitro and in vivo, which was mediated by activation of KLF4 and Sp1 and increased KLF4 binding to the Col8a1 gene promoter. Such activation, as well as POVPC-induced VSMC migration were markedly reduced in KLF4- or type VIII collagen-knockout VSMCs (65). These findings suggest that changes in VSMC phenotype, extracellular matrix composition, and resulting stimulation of VSMC migration are induced by fragmented OxPL products and may contribute to the pathogenesis of atherosclerosis.
5. Angiogenic activity of OxPLs
High concentrations of OxPLs accumulate in atherosclerotic lesions; another characteristic feature of late atheroma is presence of neovessels that make the plaque prone to rupture. Since the density of neovessels does not always correlate with the thickness of plaque and corresponding level of hypoxia, it was hypothesized that additional intraplaque factors may play a role. Available data indicate that OxPLs are likely to be one of locally accumulated mediators promoting angiogenesis within the lesion. It was shown that diacyl-OxPLs stimulate migration of ECs, induce formation of EC sprouts, and promote angiogenesis in Matrigel plug model in mice (40). The authors presented evidence that angiogenic effects of OxPLs were mediated by autocrine mechanisms. According to the model, OxPLs stimulate production by ECs of VEGF-A, IL-8, and COX-2-derived prostaglandins that act in concert to induce angiogenic switch in ECs (40) (Fig. 11). Furthermore, OxPLs stimulated expression of metalloproteinase ADAMTS-1, further suggesting that OxPLs may play a role in plaque destabilization (40). Later studies demonstrated that OxPL-induced production of VEGF-A is mediated by ATF4-dependent transcription (248).
FIG. 11.

OxPLs induce angiogenic shift in ECs via autocrine mechanisms. OxPAPC and other classes of oxidized diacyl-OxPLs, but not their unoxidized precursors, stimulate angiogenic reactions in several in vitro and in vivo models. Angiogenic effects of OxPLs are mediated by autocrine loops, including production of VEGF, IL-8, and COX2-derived prostaglandins. In parallel, OxPLs stimulate expression of metalloproteinase ADAMTS1. Together, these effects are likely to promote formation of neovessels and degradation of matrix, thus leading to destabilization of atherosclerotic plaque.
In contrast to diacyl-OxPLs, PAF-like (alkyl-acyl) lipids and PAF were shown in different models either to stimulate or inhibit angiogenic reactions. In a study describing negative effects on angiogenesis, PAF-like (alkyl-acyl) lipids present in OxLDL inhibited production of FGF2, a key growth factor crucially important for formation of neovessels (58). In addition, fragmented OxPLs such as POVPC inhibited in a CD36-dependent manner production of VEGF-A by macrophages and inhibited corneal neovascularization (232).
Taken together, available data show that depending on chemical structure and biological context, OxPLs can induce both pro- and antiangiogenic effects. Further studies are required in order to identify the factors determining the balance of these opposite activites.
6. Calcification of atherosclerotic lesions and bone
Advanced atherosclerotic lesions are characterized by calcification and express osteoblastspecific markers. Increasing evidence shows that OxPLs might represent one of the factors responsible for calcification of atheroma. Treatment of calcifying vascular cells derived from aortic smooth muscle cells with MM-LDL or OxPAPC but not unoxidized lipoprotein or lipids, stimulated expression of alkaline phosphatase, specific changes in cell morphology and formation of cellular aggregates containing calcium mineral–all characteristic features of osteoblastic differentiation (256).
Paradoxically, in many patients with atherosclerosis and calcification of arteria, parallel loss of bone mineral often leads to osteoporosis. Parhami et al. offered an explanation for this phenomenon by showing that OxLDL and OxPLs exert opposite effects on evolution of bone as compared to arterial wall. They demonstrated that MM-LDL and OxPAPC inhibited differentiation of bone-derived preosteoblasts (256). Furthermore, OxPAPC was shown to inhibit action of bone anabolic agents such as bone morphogenic protein-2 and parathyroid hormone (148). An in vivo study using LDL receptor knockout mice, characterized by increased levels of oxidized (phospho)lipids, suggested that hyperlipidemia may interfere with osteoanabolic effects of parathyroid hormone (147).
Taken together, the data suggest that OxPLs are likely to play opposite role in calcification of arterial wall and bone.
7. Anti-inflammatory activities
Products of lipid peroxidation traditionally are regarded as toxic compounds inducing tissue damage and initiating inflammation. The data on the proinflammatory action of OxPLs in atherosclerotic vessels described above are consistent with this notion. Paradoxically, under certain conditions OxPLs also demonstrate tissue-protective and anti-inflammatory activites. The best documented anti-inflammatory effect of OxPLs is their ability to block acute inflammatory reactions induced by LPS (38, 192, 364). Below follows a description of TLR-specific and general anti-inflammatory mechanisms activated by OxPLs.
a. Modulation of TLR activity
Anti-endotoxin activity is characteristic of several OxPL species, including fragmented saturated OxPLs (192), fragmented unsaturated OxPLs (327), PL-hydroperoxides, PL-hydroxides, and isoP-containing PLs (360). In addition to TLR4, OxPAPC inhibited activation of TLR2 (83, 364), but did not influence the activity of other TLRs in transfected HEK 293 cells (83). Testing in several animal models confirmed the ability of OxPLs to inhibit acute inflammatory responses to exogenous agonists of TLRs 2 and 4 in vivo (38, 83, 204).
Available evidence suggests that OxPLs inhibit several steps in activation of TLRs, including extracellular components of the cascade (LBP, CD14, etc.) as well as cell-associated machinery (360) (Fig. 12). One hypothesis postulates that OxPLs bind to LBP and CD14, and that binding of OxPLs and LPS is mutually exclusive (360). Furthermore, OxPLs inhibit binding of LPS to MD-2 (83). Since LBP and especially CD14 and MD-2 are crucially important for presentation of LPS to TLR4, inactivation of these proteins is expected to inhibit action of LPS. In addition to TLR4, CD14 presents PAMPs of gram-positive bacteria to TLR2, explaining why OxPLs inhibit activation of TLR 2 and 4 but not other TLRs that are CD14-independent (83).
FIG. 12.

Potential mechanisms of the anti-endotoxin action of OxPLs. Different classes and molecular species of OxPLs were shown to inhibit effects of LPS in vitro and in vivo. OxPLs were hypothesized to inhibit several steps in recognition of LPS by TLR4 and activation of downstream signaling events. OxPAPC and other classes of OxPLs were shown to bind to LBP, soluble and membrane-bound CD14, and MD-2, and thus to inhibit interactions of these proteins with LPS, which are critically important for activation of TLR4 (mechanisms 1–3). In addition, OxPLs were shown to disrupt lipid rafts thus preventing formation of signaling complex of TLR4 with intracellular adaptors within caveolin-rich membrane domains (mechanism 4). Thus, OxPLs inhibit action of bacterial endotoxin via a multi-hit mechanism.
Another hypothesis suggests that OxPLs inhibit formation of TLR4 signaling complex with its downstream adaptors. This effect may be due to disruption of lipid rafts by OxPLs resulting from adsorption of cholesterol or action of sphingomyelinase products (364, 365), although another work showed that action of cholesterol-disrupting agents alone is not sufficient for inhibition of TLR2 and 4 (83), thus suggesting the involvement of more complicated inhibitory mechanisms.
In summary, it is likely that OxPLs inhibit action of LPS via a multi-hit mechanism targeting several steps in LPS recognition (360) (Fig. 12).
Some studies reported TLR4-dependent proinflammatory action of OxPLs (153, 366). These data suggest that under specific conditions or in certain cell types OxPLs may be agonistic for TLR4. One possibility is that alternative co-receptors, such as 37 kDa GPI-anchored protein (366) or scavenger receptors (136), rather than CD14, present OxPLs to TLR4. It is also possible that in those cases OxPLs upregulated endogenous TLR4 agonists or induced switch of TLR4 to other signaling adaptors. These speculative mechanisms require further investigation.
b. Induction of antioxidant and anti-inflammatory genes
An additional mechanism whereby OxPLs may exert anti-inflammatory and tissue-protective action is induction of drug metabolism phase II genes mediating protection from oxidant stress. Low concentrations of OxPLs do not damage cells but induce antioxidant enzymes such as glutamate-cysteine ligase and especially HO-1, well recognized for its prominent anti-inflammatory activity (2, 161, 182). These reactions are an important part of electrophilic stress response (ESR) and are discussed in more detail in Section II.B.5.
c. Inhibition of oxidative burst
In addition to modifying inflammatory reactions, different OxPLs including OxPAPC, OxPAPG, OxPAPS, OxPLPC were shown to inhibit oxidative burst induced by fMLP or PMA in neutrophil granulocytes without influencing viability or other functions of these cells (37). The effect resulted from the inhibition of assembly of phagocyte oxidase complex. Thus, oxidation of PLs may serve as a negative feedback preventing tissue damage by uncontrolled oxidative burst.
d. Lung barrier protection
Lung edema is a common life-threatening complication of acute systemic inflammation. OxPLs prevented lung edema formation in several in vivo models, including application of PAMPs or pathological lung ventilation. These protective effects of OxPLs are described in more detail in Section III.B.3.
8. Effects of OxPLs on cytoskeleton, noninflammatory cell adhesion, and permeability of endothelium
a. Cytoskeletal mechanisms of endothelial permeability
Paracellular pathway of endothelial permeability is regulated by a balance between competing contractile forces imposed by actomyosin cytoskeleton organized into stress fibers and opposing adhesive cell–cell and cell–matrix tethering forces produced by focal adhesions, adherens junctions, and peripheral actin cytoskeletal rim (220). Activation of myosin light chain kinase, MAP kinases, tyrosine kinases, and RHO-mediated signaling by edemagenic and pro-inflammatory agents causes phosphorylation of regulatory myosin light chains (MLC), cytoskeletal changes, activation of EC contraction, and destabilization of adherens junctions, leading to paracellular gap formation and increased EC permeability (220). RHO effectors MDIA and RHO-associated kinase (RHO-kinase) appear to be required for RHO-induced assembly of stress fibers, MLC phosphorylation, and actomyosin-driven cell contraction (110, 171, 353). In contrast, RAC-mediated enhancement of peripheral actin cytoskeleton and cell–cell junctions in OxPAPC-stimulated ECs is essential for restoration of EC barrier (28, 31, 33).
b. OxPAPC and EC permeability
Treatment of pulmonary ECs with OxPAPC in the range of 5–30 μg/ml caused dose-dependent enhancement of monolayer barrier (Fig. 13A), which lasted over 12 h (28). Of note, unoxidized PLs did not affect basal or agonist-induced permeability in ECs. At higher concentrations OxPAPC induced barrier disruption (Fig. 13A). Structure-function analysis showed that different molecular species present in OxPAPC have opposite influence on endothelial barrier. Nonfragmented sn-2-oxygenated OxPLs, especially cyclopenthenone-containing species (PEIPC, PECPC) are likely to be responsible for barrier-protective and barrier-enhancing effects of OxPAPC (28). In contrast, sn-2-fragmented OxPLs such as PGPC (Fig. 13B) and POVPC, increased endothelial permeability even at low concentrations (28, 272).
FIG. 13.

Effects of oxidized phospholipids on endothelial barrier function. (A) Bi-phasic concentration-dependent effects of OxPAPC on endothelial barrier function. Transendothelial electrical resistance (TER) reflecting barrier properties of EC monolayer was recorded in pulmonary ECs exposed to OxPAPC. OxPAPC exhibited prominent barrier-protective effects at concentrations below 20 μg/ml. Higher OxPAPC concentrations (50 and 100 μg/ml) caused barrier-disruptive response. (B) Nonfragmented, but not oxidatively fragmented PLs exhibit barrier-protective effect. TER was measured in pulmonary EC monolayers exposed to HPLC-purified isoprostanes esterified in PC (upper panel), or fragmented product PGPC (lower panel). Another fragmented product POVPC exhibited disruptive effects similar to that shown for PGPC. Note that individual components of OxPAPC, such as PEIPC and PGPC, demonstrate opposite action on EC barrier. Adapted from (28). (C) OxPAPC attenuates thrombin-induced elevation of EC permeability. TER was monitored across the confluent EC monolayers treated with OxPAPC (20 μg/ml) and thrombin (0.5 U/ml) added at the times shown by arrows. Adapted from (31).
Oxidized phosphatidyl serine (OxPAPS) and phosphatidyl choline (OxPAPC) bearing negatively or positively charged polar head groups, respectively, both exhibited potent and sustained barrier-protective effects. However, OxPL lacking head group (oxidized phosphatidic acid, OxPAPA) only transiently enhanced EC barrier (31). All three classes of OxPLs attenuated agonist-induced EC permeability triggered by transient activation of RHO pathway, but only OxPAPC and OxPAPS abolished short- and long-term EC barrier disruption caused by LPS (31).
One potential clinically relevant feature of OxPAPC is its ability to suppress RHO-dependent elevation of EC permeability induced by inflammatory and edemagenic agents (Fig. 13C). OxPAPC attenuated elevation of endothelial permeability caused by thrombin, IL-6, LPS, or exposure of endothelial cells to high-magnitude cyclic stretch and thrombin (31, 244). Treatment with OxPAPC accelerates the recovery of the compromised EC barrier function (28, 31). Another important observation is the barrier-protective effect of OxPAPC in the in vivo model of ventilator-induced lung injury (VILI) (244). VILI-associated EC barrier dysfunction was also reproduced in the in vitro model in EC monolayers exposed to high magnitude cyclic stretch and thrombin (244). This study demonstrated suppression of RHO signaling as a critical mechanism of protective effect of OxPAPC in VILI.
c. OxPAPC and cytoskeletal remodeling
Barrier-protective OxPLs cause specific rearrangements of cytoskeleton. They reduce the number of central F-actin stress fibers and markedly enhance peripheral F-actin rim (Fig. 14A, shown by arrows) responsible for the maintenance of monolayer integrity and EC barrier function. These features are similar to effects of other barrier-protective agonists, such as sphingosine 1-phosphate and prostacyclin (34, 106). The unique feature of OxPAPC is formation of microspike-like actin structures at the cell–cell interface (28) mediated by simultaneous activation of RAC and CDC42 (28), which likely contributes to the sustained character of EC barrier protective responses to OxPAPC. RAC effectors, WAVE, WASP, and ARP-2,3 complex are required for actin polymerization and enhancement of peripheral actin rim (43, 59, 230). Consistent with RAC/CDC42-mediated mechanism of cytoskeletal remodeling, OxPAPC induced peripheral translocation of the regulators of actin polymerization, preferentially activated by RAC (cortactin, p21ARC), CDC42 (N-WASP), and RAC/CDC42 (ARP3, phospho-cofilin) (28).
FIG. 14.
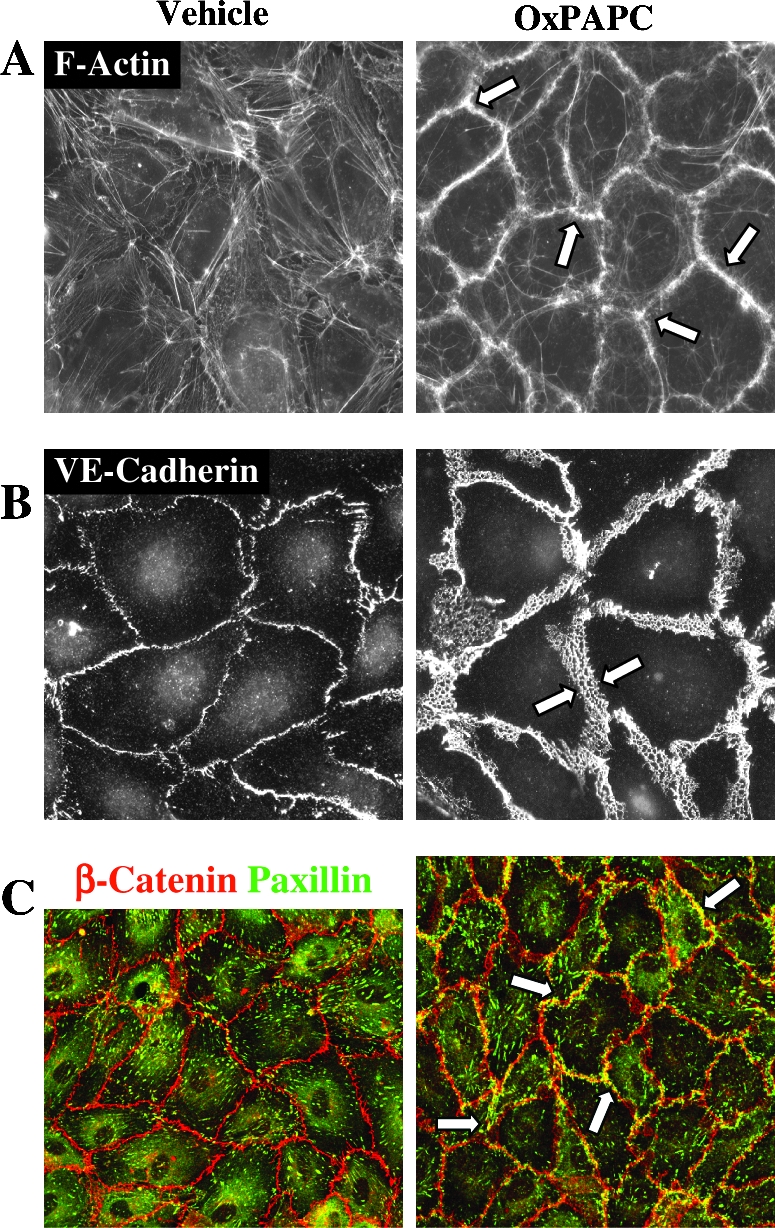
OxPAPC-induced endothelial remodeling. Enhancement of peripheral endothelial actin cytoskeleton (A, arrows), VE-cadherin positive adherens junctions (B, arrows), and peripheral colocalization of focal adhesions and adherens junctions (C, arrows) detected by double immunofluorescent staining for β-catenin (red) and paxillin (green) and confocal microscopy. ECs were stimulated with barrier-protective OxPAPC concentration (20 μg/ml). Adapted from (31, 33).
d. OxPAPC and assembly of adherens junctions
Assembly of adherens junctions (AJ) is critical for basal barrier enhancement and restoration of endothelial barrier disrupted by inflammatory agents or pathologic mechanical forces. Activated RAC stimulates association of VE-cadherin with intracellular catenin complex. This mechanism also provides physical integrity and cytoskeletal attachment of AJ complex, leading to increased EC barrier properties (76). OxPAPC at barrier-protective concentrations (5–30 pg/ml) causes enlargement of AJ areas in EC monolayers shown by arrows in Fig. 14B (33). Knockdown of AJ protein β-catenin attenuates OxPAPC-induced barrier enhancement in pulmonary EC (33).
e. OxPAPC induces FA-AJ interactions
OxPAPC induces peripheral accumulation of focal adhesion (FA) protein complexes containing paxillin (33) and leads to colocalization of FA complexes with enlarged adherens junction complexes (Fig. 14C, marked by arrows). Morphological and biochemical assays revealed novel OxPAPC-induced interactions between FA and AJ complexes via paxillin and β-catenin association mediated by RAC- and CDC42-dependent mechanisms (33). Disruption of paxillin-β-catenin interactions attenuated OxPAPC-induced barrier protective effects (33). This study suggests that novel paxillin-β-catenin interactions may form a “double rim of defense” in endothelial monolayers, which secures intercellular flux of solutes and macromolecules controlled by AJ and limits the fluid influx between EC and basal membrane by formation of peripheral FA rim. FA–AJ interactions thus may function as a double lock, preventing macromolecular transport between and under EC.
f. Gap junctions and tight junctions
As discussed above, high OxPAPC concentrations or EC treatment under serum-free conditions may cause EC barrier dysfunction. DeMaio et al. reported decreased expression of tight junction proteins occludin and ZO-1 and increased flux of 10-kDa dextran through EC monolayers indicative of EC barrier dysfunction in EC stimulated with 20–50 pg/ml OxPAPC in serum-free conditions. These effects were linked to oxidative stress induced by OxPAPC (78).
A study by Isakson et al. (155) showed OxPAPC-induced dysregulation of connexins CX37, CX40, and CX43 in the carotid artery EC and VSMC. These proteins form gap junctions that are assembled as dodecameric channels composed of a pair of connexin hexamers linking two adjacent cells. Gap junctions are involved in transduction of signal from cell to cell by intercellular transport of small signaling molecules. OxPAPC-induced changes in connexin expression markedly reduced biocytin dye transfer between ECs and VSMC, suggesting impaired communication between EC and VSMC. These results show that phospholipid oxidation products such as OxPAPC, which accumulate during the development of atherosclerotic lesions, may affect gap junction-mediated communication in the vascular wall.
In summary, OxPAPC and isoP-PLs demonstrate potent lung barrier-protective effects in cell and animal models of acute lung injury induced by inflammatory and edemagenic agonists and pathologic mechanical strain (Fig. 15). These effects are mainly mediated by activation of RAC and downregulation of RHO-mediated signaling, leading to enhancement of peripheral actin structures, cell–cell junctions, and novel interactions between cell–cell and cell–substrate adhesive protein complexes. Another important lung vascular protective mechanism is inhibition of inflammatory cascades triggered by bacterial components, which will be described below (Section III.B.3).
FIG. 15.

Protective effects of OxPLs against acute lung injury and endothelial barrier dysfunction. PL oxidation products such as OxPAPC can inhibit lung damage in acute bacterial inflammation due to their ability to inhibit activation of TLRs 2 and 4. Furthermore, PL-esterified isoprostanes activate tyrosine kinase SRC and PI3K-AKT signaling, leading to recruitment of RAC/CDC42 specific nucleotide exchange factors TIAM1 and βPIX, stimulation of RAC and CDC42 GTPases and their effectors involved in activation of peripheral actin polymerization (cortactin, p21ARC, ARP2/3, N-WASP, cofilin), focal adhesion remodeling (FAK, paxillin) and enhancement of adherens junctions (β-catenin). These cytoskeletal changes are critical for enhancement of vascular endothelial barrier properties. Finally, OxPAPC and other classes of OxPLs protect against vascular hyperpermeability caused by edemagenic, inflammatory factors, and pathologic lung overdistension. This protection involves inhibition of RHO GTPase-dependent pathway of endothelial contraction and monolayer barrier disruption via activation of RAC-dependent mechanisms triggered by OxPLs.
9. Regulation of adaptive immunity
Inflammatory signals play a key role in initiation of adaptive immune responses. Recent studies demonstrated that OxPLs can modulate adaptive immune system by acting on antigen-presenting dendritic cells as well as T cells.
a. Modulation of dendritic cell function by OxPLs
Dendritic cells (DCs) are key regulators of adaptive immunity. In the steady state, DCs reside in peripheral tissues as immature antigen-presenting cells and are considered to be tolerogenic. During infections, DCs are activated by stimulatory signals from invading pathogens. DC maturation is accompanied by translocation of MHC–peptide complexes to the cell surface and upregulation of co-stimulatory molecules such as CD40, CD80, and CD86. As a result, DCs become fully competent to activate T-cells. Recent studies demonstrated that OxPLs have no significant direct influence on phenotype of immature DCs except for modestly elevated surface expression of CD86 and MHC class II (36). Furthermore, OxPAPC alone did not have major impact on the functional behavior of DCs. However, OxPLs can strongly interfere with the activation of DCs induced by PAMPs recognized by TLRs 2, 3, and 4, as well as by CD40–CD40L interaction.
OxPAPC was found to inhibit in human monocyte-derived DCs upregulation of CD40, CD80, CD86 and CD83 induced by LPS (36) (Fig. 16). Moreover, LPS-induced surface expression of MHC classes I and II as well as production of IL-12 was significantly inhibited by OxPAPC. As a consequence, OxPAPC dramatically reduced T-cell stimulatory capacity of DCs treated with LPS (36). OxPAPC also strongly inhibited surface expression of chemokine receptor CCR7 in LPS-treated DCs. Downregulation of CCR7 is known to impair emigration of DCs from the inflamed tissue to the lymph node that is crucially important for the induction of an antigen-specific immune response (214).
FIG. 16.

Inhibitory effects of OxPLs in adaptive immunity. OxPLs can influence adaptive immune responses via several mechanisms. First, OxPLs inhibit maturation of dendritic cells (DCs) induced by LPS and CD40L (mechanism 1). Furthermore, OxPLs inhibit production of IL-12 by DCs (mechanism 2). In addition, several classes of OxPLs were shown to inhibit activation of T cell receptor (mechanism 3). Finally, OxPLs via as yet unidentified mechanisms upregulate expression in T cells of transcription factors CBL-B and EGR-3 inducing an anergy-like state (mechanism 4). Together, these effects characterize OxPLs as immunosuppressors.
It is likely that many of the abovementioned effects result from the ability of OxPLs to inhibit LPS binding to, and activation of, TLR4 that is discussed in Section III.A.7. However, low concentrations of OxPAPC did not block LPS-induced signaling (i.e., NF-κB activation) or upregulation of the DC activation marker (CD83), but suppressed LPS-induced production of IL-12 and TNFα (36) (Fig. 16). Moreover, at low concentrations of OxPAPC, the ability of LPS-treated DCs to polarize T-cells towards IFN-γ-producing Th-1 cells was abrogated (36). These results suggest that in parallel to the inhibition of TLR4 activation, OxPLs modulate Th-1-driving capacity of DCs via currently unknown mechanisms.
OxPAPC also inhibited DC maturation induced by Poly I:C, which signals through TLR3. Similarly to the effects on LPS-induced DC maturation, addition of OxPAPC prevented induction of costimulatory molecules, production of cytokines and increase in T cell-stimulatory capacity in DCs activated by Poly I:C (36). Although to a lower extent, OxPAPC also inhibited maturation of DCs induced via TLR2 or CD40. OxPAPC did not influence upregulation of co-stimulatory molecules or activation of NF-κB induced by CD40L; however, CD40L-induced elevation of TNFα and IL-12 was potently suppressed by OxPAPC (36). Similarly, in case of TLR2-mediated maturation stimulated by Pam3CSK4 or proteoglycan, OxPAPC did not interfere with upregulation of co-stimulatory molecules, but potently inhibited TNFα and/or IL-12 production (36). These data identify OxPAPC as a strong negative regulator of IL-12 production in DCs treated with various maturation-inducing stimuli, pointing to a mechanism whereby OxPLs can impair adaptive immune responses.
b. Induction of T cell anergy by OxPLs
In addition to modulating DC function, OxPLs can directly regulate T cell activation. OxPLs having different polar head groups (OxPAPC, OxPAPS, OxPAPG, and OxPAPA), but not their unoxidized precursors, strongly inhibited the proliferation of purified human T cells stimulated with anti-CD3/CD28 or anti-CD3/CD63 mAbs (302). Inhibition of T cell proliferation by OxPLs was not due to their toxicity since T cell proliferation triggered by PMA/ionomycin was not diminished by OxPLs. T cells activated by anti-CD3/CD28 in the presence of OxPLs produced and released lower amounts of IFN-γ and IL-2, whereas IL-4 was only slightly diminished. OxPL-treated T cells produced only low amounts of IL-10, a well-established immunosuppressive cytokine. Thus, it is unlikely that the inhibitory effects of OxPLs on T cells were due to elevated production of IL-10. OxPLs prevented the expression of de novo- synthesized activation markers (CD25, MHC-II) but not expression of CD63 or CD69, which translocate from preformed intracellular pools to the cell surface of anti-CD3/CD28 mAb-stimulated T cells. Furthermore, T cells stimulated in the presence of OxPLs were characterized by low cytotoxicity of CD8+ T cell, which is pivotal mechanism in removing pathogens. Most importantly, T cells that were activated in the presence of OxPLs failed to proliferate in response to restimulation, a phenomenom called anergy. This hypoproliferative state was accompanied with an upregulation of the anergy-related genes early growth response gene 3 (EGR-3) and the E3 ligase Casitas-B-lineage lymphoma protein-B (CBL-B), which are both well-defined negative regulators of T cell activation (88). In summary, available data suggest that treatment of T cells with OxPLs rather modulates TCR/CD3 signaling than completely blocks it. In contrast to IL-10 and immunosuppressive drugs such as FK506, cyclosporine A, glucocorticoids, or rapamycin, OxPLs are known to repress the cytolytic function of cytotoxic T cells (307, 325, 388). These observations show that OxPLs demonstrate unique immunosuppressive characteristics and allow hypothesizing that direct inhibitory effects of OxPLs on T cells might contribute to the control of T cell function necessary to avoid overwhelming or even detrimental Th1-driven immune responses at sites of inflammation.
B. Accumulation and role of OxPLs in specific pathologies
1. Atherosclerosis
Quantification of OxPCs using liquid chromatography coupled to tandem mass spectrometry showed that atherosclerotic vessels contain high concentrations of OxPCs. Different species of OxPCs were detected in atherosclerotic vessels including PL-hydroperoxides and hydroxides (362), oxidatively fragmented PL species containing saturated and unsaturated truncated residues (137, 264, 328, 368), PL-esterified isoPs (118, 269, 328, 368, 370), and PL-isolevuglandins (267, 292). The analysis of OxPL spectra at different stages of human atherosclerosis (fatty streak, fibrous plaque, necrotic core, and calcified tissue) identified similar OxPL species at different stages of the disease, including early oxidation species (hydroperoxides) and advanced products (fragmented OxPLs) (276). These data suggest that during the evolution of atherosclerotic lesion, OxPLs are continuously generated and degraded.
In parallel with elevated levels of OxPLs, atherosclerotic vessels express high amounts of proteins known to be induced by OxPLs in vitro. These include for example MKP-1 (279), ATF3, ATF4 (107), SREBP-1 (380), HO-1 and IL-8 (64), MCP-1 and COX-2 (203), and GROα (253). Furthermore, quantification of OxPLs in atherosclerotic vessels showed that total concentration of active OxPLs reach locally in atherosclerotic vessels high levels comparable with those inducing biological effects in vitro (Table 4). These data support the view that OxPLs not only accumulate in atheroma but play mechanistic role in atherogenesis.
Table 4.
In vivo Levels of OxPLs
| Sample type | OxPL species | Control conditions | Conditions inducing elevation of OxPLs | References | |
|---|---|---|---|---|---|
| Thoracic aorta of Watanabe heritable hyperlipidemic rabbits | Young rabbits | Rabbits >>6 months old (hypercholesterolemia, atherosclerosis) | 264 | ||
| PC-esterified: HOOA, KOOA, HOdiA, KOdiA | 0.2–1 ng/mg PAPC (each) | 1–3 ng/mg PAPC (each) | |||
| PC-esterified: HODA, KODA, HDdiA, KDdiA | < 0.2 ng/mg PLPC (each) | 0.5–2 ng/mg PLPC (each) | |||
| POVPC, PGPC | 1 ng/mg PAPC (each) | 3–4 ng/mg PAPC (each) | |||
| PONPC, PAzPC | 2–3 ng/mg PLPC (each) | 6–10 ng/mg PLPC (each) | |||
| Rabbit aorta | Normal chow diet | Atherogenic diet (hypercholesterolemia, atherosclerosis) | 368 | ||
| POVPC, PGPC, PEIPC | 20 μg/g wet tissue (each) | 40–60 μg/g wet tissue (each) | |||
| Rabbit aorta | Normal chow diet | Atherogenic diet (hypercholesterolemia, atherosclerosis) | 328 | ||
| POVPC, PGPC, PEIPC, SOVPC, SGPC, SEIPC | 15–25 μg/g wet tissue (each) | 50–100 μg/g wet tissue (each) | |||
| Human carotid artery plaque | Several major species of fragmented and full-length OxPC | – | 0.1–2% of total PC (each) | 73 | |
| Rat plasma | Vitamin E supplemented, normoxia | Vitamin E depleted, hyperoxia | 97 | ||
| Fragmented ω-carboxylic PCs | 1 μm | 3 μM | |||
| Human plasma | Young, healthy donors | Elderly people with CHD | 97 | ||
| Fragmented ω-carboxylic PCs | 2.5 μM | 4 μM | |||
|
Before smoking 3 μM |
After smoking 6 μM |
||||
| Mouse plasma | Wild type mice, chow diet | Apoe−/−mice, western diet | 263 | ||
| PC-esterified: HOdiA, KOdiA, HOOA, KOOA, HDdiA, KDdiA, HODA, KODA |
0.04–0.36 μM (each) | 0.64–3.00 μM (each) | |||
| Wild type mice, chow diet | Ldlr−/−mice, western diet | ||||
| PC-esterified: HOdiA, KOdiA, HOOA, KOOA, HDdiA, KDdiA, HODA, KODA |
0.10–0.40 μM (each) | 0.50–2.21 μM (each) | |||
| Human plasma | Normotriglyceridemic donors with variable LDL cholesterol levels | 263 | |||
| PC-esterified: HOdiA, KOdiA, HOOA, KOOA, HDdiA, KDdiA, HODA, KODA |
Undetectable to 1.8 μM (each) 0.7–4.6 μM (combined) |
||||
| Human plasma | Normotriglyceridemic donors with variable LDL cholesterol levels | 263 | |||
| PC-esterified: HOdiA, KOdiA, HOOA, KOOA, HDdiA, KDdiA, HODA, KODA POVPC, PGPC |
5.4–51 μM (combined) | 263 | |||
| Small intestine of mice | No irradiation | Total body gamma-irradiation | 347 | ||
| CL-OOH PS-OOH PE-OOH PC-OOH PI-OOH |
6.9 ± 1.4 mmol/mol of CL 4.1 ± 1.8 mmol/mol of PS ∼4 mmol/mol of PE ∼5 mmol/mol of PC ∼5 mmol/mol of PC |
60.1 ± 13 mmol/mol of CL 37.4 ± 6.0 mmol/mol of PS ∼15 mmol/mol of PE ∼15 mmol/mol of PC ∼21 mmol/mol of PC |
|||
| Human dermal fibroblasts | No irradiation | UVA-1 irradiation | 125 | ||
| PEIPC | 0.8 μM | 7.7 μM | |||
| Mouse brain tissue | Sham-operated brain | Ischemic cerebral tissue | 105 | ||
| OxPC-furan (2) OxPC-furan (3) OxPC-furan (7) |
< 0.018 mmol/mol precursor PC 0.4 mmol/mol precursor PC 4 mmol/mol precursor PC |
0.18 mmol/mol precursor PC 4.0 mmol/mol precursor PC 60 mmol/mol precursor PC |
|||
| Cultured HAECs | Control | Treatment with IL-1β | 329 | ||
| POVPC PGPC PEIPC PECPC |
12.5 ng/μg lipid phosphorus 30 ng/μg lipid phosphorus 13 ng/μg lipid phosphorus 6 ng/μg lipid phosphorus |
22.5 ng/μg lipid phosphorus 42.5 ng/μg lipid phosphorus 22.5 ng/μg lipid phosphorus 11 ng/μg lipid phosphorus |
|||
| Rat retinas | Control animals | Rats exposed to light | 331 | ||
| PGPC POVPC PONPC PAzPC OBPC |
1 mmol/mol PAPC 17 mmol/mol PAPC 60 mmol/mol PLPC 25 mmol/mol PLPC 10 mmol/mol DHA-PC |
2 mmol/mol PAPC 36 mmol/mol PAPC 150 mmol/mol PLPC 50 mmol/mol PLPC 22 mmol/mol DHA-PC |
Several activities of OxPLs described above suggest that OxPLs are likely to be involved into all stages of atherogenesis. OxPLs can play a role in initiation of arterial inflammation (monocyte–endothelial interactions, production of chemokines), progression of disease (foam cell formation, smooth muscle cell phenotypic modulation and migration, extracellular matrix production, and calcification) and development of complications (angiogenesis, production of metalloproteinases, thrombogenic shift in endothelium, and activation of platelets). OxPLs act on all major cell types involved in atherogenesis including monocytes, endothelial and vascular smooth muscle cells, lymphocytes, and platelets. These findings raise a possibility that regulation of OxPL levels and inhibition of their deleterious effect may become a useful strategy for prevention and treatment of atherosclerosis. Current research in this direction is described in Section IV.B.
2. Acute inflammation
Acute inflammation is accompanied by massive production of ROS, which in parallel with other antibacterial mechanisms kill invading microorganisms. As a result of “friendly fire,” also host lipoproteins and membrane lipids become oxidized. The elevation of circulating levels of OxLDL in response to inflammatory stimuli has been shown in vivo in models of acute inflammation induced in hamsters by injecting LPS, zymozan, or turpentine (221). Elevated plasma levels of PL-hydroxides were detected in plasma of patients with dengue fever (188).
The production of OxPLs in response to inflammation may be induced in different cell types including leukocytes. Phorbol ester-stimulated neutrophils and monocytes incubated with PUFA-PCs produced mono- and bishydroperoxides of PC, as well as isoP–PC, thus suggesting that activated phagocytes can oxidize lipids in the surrounding medium (159). Furthermore, macrophages infected with mycobacteria accumulated isoP–PC (PEIPC) (70). Nonleukocyte cell types also generate OxPLs in response to inflammatory stimuli. It has been shown that accumulation of fragmented OxPLs (POVPC, PGPC), as well as OxPLs containing esterified epoxyisoprostanes (PEIPC, PECPC) was induced by IL-1 treatment of human aortic ECs (329). In addition, infection with influenza A virus stimulated production of POVPC, PGPC, and PEIPC by human alveolar type II cell line (352). Moreover, accumulation of OxPCs recognized by E06 mAb was observed in the lungs of humans and animals infected with H5N1 avian flu, anthrax, or SARS (153). These data demonstrate that accumulation of OxPLs is characteristic of different types of inflammatory reactions.
Functional importance of OxPLs accumulating during acute inflammation is insufficiently understood. OxPLs both positively and negatively regulate multiple inflammatory reactions and therefore may exert variable and context-dependent effects on the outcome of inflammation. Due to their ability to upregulate inflammatory cell adhesion molecules and chemokines (see Section III.A.1), OxPLs can promote inflammation. Proinflammatory action of IL-6 induced by OxPLs was suggested to play important role in the pathogenesis of acute lung injury induced by viruses, bacteria, and application of acid (see Section III.B.3). In addition, OxPLs inhibit activation of TLRs 2 and 4 by bacterial PAMPs (see Section III.A.7) and thus may interfere with innate immune responses to bacteria. Furthermore, OxPLs were shown to inhibit phagocytosis of bacteria by neutrophils and macrophages, leading to enhanced mortality of animals from abdominal sepsis induced by E. coli (178). Impaired clearance of bacteria can be aggravated by the ability of OxPLs to inhibit oxidative burst in neutrophils and to suppress adaptive immune reactions (see Sections III.A.7 and III.A.9). These deleterious effects characterize OxPLs as potential immunosuppressors impairing various facets of antibacterial defense. On the other hand, inhibition of TLRs and oxidative burst by OxPLs may serve as a negative feedback preventing excessive activation of antibacterial mechanisms in order to avoid damage to host tissue. This hypothesis is supported by several studies showing protective effects of OxPLs under the conditions of acute “sterile” inflammation induced by injection of purified PAMPs (see Section III.A.7). Further studies are required in order to determine factors influencing the balance of pro- and anti-inflammatory activities of OxPLs in vivo.
3. Lung injury
a. Oxidative stress and generation of OxPLs in lungs
The epithelial lining pulmonary surfactant is permanently exposed to high concentrations of oxygen and other oxidants present in the air. Ozone, a common pollutant in urban air, is a highly reactive gas readily attacking double bonds and producing oxidatively truncated PLs (349). PLs (mainly PCs) represent the major component of lung surfactant comprising 80% of its weight. Under physiological conditions, surfactant is protected from oxidation by low contents of PUFAs, antioxidant action of glutathione present in the lining fluid, and by surfactant proteins A and D (185, 187). However, in pathological states oxidation of surfactant PCs, membrane lipids and apoptosis of bronchial cells may result in accumulation of biologically active OxPL products.
Several types of lung injury are accompanied by oxidative stress. Infection of type II pneumocytes with influenza A virus dramatically increased secretion of PAPC oxidation products that induced MCP-1 expression (352). Imai et al. (153) used OxPC-specific E06 antibody to document elevated lung levels of OxPLs in murine models of lung injury caused by H5N1 avian influenza virus and acid instillation. Formation of OxPLs was also observed in inflammatory exudates and alveolar macrophages of H5N 1-infected human patients, anthrax-infected rabbits, and anthrax-, pox- and Yersinia pestis-infected monkeys. Oxidative stress is also characteristic of idiopathic interstitial pneumonia, as illustrated by accumulation of OxPC-positive cells in alveolar spaces of the lungs with desquamative or usual interstitial pneumonia (385). These cells were characterized as macrophages expressing high levels of CD36, suggesting that they play a role in scavenging of OxPLs generated in the inflamed lung (385).
b. Proinflammatory effects of OxPLs in the lungs: SARS, anthrax, H5N1 avian influenza virus, and acid-induced lung injury
Accumulation of OxPLs in the murine models of H5N1 avian influenza virus and acid-induced lung injury (153) was accompanied by increases in lung elastance reflecting compromised lung function. Mechanistic involvement of OxPLs in lung inflammation was demonstrated by inhibitory effect of OxPC-specific E06 antibody on production of cytokines by alveolar macrophages in response to bronchoalveolar lavage fluid (BAL) from injured lungs. Oxidized lung surfactant preparations increased lung elastance and IL-6 production. Similarly, OxPAPC increased lung elastance within an hour after intratracheal application. Based on amelioration of OxPL- and acid-induced increases in lung elastance in il-6, tlr4, and trif knockout mice, the authors proposed that IL-6 induced by OxPLs via the TLR4-TRIF pathway plays a crucial role in deleterious effects of OxPLs on lungs (153).
On the other hand, under certain conditions OxPLs can also exert anti-inflammatory effects and protect pulmonary vascular endothelial barrier. The lung-protective activity of OxPLs is discussed below.
c. Anti-inflammatory and barrier-protective effects of OxPAPC in the models of acute lung injury
Several studies showed that OxPLs protect lung function in animal models of acute lung injury (ALI) induced by application of pure PAMPs such as LPS and CpG DNA. Ma et al. showed that OxPAPC inhibits elevation of TNFα in mice upon intratracheal or systemic administration of LPS or CpG DNA (204). Another work demonstrated that lung inflammation and vascular leak induced by intratracheal LPS aerosol administration were markedly attenuated by OxPAPC (245). These results are in good correlation with and are likely to be explained by the ability of OxPAPC to reverse LPS-induced cytoskeleton remodeling and disruption of monolayer integrity (see Section III.A.8).
In addition to PAMP-induced models of acute lung injury, OxPAPS and OxPAPC markedly reduced lung vascular leak associated with ventilator-induced injury (244). Murine and rat models of ventilator-induced lung injury (VILI) demonstrated that intravenous injection of OxPAPC markedly attenuated protein and inflammatory cell accumulation in bronchoalveolar lavage fluid and lung tissue (244). An in vitro model of VILI showed that high magnitude stretch enhanced endothelial paracellular gap formation induced by thrombin via synergistic effects on RHOGTPase activation. Stretch-induced endothelial barrier disruption was markedly attenuated by OxPAPC and was accompanied by activation of RAC and suppression of RHO (244). Taken together, these findings suggest lung-protective effects of OxPAPC in “aseptic” type of ALI.
d. Summary: Dual effects of OxPLs in lung pathology
The data discussed above show that OxPLs may induce either beneficial or detrimental effects on lungs under different circumstances. Several factors may determine the balance of pro- and anti-inflammatory effects of OxPLs in the lung. First, due to the ability of OxPLs to inhibit inflammatory effects of PAMPs mediated by TLRs 2 and 4 (see Section III.A.7), OxPLs are likely to exhibit anti-inflammatory effects in lung injury induced by a variety of PAMPs from gram-negative and gram-positive bacteria. On the other hand, study by Imai et al. suggests that high OxPL levels may directly or indirectly engage TLR signaling, leading to lung injury progression (153). Second, due to direct effects on lung endothelial permeability, specific OxPLs can suppress lung vascular leak induced by aseptic edemagenic molecules (thrombin) or excessive lung distension during mechanical ventilation. Third, the action of OxPLs on the lungs may depend on their concentrations: whereas low levels of OxPLs protect endothelial barrier, high concentrations of the same OxPLs induce barrier-disruptive effects (28, 78). This biphasic action may be explained by the overwhelming action of barrier-disruptive fragmented and lyso-species at high concentrations of OxPLs (28, 272). Different mechanisms of oxidation produce different proportions of barrier-protective and -disruptive species. For example, oxidation of monounsaturated fatty acids in the lung surfactant by ozone selectively generates barrier-disruptive fragmented OxPLs such as POVPC (see Section III.A.8). In contrast, oxidation of PLs in membranes and lipoproteins by cell-generated free radicals produces a large proportion of esterified isoPs demonstrating prominent barrier-protective effects (28). Therefore, the net pro- vs anti-inflammatory effect of endogenous OxPLs likely depends on the intensity, duration, and mechanism of oxidative stress and efficiency of OxPL clearance since these factors determine total concentrations of OxPLs as well as contents of individual species. Quantitative analysis of systemic and tissue concentrations of molecular OxPL species is required in order to get more insight into the action of OxPLs in the lungs under different inflammatory states.
4. Ischemia
Ischemia and ischemia followed by reperfusion are characterized by massive production of ROS. Several studies demonstrated elevated tissue and systemic levels of OxPLs in response to ischemia/reperfusion. Plasma levels of esterified hydroxides and F2-isoPs were elevated in patients with acute ischemic stroke (188). Systemic and local levels of hydroperoxides of PE and PC were elevated upon ischemia-reperfusion of rat liver (101, 333). In addition, PAF-like (alkyl-acyl) OxPLs were detected within the first minutes after reperfusion of kidneys after warm ischemia (200). Furthermore, plasma concentrations of fragmented OxPCs were increased in patients during the reperfusion period after coronary surgery with cardiopulmonary bypass (97). Stroke induced by ligation of the middle cerebral artery in mice resulted in accumulation of enhanced levels of furan-PCs in infarcted brain tissue (105). Furan-PCs accumulated in ischemic hemisphere but not in contralateral control hemisphere (105). Myocardial ischemia-reperfusion models demonstrated elevated levels of PC-hydroperoxides and oxidatively fragmented PCs in the heart ventricles 6 hours after reperfusion that was preceded by 30 minutes of coronary artery occlusion (235). In addition, oxidation of mitochondrial CL was stimulated by ischemia-reperfusion in aged rat hearts (194).
In summary, accumulated data show that ischemia/reperfusion is a pathological state characterized by elevated local and circulating levels of OxPLs. In some of these studies, amelioration of reperfusion-induced symptoms was achieved by application of antioxidants (200), suggesting that oxidized lipids and OxPLs in particular may play a role in the pathogenesis of this condition.
5. Light- and radiation-induced stress
Formation of OxPLs can be stimulated by visual and UV-light. Several oxidatively fragmented OxPEs including sn-2-succinyl-, sn-2-(9-oxo) nonanoyl-, and sn-2-(4-oxo)butaroyl-PE were found in rat retina (128). Furthermore, acute exposure of rats to green light induced accumulation in the retinas of saturated and unsaturated fragmented OxPCs that are ligands for scavenger receptor CD36 (331). OxPLs accumulating in retinas serve as ligands for CD36-dependent phagocytosis of shed photoreceptor outer segments by retinal pigment epithelium; this process is necessary for normal function of the retina (331).
Generation of OxPLs induced by light exposure was also demonstrated in skin cells. UVA-1 ultraviolet radiation (340–390 nm) stimulated accumulation of E06-reactive OxPCs in dermal fibroblasts via singlet oxygen-dependent mechanism (125). UVA-1-irradiated PAPC containing several OxPL species induced expression of antioxidant and anti-inflammatory enzyme heme oxygenase-1 in dermal fibroblasts, keratinocytes, and in a three-dimensional epidermal equivalent model (125). Thus, OxPLs are likely to play a protective role in UVA-irradiated skin by inducing HO-1. In addition, UVB radiation was shown to stimulate production in skin of PAF and PAF-like (alkyl-acyl) OxPLs that are likely to play a role in UVB-induced hyperalgesia (390). Furthermore, UVB irradiation of cultured epidermal cells stimulated production of fragmented alkyl-PCs such as hexadecyl-azelaoyl-PC that is a ligand for PPARγ (391). PAF-like (alkyl-acyl) lipids generated in response to irradiation promoted in a PPARγ-dependent manner expression of cyclooxygenase-2 and production of PGE2—events potentially relevant to UVB-induced immuno-suppression (391).
Tissue injury induced by gamma-irradiation was shown to induce oxidation of PLs. The analysis of OxPLs in small intestine upon whole body gamma-irradiation of mice demonstrated selective oxidation of CL in mitochondria and PS outside of mitochondria, leading to formation of several hydroperoxides of PS and CL. The authors hypothesized that elevated levels of these OxPLs may result from radiation-induced apoptosis, which is characterized by selective oxidation of CL and PS (347).
6. Leprosy
Oxidized PCs recognized by E06 mAb were detected in lepromatous (disseminated) leprosy lesions, but not in tuberculous leprosy characterized by stronger host immune response and self-contained infection (70). OxPLs mainly accumulated in macrophage-derived foam cells. Analysis by mass spectrometry confirmed elevated levels of isoP-containing PEIPC in mycobacteria-infected human macrophages (70).
In order to demonstrate functional importance of OxPLs accumulated in leprosy lesions, the effects of PEIPC on innate immune responses were analyzed. It was shown that PEIPC prevented upregulation on differentiating dendritic cells of CD1b, which is important for presentation of lipid antigens to T cells (70). The effect was accompanied by impaired activation of M. leprae-reactive CD1b-restricted T cells. Furthermore, OxPLs modulated the activity of TLR2/1 known to recognize mycobacterial triacylated lipoproteins. PEIPC inhibited secretion of proinflammatory IL-12, but stimulated production of anti-inflammatory IL-10 induced by TLR2/1 ligand. Furthermore, PEIPC inhibited TLR2/1-induced production of antimicrobial peptide cathelicidin, known to protect human monocytes and macrophages against myco-bacteria (70).
In summary, lepromatous leprosy lesions are characterized by accumulation of OxPLs, which can counteract innate and specific immune responses to mycobacteria and thus promote their survival.
7. Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease of the brain, which targets the myelin sheath of the central nervous system and results in neurodegeneration. A number of potential culprit protein antigens have been identified, but there is also increasing evidence that lipid-specific autoimmune responses are equally involved. With more than 70%, lipids comprise a major part of myelin sheaths. Kanter et al. have tested this hypothesis by analyzing cerebrospinal fluids of MS patients for the presence of antibodies to specific lipids using a newly developed large-scale lipid microarray (167). Antibodies in MS serum samples showed strong reactivity to various lipids, such as sulfatide, sphingomyelin, and PLs, but also to three forms of OxPC (PAzPC, PONPC, and PGPC). While the authors show that sulfatide-specific antibodies can worsen disease in mice in the experimental allergic encephalitis (EAE) model, the pathogenic role of antibodies to OxPLs is not clear. Nevertheless, the importance of OxPLs in MS was further supported in a later report by Qin et al., who demonstrated the presence of OxPLs (alone and conjugated to a 15 kDa protein) in extracts of MS lesions directly by Western blot analyses using the E06 antibody (273). Interestingly, the authors also demonstrated the presence of natural E06 antibodies themselves using an anti-idiotypic Ab that specifically recognizes the idiotope of E06/T15. Thus, both OxPLs and OxPL-reactive antibodies are found in plaques of MS patients and mouse models. Likely, OxPLs promote the inflammatory process in MS lesions, whereas the role of specific anti-OxPL antibodies is more complex and requires further evaluation.
IV. OxPLs as Biomarkers of Disease and Targets for Therapy
A. Association of OxPL levels with disease
1. Methods of quantification of OxPLs
Quantification of OxPLs in vivo is a challenging analytical task since OxPLs are significantly more variable as compared to unoxidized precursors (see Fig. 2), and each of multiple oxidized species is present in vivo in significantly lower amounts than precursor PL (Table 4). OxPLs can be detected using specific antibodies (337a) or a combination of HPLC with reagents specific for oxidized groups (e.g., carbonyls or hydroperoxides) (97, 101, 109). However, the most universal and sensitive method for quantification of OxPLs is mass spectrometry. Since PLs are thermolabile and nonvolatile molecules, their analysis is performed using soft ionization techniques such as electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI). Complex biological samples such as animal tissues and blood plasma contain multiple molecules having mass-to-charge (m/z) ratios very close to OxPLs, so-called isobaric compounds. As a result, OxPLs cannot be detected in such samples by the routine methods of “shot-gun” lipidomics. Typically, reliable identification of OxPLs requires separation of isobaric lipids by high-performance liquid chromatography followed by tandem mass spectrometry. Each molecular species is characterized by a “signature” consisting of its m/z value, chromatography retention time, and m/z value(s) of its characteristic degradation product(s). Detailed description of mass spectrometry of OxPLs can be found in the review of Domingues et al. (81).
OxPLs were detected in a variety of normal human and animal tissues, and their elevated levels were found in different pathological states (Table 4). High levels of OxPLs reaching 1% of precursor PL were observed in plasma and arterial wall in patients and experimental animals with hypercholesterolemia and atherosclerosis. Normalization of OxPL amounts to the volume of plasma or wet weight of tissue showed that under physiological conditions individual species of OxPLs are either nondetectable, or present at low concentrations (Table 4). However, in pathology their concentrations can reach the levels of micromoles or even tens of micromoles per liter. Importantly, the majority of biological effects of OxPLs that were detected in vitro (e.g., monocyte binding to ECs, production of IL-8, induction of electrophilic genes, activation of UPR, the angiogenic switch of ECs, inhibition of LPS effects, enhancement of endothelial barrier, etc.) are also observed in the range of micromoles to tens of micromoles per liter, and on the other hand these effects are induced by different classes and molecular species of OxPLs. Therefore, combined concentration of active species, rather than their individual levels, should be taken into account. Furthermore, Table 4 mainly summarizes data on OxPLs generated from palmitoyl-arachidonoyl- and palmitoyl-linoleoyl-PC, while other molecular species and classes of OxPLs, which are likely to have similar biological activities, were not analyzed. Thus, concentrations of OxPLs detected in certain pathologies are close to those inducing biological effects in vitro, and are likely to be of biological importance.
Which of multiple species of OxPLs is the most convenient and informative marker of general oxidative stress, PL oxidation, and disease is currently under investigation. Two groups of OxPLs look promising due to their relatively long half-life as compared to other species that are rapidly reduced or cleaved by phospholipases and other enzymes. The measurement of isolevuglandin/protein adducts generated after cleavage of PL–isolevuglandin/protein complexes by PLA2 may be preferred as compared to quantification of isoPs due to slower clearance of protein adducts from the circulation (71). It was suggested that isolevuglandin/protein complexes may serve as integral markers of oxidative stress (266). Significantly elevated levels of isolevuglandin–protein adducts were found under different pathological conditions, including atherosclerosis (392), renal disease (292), Alzheimer disease (387), mouse sepsis model (266), and glaucoma (120). In addition to isolevuglandin–protein adducts, also isofuran–PLs are relatively stable and therefore can be used as conventional biomarkers of lipid oxidation. Elevated levels of PL-isofurans were found in a number of human and experimental pathologies, including hyperoxia-induced lung injury (94), Parkinson disease (93), and a mouse model of Alzheimer disease (314). Although these data characterize isolevuglandins and isofurans as potential biomarkers of disease, clinical relevance of their quantification is insufficiently studied and requires further analysis.
The most convincing data characterizing OxPLs as potential disease biomarkers were obtained in clinical studies using an ELISA test (343) based on detection of OxPCs (but not OxPLs containing other polar head groups) by mAb E06 that specifically recognizes phosphocholine group present in oxidized PCs. The mAb also binds to bacterial products such as capsular polysaccharide of Streptococcus pneumoniae but does not interact with the phosphocholine group present in unoxidized choline-containing PLs (303). E06 recognizes a variety of, but not all, oxidized species of PC, including polymerized species and protein adducts (98), and thus may serve as an integral marker of PC oxidation. Drawbacks of E06 represent the opposite side of its advantages, including complex and still not exhaustively characterized antigen specificity, cross-reactivity with bacterial wall antigens, as well as the lack of reactivity with other classes of OxPLs (98). In spite of these limitations, clinical studies reproducibly showed that E06 reactivity normalized to apoB-100 levels (OxPL/apoB-100 ratio) demonstrates good correlation with the presence, extent, and prognosis of cardiovascular disease. Clinical studies on ∼10,000 individuals showed that OxPL/apoB-100 levels are elevated in patients with coronary, carotid, and femoral atherosclerosis, during acute coronary syndrome and after percutaneous coronary intervention (338, 340, 341). Furthermore, increased levels of OxPL/apoB-100 predict myocardial infarction, stroke, and cardiovascular death (175). Importantly, the prediction of cardiovascular events by OxPL/apoB-100 levels was independent of Framingham atherosclerosis risk factors (175). In addition, OxPL/apoB-100 levels were increased in patients treated with low-fat diet or statins, raising the possibility that OxPL/apoB-100 ratio may serve as a biomarker of efficiency of hypolipidemic therapy and removal of atherogenic OxPLs from the vessel wall (95, 186, 284, 308).
It was found that values of OxPL/apoB-100 demonstrated high and reproducible correlation with the levels of Lp(a) and especially its forms containing the lowest number of kringle IV type 2 repeats (340). This correlation is likely due to the fact that the vast majority of E06-reactive lipids in circulation is bound to the lipid phase and apo(a) component of Lp(a) particles (17) (see Section I.E). Taking into account close correlation between OxPL/apoB-100 and Lp(a) levels, further clinical studies are required in order to establish which additional diagnostic information can be provided by quantification of OxPL/apoB-100 using E06 antibody as compared to existing Lp(a) tests.
B. Experimental therapies involving inactivation of OxPLs
Identification of important pathological activities of OxPLs raises a question whether inactivation or removal of such lipids could be used for therapy. Currently, there are no approaches selectively targeting OxPLs in vivo. However, several experimental therapies in parallel to other beneficial mechanisms also interfere with the generation or action of OxPLs.
1. HDL and apolipoprotein peptide mimetics
HDL and its mimetics are regarded as potential therapeutic agents against atherosclerosis (373). In addition to the reverse cholesterol transport, beneficial effects of HDL particles may be explained by multiple mechanisms including inactivation of OxPLs by HDL-associated PAF-AH and LCAT (121, 369). Apart from enhancing degradation of OxPLs, HDL can antagonize their effects by decreasing sensitivity of cells. Pretreatment of aortic ECs with HDL was shown to inhibit activation of several signaling pathways and reduce induction of chemotactic activity and monocyte binding in response to OxPLs (115). At the same time, HDL did not inhibit induction of antioxidant genes (115). The authors hypothesized that the protective effects of HDL may be partially explained by stabilization of caveoli and inhibition of OxPAPC-induced production of superoxide radicals (115).
Interestingly, the ability of HDL to inhibit formation and biological activity of OxPLs may be compromised as a result of disease. The loss of protective properties of HDL was observed in patients with coronary disease, diabetes, and lepromatous leprosy, inter alia (70, 215, 239). Quantification of the anti-OxPL activity of HDL was suggested as a clinical test for prediction of atherosclerosis (239).
Anti-inflammatory and anti-atherogenic effects of HDL can be mimicked by short peptide fragments of apoA-I or apoJ (238). An advantage of short peptides as compared to HDL or apoA-I to be used as therapeutic agents is that they can be applied per os. Such peptides that were chemically modified in order to increase stability in vivo, were shown to protect vessels in animal models of atherosclerosis, diabetes, and other diseases (236). Moreover, the reversal of atherosclerosis was achieved by combined treatment of apoE knockout mice with apoA-I peptide mimetics and statins (237). Anti-atherosclerotic activity of apoA-I peptide mimetics is based on multiple mechanisms including direct binding of OxPLs. Interaction of most active peptide mimetics with OxPLs was characterized by very high affinity that was several orders of magnitude higher as compared to the interaction with apoA-I (351). This binding is likely to inhibit biological effects of OxPLs as suggested by a correlation between the ability of different apoA-I mimetic peptides to bind OxPLs and inhibit development of atherosclerosis in mice (351).
In summary, inactivation of OxPLs is a recently identified mechanism likely to play a role in the anti-atherogenic and anti-inflammatory activity of HDL independently of reverse cholesterol transport. OxPL-binding peptide mimetics of apoA-I are promising pharmacological agents for prophylactics and treatment of atherosclerosis.
2. CS-1 fibronectin peptide mimetics
An alternative peptide-based approach used synthetic peptide fragments of CS-1 fibronectin (CS-1 FN). CS-1 FN mediates VLA-4-dependent binding of monocytes to OxPL-treated ECs (see Section III.A.1). It was shown that long-term infusion of CS-1 peptide mimetic applied using osmotic pump inhibited monocyte accumulation and fatty streak formation in aortic sinus of LDL receptor knockout mice (305). This study provided a proof-of-principle that peptide mimetics can be used for inhibition of OxPL-induced CS-1 FN-dependent monocytic inflammation.
3. Immunization against OxPLs: Vaccine against atherosclerosis?
Immunization studies of atherosclerosis-prone rabbits and mice with models of autologous OxLDL have demonstrated that OxLDL-specific immune responses are accompanied by atheroprotective effects, as all these interventions led to decreased lesion formation (3, 23, 96, 111, 243, 252, 395). Interestingly, vaccination with adjuvants alone, without antigen, also provides protection against atherosclerosis, which may result from normalization of lipoprotein profile, stimulation of Th2 responses or increased serum titers of antibodies to MDA-LDL (174). However, as discussed below, induction of specific response to OxLDL or its components is much more effective in preventing vessel disease. The exact mechanisms by which immunization with OxLDL is atheroprotective are still unknown, which is in part due to the heterogeneity of the model antigens used, which make the analyses of the induced responses more difficult to interpret. In this regard, we have focused on the role of phosphocholine-specific immune responses, as phosphocholine-specific antibodies constitute a part of anti-OxLDL antibodies.To test this, the molecular mimicry between phosphocholine groups present on the surface of pneumococci and phosphocholine present in OxPC was exploited. Cholesterol-fed ldlr−/− mice were immunized with phosphocholine-containing pneumococcal extracts. This immunization led to a near monoclonal expansion of E06/T15 idiotypic IgM antibodies, but not IgG antibodies to OxLDL or phosphocholine, and significantly decreased lesion formation (24). Thus, expansion of the phosphocholine-specific IgM E06, which specifically binds OxPL, provided atheroprotection. The atheroprotective effect of E06/T1 5 was further corroborated in a study by Faria–Neto et al., in which the authors showed that passive transfusion of purified T15 idiotypic IgM decreased lesion formation in a carotid artery vein graft model in apoe−/− mice (87). There are multiple potential mechanisms by which E06/T15 can mediate atheroprotection (25). E06 IgM has been shown to block the uptake and degradation of OxLDL by macrophages which in vivo would prevent foam cell formation (141). In addition, E06/T15 may neutralize the many proinflammatory effects of OxPLs, as has been shown for the activation of ECs in response to apoptotic cells (56) and blebs (151) carrying OxPLs, as well as the production of IL- 6 by macrophages stimulated with OxPLs (153). Many of these effects may be dependent on the isotype of the induced antibodies, as potential pro-inflammatory effects mediated by binding to Fcγ receptors are absent with IgM Abs. Nevertheless, Caligiuri et al. have followed another strategy to induce phosphocholine-specific antibodies by immunization of ldlr−/− mice with phosphocholine conjugated to KLH (52). In their study the immunization led to development of both IgG and IgM Abs specific for phosphocholine and OxLDL, and mice still developed less atherosclerosis. The contribution of the induced IgG antibodies in this study is unclear, as the IgG subclasses, which have different affinities for Fcγ receptors and different Fcγ-mediated effects, were not differentiated. Humans also have IgM and mostly IgG2 antibodies against phosphocholine, which exhibit cross-reactivity to OxLDL (24, 296). However, their role in cardiovascular disease is unknown. Identification of the protective subset of anti-phosphocholine antibodies and development of strategies to selectively induce them, may enable the future development of vaccines against atherosclerosis.
C. OxPLs as targets for imaging techniques
Advanced atherosclerotic lesions are rich in oxidized lipoproteins, thus suggesting that antibodies specific for oxidized epitopes can be used for detection of plaques in vivo. Indeed, several studies described selective accumulation of radioactively labeled antibodies specific for malondialdehyde-LDL in atherosclerotic lesions of rabbits and mice (304, 342). Recent studies showed that in addition to MDA-LDL-specific antibodies, also E06 mAb, which recognizes a variety of OxPCs, selectively interacted with arteria of hypercholesterolemic apoe−/− mice. Magnetic resonance imaging was performed after injection of antibodies coupled to micelles containing gadolinium (49). The study demonstrated significant and specific enhancement of signal in atherosclerotic vessels of apoe−/− but not wild-type mice, suggesting that OxPL-specific antibodies may be used for noninvasive detection and quantification of atherosclerosis.
V. Open Questions and Perspectives
It is increasingly recognized that OxPLs are markers of disease of the “modified-self” type that are recognized by several PRRs including membrane receptors (e.g., CD36) and soluble proteins such as CRP, CD14, and natural antibodies. OxPLs can be easily distinguished from normal membrane PLs since truncated oxidized residues protrude into the water phase where they have spatial access to PRRs. Since lipid oxidation in vivo is strictly controlled, and on the other hand oxidized molecules are rapidly eliminated, accumulation of OxPLs in cell membranes and lipoproteins (formation of “lipid whiskers” (122)) may be a universal marker of senescence and disease, processes that typically are accompanied by enhanced production of ROS and impaired antioxidant defense. Importantly, recognition of OxPLs by PRRs and other receptors not only targets OxPL-containing structures for elimination, but also modulates cellular functions.
A common theme in this review is that OxPLs often exert functionally different, and even opposite, effects on various steps within one functional pathway. Examples are pro- and anti-inflammatory, pro- and anti-angiogenic, and pro- and antioxidant effects of OxPLs. Apparently, the net outcome depends on the biological context. One explanation for variability of biological effects of OxPLs is activation of several receptor-mediated and nonreceptor signaling mechanisms. In this respect, OxPLs are analogous to the family of prostanoids known to act via multiple plasma membrane and nuclear receptors, and also capable of inducing electrophilic and unfolded protein stress responses.
The data on biological activities of OxPLs were mainly obtained in vitro after treatment of cells with artificially oxidized pure lipids that were added as emulsions. Therefore, physical state of OxPLs was different from that in lipoproteins and cell membranes. However, available evidence suggests that published effects of pure OxPLs adequately mimic the action of their more physiological forms. As mentioned in corresponding sections, the majority of biological effects of OxPLs were reproduced using OxLDL or its more physiological form MM-LDL. Furthermore, apoptotitc blebs and apoptotic cells were shown to induce proinflammatory effects similar with those of pure OxPLs (56, 151). In addition, a close parallel exists between induction of several genes by OxPLs in vitro, and enhanced expression of these genes in atherosclerotic lesions known to contain high levels of OxPLs (Section III.B.1). In summary, accumulating evidence supports the notion that biological activities of OxPLs are not artifacts of in vitro experimentation but correclty reflect their effects in vivo. The influence of the physical state of OxPLs on their biological activity deserves further investigation because it may give a clue to the mechanisms initiating cellular responses to OxPLs.
It has to be established to which extent pleiotropic effects induced by OxPLs depend on their structural variability. The data on the structure–function relationship show that while some activities are specific for molecular species containing certain chemical groups (e.g., activation of PAF receptor by PAF-like OxPCs, induction of electrophilic stress, enhancement versus disruption of endothelial barrier, and activation of prostaglandin receptors), other important effects such as monocyte adhesion and induction of IL-8 in ECs, inhibition of LPS effects or stimulation of unfolded protein response are induced by essentially all studied molecular species of OxPLs. Therefore, further analysis of structure–function relationships in combination with quantification of individual and combined species of OxPLs is required in order to understand the biological role of these lipid mediators.
Significant complexity of signaling mechanisms activated by OxPLs hampers evaluation of their role in vivo. Variable chemical structures of OxPLs, enzymatic and nonenzymatic mechanisms of their generation and inactivation, multiple receptors and nonreceptor mechanisms mediating action of OxPLs together dictate the necessity of using a broad panel of gene knockout and knockdown models, as well as enzyme inhibitors and receptor antagonists. The progress in understanding the role of OxPLs in vivo may be promoted by the success of novel approaches to modulation of effects of OxPL (see Section IV.B), which potentially may evolve into the methods of experimental therapy. In addition, potential therapeutic applications of OxPLs such as inhibition of TLR2 and 4 and protection of lung vessel endothelial barrier deserve further validation. Since effects of the same OxPL may be pathologic in one setting (e.g., induction of proinflammatory changes in ECs) and beneficial in another setting (protection from lung edema during acute aceptic inflammation), possible use of OxPLs must be considered in relation to particular pathology. Last but not least, OxPLs are increasingly recognized as promising diagnostic markers.
In summary, OxPLs represent an emerging family of lipid mediators. Analysis of gene expression in OxPL-treated cells identified very high number of regulated genes (107, 108), suggesting that our current knowledge on OxPLs is just the tip of the iceberg, and that future studies will discover new biological activities of OxPLs and improve our understanding of the role of OxPLs in health and disease.
Abbreviations Used
- AJ
adherens junction
- ALl
acute lung injury
- aPLs
antiphospholipid antibodies
- APS
antiphospholipid syndrome
- ARE
antioxidant response element
- ATF
activating transcription factor
- CL
cardiolipin
- COX
cyclooxygenase
- CRP
C-reactive protein
- cyt c
cytochrome c
- DC
dendritic cell
- EC
endothelial cell
- ESR
electrophilic stress response
- FA
focal adhesion
- GCLC
catalytic subunit of glutamate-cysteine ligase
- GCLM
modifier subunit of glutamate-cysteine ligase
- GPx4
phospholipid glutathione peroxidase
- HAEC
human aortic endothelial cell
- HDL
high-density lipoprotein
- HIF
hypoxia-inducible factor
- HMEC
human microvascular endothelial cell
- Ho1
heme oxygenase-1
- IL-8
interleukin 8
- isoP
isoprostane
- LBP
LPS-binding protein
- LDL
low-density lipoprotein
- LOX
lypoxygenase
- Lp-PLA2
lipoprotein-associated PLA2
- LPS
lipopolysaccharide (endotoxin)
- mCD14
membrane-bound CD14
- MDA
malondialdehyde
- MKP-1
mitogen-activated protein kinase phosphatase 1
- MM-LDL
minimally modified LDL
- OxCL
oxidized cardiolipin
- OxLDL
oxidized LDL
- OxPAPA
oxidized 1-palm itoyl-2-arachidonoyl-sn-glycero-3-phosphate (phosphatidic acid)
- OxPAPC
oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- OxPAPE
oxidized 1-palm itoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine
- OxPAPS
oxidized 1-palm itoyl-2-arachidonoyl-sn-glycero-3-phosphoserine
- OxPE
oxidized phosphatidylethanolamine
- OxPL
oxidized phospholipid
- PAF
platelet-activating factor
- PAF-AH
PAF-acetylhydrolase
- PAMP
pathogen-associated molecular pattern
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PECPC
1-palm itoyl-2-(epoxycyclopentenone isoprostane)-sn-glycero-3-phosphocholine
- PEIPC
1-palm itoyl-2- (5,6-epoxy isoprostane E2)-sn-glycero-3-phosphochol ine (isoprostane-PC)
- PG
prostaglandin
- PGPC
1-palmitoyl-2-gl utaroyl-sn-glycero-3-phosphocholine
- PLPC
1-palmitoyl-2-l inoleoyl-sn-glycero-3-phosphocholine
- POVPC
1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine
- PPAR
peroxisome proliferator-activated receptor
- PS
phosphatidylserine
- ROS
reactive oxygen species
- sCD14
soluble CD14
- SP1
sphingosine-1-phosphate
- TLR
Toll-like receptor
- UPR
unfolded protein response
- VEGF
vascular endothelial growth factor
- VILI
ventilation-induced lung injury
- VSMCs
vascular smooth muscle cells
Footnotes
Reviewing Editors: Judith Berliner, Denis Blache, Clett Erridge, Valerian Kagan, Vishwanathan Natarajan, Sampath Parthasarathy, Yulia Tyurina, and Fulvio Ursini
Acknowledgments
This work was supported by grants from Fonds zur Förderung wissenschaftlicher Forschung (FWF, projects P20801-B11 and P18232-B11 to VNB) and Österreichischer Forschungsförderungsgesellschaft (project 815445 to VNB).
References
- 1.Ago T. Kitazono T. Ooboshi H. Iyama T. Han YH. Takada J. Wakisaka M. Ibayashi S. Utsumi H. Iida M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004;109:227–233. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]
- 2.Alcaraz MJ. Fernandez P. Guillen MI. Anti-inflammatory actions of the heme oxygenase-1 pathway. Curr Pharm Des. 2003;9:2541–2551. doi: 10.2174/1381612033453749. [DOI] [PubMed] [Google Scholar]
- 3.Ameli S. Hultgardh-Nilsson A. Regnstrom J. Calara F. Yano J. Cercek B. Shah PK. Nilsson J. Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arterioscler Thromb Vasc Biol. 1996;16:1074–1079. doi: 10.1161/01.atv.16.8.1074. [DOI] [PubMed] [Google Scholar]
- 4.Androulakis N. Durand H. Ninio E. Tsoukatos DC. Molecular and mechanistic characterization of platelet-activating factor-like bioactivity produced upon LDL oxidation. J Lipid Res. 2005;46:1923–1932. doi: 10.1194/jlr.M500074-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Anton R. Camacho M. Puig L. Vila L. Hepoxilin B3 and its enzymatically formed derivative trioxilin B3 are incorporated into phospholipids in psoriatic lesions. J Invest Dermatol. 2002;118:139–146. doi: 10.1046/j.0022-202x.2001.01593.x. [DOI] [PubMed] [Google Scholar]
- 6.Arai M. Imai H. Metori A. Nakagawa Y. Preferential esterification of endogenously formed 5-hydroxyeicosatetraenoic acid to phospholipids in activated polymorphonuclear leukocytes. Eur J Biochem. 1997;244:513–519. doi: 10.1111/j.1432-1033.1997.00513.x. [DOI] [PubMed] [Google Scholar]
- 7.Arthur WT. Quilliam LA. Cooper JA. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J Cell Biol. 2004;167:111–122. doi: 10.1083/jcb.200404068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arumugam TV. Okun E. Tang SC. Thundyil J. Taylor SM. Woodruff TM. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32:4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 9.Asai A. Okajima F. Nakagawa K. Ibusuki D. Tanimura K. Nakajima Y. Nagao M. Sudo M. Harada T. Miyazawa T. Oikawa S. Phosphatidylcholine hydroperoxide-induced THP-1 cell adhesion to intracellular adhesion molecule-1. J Lipid Res. 2009;50:9557–965. doi: 10.1194/jlr.M800582-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashraf MZ. Kar NS. Chen X. Choi J. Salomon RG. Febbraio M. Podrez EA. Specific oxidized phospholipids inhibit scavenger receptor bi-mediated selective uptake of cholesteryl esters. J Biol Chem. 2008;283:10408–10414. doi: 10.1074/jbc.M710474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashraf MZ. Kar NS. Podrez EA. Oxidized phospholipids: Biomarker for cardiovascular diseases. Int J Biochem Cell Biol. 2009;41:1241–1244. doi: 10.1016/j.biocel.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baier J. Maisch T. Maier M. Landthaler M. Baumler W. Direct detection of singlet oxygen generated by UVA irradiation in human cells and skin. J Invest Dermatol. 2007;127:1498–1506. doi: 10.1038/sj.jid.5700741. [DOI] [PubMed] [Google Scholar]
- 13.Baker PR. Schopfer FJ. Sweeney S. Freeman BA. Red cell membrane and plasma linoleic acid nitration products: Synthesis, clinical identification, and quantitation. Proc Natl Acad Sci USA. 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barleon B. Sozzani S. Zhou D. Weich HA. Mantovani A. Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 15.Bayir H. Tyurin VA. Tyurina YY. Viner R. Ritov V. Amoscato AA. Zhao Q. Zhang XJ. Janesko-Feldman KL. Alexander H. Basova LV. Clark RS. Kochanek PM. Kagan VE. Selective early cardiolipin peroxidation after traumatic brain injury: An oxidative lipidomics analysis. Ann Neurol. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- 16.Belkner J. Wiesner R. Rathman J. Barnett J. Sigal E. Kuhn H. Oxygenation of lipoproteins by mammalian lipoxygenases. Eur J Biochem. 1993;213:251–261. doi: 10.1111/j.1432-1033.1993.tb17755.x. [DOI] [PubMed] [Google Scholar]
- 17.Bergmark C. Dewan A. Orsoni A. Merki E. Miller ER. Shin MJ. Binder CJ. Horkko S. Krauss RM. Chapman MJ. Witztum JL. Tsimikas S. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–2239. doi: 10.1194/jlr.M800174-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Berliner JA. Gharavi NM. Endothelial cell regulation by phospholipid oxidation products. Free Radic Biol Med. 2008;45:119–123. doi: 10.1016/j.freeradbiomed.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berliner JA. Leitinger N. Tsimikas S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res. 2009;50:S207–S212. doi: 10.1194/jlr.R800074-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berliner JA. Subbanagounder G. Leitinger N. Watson AD. Vora D. Evidence for a role of phospholipid oxidation products in atherogenesis. Trends Cardiovasc Med. 2001;11:142–147. doi: 10.1016/s1050-1738(01)00098-6. [DOI] [PubMed] [Google Scholar]
- 21.Berliner JA. Territo MC. Sevanian A. Ramin S. Kim JA. Bamshad B. Esterson M. Fogelman AM. Mini-mally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. 1990;85:1260–1266. doi: 10.1172/JCI114562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Binder CJ. Chang MK. Shaw PX. Miller YI. Hartvigsen K. Dewan A. Witztum JL. Innate and acquired immunity in atherogenesis. Nat Med. 2002;8:1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- 23.Binder CJ. Hartvigsen K. Chang MK. Miller M. Broide D. Palinski W. Curtiss LK. Corr M. Witztum JL. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. 2004;114:427–437. doi: 10.1172/JCI20479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binder CJ. Horkko S. Dewan A. Chang MK. Kieu EP. Goodyear CS. Shaw PX. Palinski W. Witztum JL. Silverman GJ. Pneumococcal vaccination decreases atherosclerotic lesion formation: Molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. 2003;9:736–743. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]
- 25.Binder CJ. Shaw PX. Chang MK. Boullier A. Hartvigsen K. Horkko S. Miller YI. Woelkers DA. Corr M. Witztum JL. The role of natural antibodies in atherogenesis. J Lipid Res. 2005;46:1353–1363. doi: 10.1194/jlr.R500005-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Bird DA. Gillotte KL. Horkko S. Friedman P. Dennis EA. Witztum JL. Steinberg D. Receptors for oxidized low-density lipoprotein on elicited mouse peritoneal macrophages can recognize both the modified lipid moieties and the modified protein moieties: Implications with respect to macrophage recognition of apoptotic cells. Proc Natl Acad Sci USA. 1999;96:6347–6352. doi: 10.1073/pnas.96.11.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birkle DL. Bazan NG. Effect of K+ depolarization on the synthesis of prostaglandins and hydroxyeicosatetra-(5,8,11,14)enoic acids (HETE) in the rat retina. Evidence for esterification of 1 2-HETE in lipids. Biochim Biophys Acta. 1984;795:564–573. doi: 10.1016/0005-2760(84)90187-5. [DOI] [PubMed] [Google Scholar]
- 28.Birukov KG. Bochkov VN. Birukova AA. Kawkitinarong K. Rios A. Leitner A. Verin AD. Bokoch GM. Leitinger N. Garcia JG. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ Res. 2004;95:892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 29.Birukov KG. Leitinger N. Bochkov VN. Garcia JG. Signal transduction pathways activated in human pulmonary endothelial cells by OxPAPC, a bioactive component of oxidized lipoproteins. Micro vasc Res. 2004;67:18–28. doi: 10.1016/j.mvr.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Birukova AA. Chatchavalvanich S. Oskolkova O. Bochkov VN. Birukov KG. Signaling pathways involved in OxPAPC-induced pulmonary endothelial barrier protection. Micro vasc Res. 2007;73:173–181. doi: 10.1016/j.mvr.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Birukova AA. Fu P. Chatchavalvanich S. Burdette D. Oskolkova O. Bochkov VN. Birukov KG. Polar head groups are important for barrier-protective effects of oxidized phospholipids on pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2007;292:L924–L935. doi: 10.1152/ajplung.00395.2006. [DOI] [PubMed] [Google Scholar]
- 32.Birukova AA. Malyukova I. Mikaelyan A. Fu P. Birukov KG. Tiam1 and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J Cell Physiol. 2007;211:608–617. doi: 10.1002/jcp.20966. [DOI] [PubMed] [Google Scholar]
- 33.Birukova AA. Malyukova I. Poroyko V. Birukov KG. Paxillin-beta-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2007;293:L199–L211. doi: 10.1152/ajplung.00020.2007. [DOI] [PubMed] [Google Scholar]
- 34.Birukova AA. Zagranichnaya T. Fu P. Alekseeva E. Chen W. Jacobson JR. Birukov KG. Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res. 2007;313:2504–2520. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bishop AL. Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–255. [PMC free article] [PubMed] [Google Scholar]
- 36.Bluml S. Kirchberger S. Bochkov VN. Kronke G. Stuhlmeier K. Majdic O. Zlabinger GJ. Knapp W. Binder BR. Stockl J. Leitinger N. Oxidized phospholipids negatively regulate dendritic cell maturation induced by TLRs and CD40. J Immunol. 2005;175:501–508. doi: 10.4049/jimmunol.175.1.501. [DOI] [PubMed] [Google Scholar]
- 37.Bluml S. Rosc B. Lorincz A. Seyerl M. Kirchberger S. Oskolkova O. Bochkov VN. Majdic O. Ligeti E. Stockl J. The oxidation state of phospholipids controls the oxidative burst in neutrophil granulocytes. J Immunol. 2008;181:4347–4353. doi: 10.4049/jimmunol.181.6.4347. [DOI] [PubMed] [Google Scholar]
- 38.Bochkov VN. Kadl A. Huber J. Gruber F. Binder BR. Leitinger N. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419:77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- 39.Bochkov VN. Mechtcheriakova D. Lucerna M. Huber J. Malli R. Graier WF. Hofer E. Binder BR. Leitinger N. Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of ERK/EGR-1 and Ca(++)/NFAT. Blood. 2002;99:199–206. doi: 10.1182/blood.v99.1.199. [DOI] [PubMed] [Google Scholar]
- 40.Bochkov VN. Philippova M. Oskolkova O. Kadl A. Furnkranz A. Karabeg E. Afonyushkin T. Gruber F. Breuss J. Minchenko A. Mechtcheriakova D. Hohensinner P. Rychli K. Wojta J. Resink T. Erne P. Binder BR. Leitinger N. Oxidized phospholipids stimulate angiogenesis via autocrine mechanisms, implicating a novel role for lipid oxidation in the evolution of atherosclerotic lesions. Circ Res. 2006;99:900–908. doi: 10.1161/01.RES.0000245485.04489.ee. [DOI] [PubMed] [Google Scholar]
- 41.Boguski MS. McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 42.Bonser RW. Siegel MI. Chung SM. McConnell RT. Cuatrecasas P. Esterification of an endogenously synthesized lipoxygenase product into granulocyte cellular lipids. Biochemistry. 1981;20:5297–5301. doi: 10.1021/bi00521a032. [DOI] [PubMed] [Google Scholar]
- 43.Borisy GG. Svitkina TM. Actin machinery: pushing the envelope. Curr Opin Cell Biol. 2000;12:104–112. doi: 10.1016/s0955-0674(99)00063-0. [DOI] [PubMed] [Google Scholar]
- 44.Borst JW. Visser NV. Kouptsova O. Visser AJ. Oxidation of unsaturated phospholipids in membrane bilayer mixtures is accompanied by membrane fluidity changes. Biochim Biophys Acta. 2000;1487:61–73. doi: 10.1016/s1388-1981(00)00084-6. [DOI] [PubMed] [Google Scholar]
- 45.Boulanger CM. Amabile N. Tedgui A. Circulating microparticles: A potential prognostic marker for atherosclerotic vascular disease. Hypertension. 2006;48:180–186. doi: 10.1161/01.HYP.0000231507.00962.b5. [DOI] [PubMed] [Google Scholar]
- 46.Boullier A. Friedman P. Harkewicz R. Hartvigsen K. Green SR. Almazan F. Dennis EA. Steinberg D. Witztum JL. Quehenberger O. Phosphocholine as a pattern recognition ligand for CD36. J Lipid Res. 2005;46:969–976. doi: 10.1194/jlr.M400496-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Brame CJ. Boutaud O. Davies SS. Yang T. Oates JA. Roden D. Roberts LJ. Modification of proteins by isoketal-containing oxidized phospholipids. J Biol Chem. 2004;279:13447–13451. doi: 10.1074/jbc.M313349200. [DOI] [PubMed] [Google Scholar]
- 48.Briles DE. Forman C. Hudak S. Claflin JL. Anti-phosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae. J Exp Med. 1982;156:1177–1185. doi: 10.1084/jem.156.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briley-Saebo KC. Shaw PX. Mulder WJ. Choi SH. Vucic E. Aguinaldo JG. Witztum JL. Fuster V. Tsimikas S. Fayad ZA. Targeted molecular probes for imaging atherosclerotic lesions with magnetic resonance using antibodies that recognize oxidation-specific epitopes. Circulation. 2008;117:3206–3215. doi: 10.1161/CIRCULATIONAHA.107.757120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brooks JD. Milne GL. Yin H. Sanchez SC. Porter NA. Morrow JD. Formation of highly reactive cyclopentenone isoprostane compounds (A3/J3-isoprostanes) in vivo from eicosapentaenoic acid. J Biol Chem. 2008;283:12043–12055. doi: 10.1074/jbc.M800122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Byun J. Mueller DM. Fabjan JS. Heinecke JW. Nitrogen dioxide radical generated by the myeloperoxidase-hydrogen peroxide-nitrite system promotes lipid peroxidation of low density lipoprotein. FEBS Lett. 1999;455:243–246. doi: 10.1016/s0014-5793(99)00893-5. [DOI] [PubMed] [Google Scholar]
- 52.Caligiuri G. Khallou-Laschet J. Vandaele M. Gaston AT. Delignat S. Mandet C. Kohler HV. Kaveri SV. Nicoletti A. Phosphorylcholine-targeting immunization reduces atherosclerosis. J Am Coll Cardiol. 2007;50:540–546. doi: 10.1016/j.jacc.2006.11.054. [DOI] [PubMed] [Google Scholar]
- 53.Carr AC. Winterbourn CC. van den Berg JJ. Peroxidase-mediated bromination of unsaturated fatty acids to form bromohydrins. Arch Biochem Biophys. 1996;327:227–233. doi: 10.1006/abbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 54.Chahdi A. Miller B. Sorokin A. Endothelin 1 induces beta 1 Pix translocation and Cdc42 activation via protein kinase A-dependent pathway. J Biol Chem. 2005;280:578–584. doi: 10.1074/jbc.M411130200. [DOI] [PubMed] [Google Scholar]
- 55.Chang MK. Bergmark C. Laurila A. Horkko S. Han KH. Friedman P. Dennis EA. Witztum JL. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: Evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci USA. 1999;96:6353–6358. doi: 10.1073/pnas.96.11.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang MK. Binder CJ. Miller YI. Subbanagounder G. Silverman GJ. Berliner JA. Witztum JL. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. 2004;200:1359–1370. doi: 10.1084/jem.20031763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang MK. Binder CJ. Torzewski M. Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci USA. 2002;99:13043–13048. doi: 10.1073/pnas.192399699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang PY. Luo S. Jiang T. Lee YT. Lu SC. Henry PD. Chen CH. Oxidized low-density lipoprotein downregulates endothelial basic fibroblast growth factor through a pertussis toxin-sensitive G-protein pathway: Mediator role of platelet-activating factor-like phospholipids. Circulation. 2001;104:588–593. doi: 10.1161/hc3101.092213. [DOI] [PubMed] [Google Scholar]
- 59.Chen H. Bernstein BW. Bamburg JR. Regulating actin-filament dynamics in vivo. Trends Biochem Sci. 2000;25:19–23. doi: 10.1016/s0968-0004(99)01511-x. [DOI] [PubMed] [Google Scholar]
- 60.Chen K. Febbraio M. Li W. Silverstein RL. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res. 2008;102:1512–1519. doi: 10.1161/CIRCRESAHA.108.172064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen R. Yang L. McIntyre TM. Cytotoxic phospholipid oxidation products. Cell death from mitochondrial damage and the intrinsic caspase cascade. J Biol Chem. 2007;282:24842–24850. doi: 10.1074/jbc.M702865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen Y. Morrow JD. Roberts LJ. Formation of reactive cyclopentenone compounds in vivo as products of the isoprostane pathway. J Biol Chem. 1999;274:10863–10868. doi: 10.1074/jbc.274.16.10863. [DOI] [PubMed] [Google Scholar]
- 63.Chen Y. Wang Y. Yu H. Wang F. Xu W. The cross talk between protein kinase A- and RhoA-mediated signaling in cancer cells. Exp Biol Med (Maywood ) 2005;230:731–741. doi: 10.1177/153537020523001006. [DOI] [PubMed] [Google Scholar]
- 64.Cheng C. Noordeloos AM. Jeney V. Soares MP. Moll F. Pasterkamp G. Serruys PW. Duckers HJ. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation. 2009;119:3017–3027. doi: 10.1161/CIRCULATIONAHA.108.808618. [DOI] [PubMed] [Google Scholar]
- 65.Cherepanova OA. Pidkovka NA. Sarmento OF. Yoshida T. Gan Q. Adiguzel E. Bendeck MP. Berliner J. Leitinger N. Owens GK. Oxidized phospholipids induce type VIII collagen expression and vascular smooth muscle cell migration. Circ Res. 2009;104:609–618. doi: 10.1161/CIRCRESAHA.108.186064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chisolm GM. Steinberg D. The oxidative modification hypothesis of atherogenesis: An overview. Free Radic Biol Med. 2000;28:1815–1826. doi: 10.1016/s0891-5849(00)00344-0. [DOI] [PubMed] [Google Scholar]
- 67.Cole AL. Subbanagounder G. Mukhopadhyay S. Berliner JA. Vora DK. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMPdependent R-Ras/PI3-kinase pathway. Arterioscler Thromb Vasc Biol. 2003;23:1384–1390. doi: 10.1161/01.ATV.0000081215.45714.71. [DOI] [PubMed] [Google Scholar]
- 68.Conrad M. Schneider M. Seiler A. Bornkamm GW. Physiological role of phospholipid hydroperoxide glutathione peroxidase in mammals. Biol Chem. 2007;388:1019–1025. doi: 10.1515/BC.2007.130. [DOI] [PubMed] [Google Scholar]
- 69.Costello PB. Baer AN. Green FA. Saturability of esterification pathways of major monohydroxyeicosatetraenoic acids in rat basophilic leukemia cells. Inflammation. 1991;15:269–279. doi: 10.1007/BF00917312. [DOI] [PubMed] [Google Scholar]
- 70.Cruz D. Watson AD. Miller CS. Montoya D. Ochoa MT. Sieling PA. Gutierrez MA. Navab M. Reddy ST. Witztum JL. Fogelman AM. Rea TH. Eisenberg D. Berliner J. Modlin RL. Host-derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J Clin Invest. 2008;118:2917–2928. doi: 10.1172/JCI34189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davies SS. Amarnath V. Brame CJ. Boutaud O. Roberts LJ. Measurement of chronic oxidative and inflammatory stress by quantification of isoketal/levuglandin gamma-ketoaldehyde protein adducts using liquid chromatography tandem mass spectrometry. Nat Protoc. 2007;2:2079–2091. doi: 10.1038/nprot.2007.298. [DOI] [PubMed] [Google Scholar]
- 72.Davies SS. Pontsler AV. Marathe GK. Harrison KA. Murphy RC. Hinshaw JC. Prestwich GD. Hilaire AS. Prescott SM. Zimmerman GA. McIntyre TM. Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor gamma ligands and agonists. J Biol Chem. 2001;276:16015–16023. doi: 10.1074/jbc.M100878200. [DOI] [PubMed] [Google Scholar]
- 73.Davis B. Koster G. Douet LJ. Scigelova M. Woffendin G. Ward JM. Smith A. Humphries J. Burnand KG. Macphee CH. Postle AD. Electrospray ionization mass spectrometry identifies substrates and products of lipoprotein-associated phospholipase A2 in oxidized human low density lipoprotein. J Biol Chem. 2008;283:6428–6437. doi: 10.1074/jbc.M709970200. [DOI] [PubMed] [Google Scholar]
- 74.Deguchi H. Fernandez JA. Hackeng TM. Banka CL. Griffin JH. Cardiolipin is a normal component of human plasma lipoproteins. Proc Natl Acad Sci USA. 2000;97:1743–1748. doi: 10.1073/pnas.97.4.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deigner HP. Hermetter A. Oxidized phospholipids: Emerging lipid mediators in pathophysiology. Curr Opin Lipidol. 2008;19:289–294. doi: 10.1097/MOL.0b013e3282fe1d0e. [DOI] [PubMed] [Google Scholar]
- 76.Dejana E. Endothelial cell-cell junctions: Happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 77.Delerive P. Furman C. Teissier E. Fruchart J. Duriez P. Staels B. Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner. FEBS Lett. 2000;471:34–38. doi: 10.1016/s0014-5793(00)01364-8. [DOI] [PubMed] [Google Scholar]
- 78.DeMaio L. Rouhanizadeh M. Reddy S. Sevanian A. Hwang J. Hsiai TK. Oxidized phospholipids mediate occludin expression and phosphorylation in vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2006;290:H674–H683. doi: 10.1152/ajpheart.00554.2005. [DOI] [PubMed] [Google Scholar]
- 79.Dever GJ. Benson R. Wainwright CL. Kennedy S. Spickett CM. Phospholipid chlorohydrin induces leukocyte adhesion to ApoE−/− mouse arteries via upregulation of P-selectin. Free Radic Biol Med. 2008;44:452–463. doi: 10.1016/j.freeradbiomed.2007.10.038. [DOI] [PubMed] [Google Scholar]
- 80.Dinkova-Kostova AT. Holtzclaw WD. Cole RN. Itoh K. Wakabayashi N. Katoh Y. Yamamoto M. Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Domingues MR. Reis A. Domingues P. Mass spectrometry analysis of oxidized phospholipids. Chem Phys Lipids. 2008;156:1–12. doi: 10.1016/j.chemphyslip.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 82.Edelstein C. Pfaffinger D. Hinman J. Miller E. Lipkind G. Tsimikas S. Bergmark C. Getz GS. Witztum JL. Scanu AM. Lysine-phosphatidylcholine adducts in kringle V impart unique immunological and potential pro-inflammatory properties to human apolipoprotein(a) J Biol Chem. 2003;278:52841–52847. doi: 10.1074/jbc.M310425200. [DOI] [PubMed] [Google Scholar]
- 83.Erridge C. Kennedy S. Spickett CM. Webb DJ. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: Roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J Biol Chem. 2008;283:24748–24759. doi: 10.1074/jbc.M800352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Esterbauer H. Schaur RJ. Zollner H. Chemistry and biochemistry of 4- hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 85.Etienne-Manneville S. Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 86.Fang X. Kaduce TL. Weintraub NL. Spector AA. Cytochrome P450 metabolites of arachidonic acid: Rapid incorporation and hydration of 14,15-epoxyeicosatrienoic acid in arterial smooth muscle cells. Prostaglandins Leukot Essent Fatty Acids. 1997;57:367–371. doi: 10.1016/s0952-3278(97)90412-9. [DOI] [PubMed] [Google Scholar]
- 87.Faria-Neto JR. Chyu KY. Li X. Dimayuga PC. Ferreira C. Yano J. Cercek Shah PK. Passive immunization with monoclonal IgM antibodies against phosphorylcholine reduces accelerated vein graft atherosclerosis in apolipoprotein E-null mice. Atherosclerosis. 2006;189:83–90. doi: 10.1016/j.atherosclerosis.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 88.Fathman CG. Lineberry NB. Molecular mechanisms of CD4+ T-cell anergy. Nat Rev Immunol. 2007;7:599–609. doi: 10.1038/nri2131. [DOI] [PubMed] [Google Scholar]
- 89.Febbraio M. Hajjar DP. Silverstein RL. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Febbraio M. Podrez EA. Smith JD. Hajjar DP. Hazen SL. Hoff HF. Sharma K. Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fei H. Berliner JA. Parhami F. Drake TA. Regulation of endothelial cell tissue factor expression by minimally oxidized LDL and lipopolysaccharide. Arterioscler Thromb. 1993;13:1711–1717. doi: 10.1161/01.atv.13.11.1711. [DOI] [PubMed] [Google Scholar]
- 92.Feng B. Yao PM. Li Y. Devlin CM. Zhang D. Harding HP. Sweeney M. Rong JX. Kuriakose G. Fisher EA. Marks AR. Ron D. Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 93.Fessel JP. Hulette C. Powell S. Roberts LJ. Zhang J. Isofurans, but not F2-isoprostanes, are increased in the substantia nigra of patients with Parkinson's disease and with dementia with Lewy body disease. J Neurochem. 2003;85:645–650. doi: 10.1046/j.1471-4159.2003.01709.x. [DOI] [PubMed] [Google Scholar]
- 94.Fessel JP. Porter NA. Moore KP. Sheller JR. Roberts LJ. Discovery of lipid peroxidation products formed in vivo with a substituted tetrahydrofuran ring (isofurans) that are favored by increased oxygen tension. Proc Natl Acad Sci USA. 2002;99:16713–16718. doi: 10.1073/pnas.252649099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fraley AE. Schwartz GG. Olsson AG. Kinlay S. Szarek M. Rifai N. Libby P. Ganz P. Witztum JL. Tsimikas S. Relationship of oxidized phospholipids and biomarkers of oxidized low-density lipoprotein with cardiovascular risk factors, inflammatory biomarkers, and effect of statin therapy in patients with acute coronary syndromes: Results from the MIRACL (Myocardial Ischemia Reduction With Aggressive Cholesterol Lowering) trial. J Am Coll Cardiol. 2009;53:2186–2196. doi: 10.1016/j.jacc.2009.02.041. [DOI] [PubMed] [Google Scholar]
- 96.Freigang S. Horkko S. Miller E. Witztum JL. Palinski W. Immunization of LDL receptor-deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arterioscler Thromb Vasc Biol. 1998;18:1972–1982. doi: 10.1161/01.atv.18.12.1972. [DOI] [PubMed] [Google Scholar]
- 97.Frey B. Haupt R. Alms S. Holzmann G. Konig T. Kern H. Kox W. Rustow B. Schlame M. Increase in fragmented phosphatidylcholine in blood plasma by oxidative stress. J Lipid Res. 2000;41:1145–1153. [PubMed] [Google Scholar]
- 98.Friedman P. Horkko S. Steinberg D. Witztum JL. Dennis EA. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. Importance of Schiff base formation and aldol condensation. J Biol Chem. 2002;277:7010–7020. doi: 10.1074/jbc.M108860200. [DOI] [PubMed] [Google Scholar]
- 99.Frostegard J. Svenungsson E. Wu R. Gunnarsson I. Lundberg IE. Klareskog L. Horkko S. Witztum JL. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005;52:192–200. doi: 10.1002/art.20780. [DOI] [PubMed] [Google Scholar]
- 100.This reference has been deleted
- 101.Fukai M. Hayashi T. Yokota R. Shimamura T. Suzuki T. Taniguchi M. Matsushita M. Furukawa H. Todo S. Lipid peroxidation during ischemia depends on ischemia time in warm ischemia and reperfusion of rat liver. Free Radic Biol Med. 2005;38:1372–1381. doi: 10.1016/j.freeradbiomed.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 102.Furnkranz A. Schober A. Bochkov VN. Bashtrykov P. Kronke G. Kadl A. Binder BR. Weber C. Leitinger N. Oxidized phospholipids trigger atherogenic inflammation in murine arteries. Arterioscler Thromb Vasc Biol. 2005;25:633–638. doi: 10.1161/01.ATV.0000153106.03644.a0. [DOI] [PubMed] [Google Scholar]
- 103.Gallon AA. Pryor WA. The reaction of low levels of nitrogen dioxide with methyl linoleate in the presence and absence of oxygen. Lipids. 1994;29:171–176. doi: 10.1007/BF02536725. [DOI] [PubMed] [Google Scholar]
- 104.Gao L. Zackert WE. Hasford JJ. Danekis ME. Milne GL. Remmert C. Reese J. Yin H. Tai HH. Dey SK. Porter NA. Morrow JD. Formation of prostaglandins E2 and D2 via the isoprostane pathway: A mechanism for the generation of bioactive prostaglandins independent of cyclooxygenase. J Biol Chem. 2003;278:28479–28489. doi: 10.1074/jbc.M303984200. [DOI] [PubMed] [Google Scholar]
- 105.Gao S. Zhang R. Greenberg ME. Sun M. Chen X. Levison BS. Salomon RG. Hazen SL. Phospholipid hydroxyalkenals, a subset of recently discovered endogenous CD36 ligands, spontaneously generate novel furan-containing phospholipids lacking CD36 binding activity in vivo. J Biol Chem. 2006;281:31298–31308. doi: 10.1074/jbc.M604039200. [DOI] [PubMed] [Google Scholar]
- 106.Garcia JG. Liu F. Verin AD. Birukova A. Dechert MA. Gerthoffer WT. Bamberg JR. English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gargalovic PS. Gharavi NM. Clark MJ. Pagnon J. Yang WP. He A. Truong A. Baruch-Oren T. Berliner JA. Kirchgessner TG. Lusis AJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 108.Gargalovic PS. Imura M. Zhang B. Gharavi NM. Clark MJ. Pagnon J. Yang WP. He A. Truong A. Patel S. Nelson SF. Horvath S. Berliner JA. Kirchgessner TG. Lusis AJ. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA. 2006;103:12741–12746. doi: 10.1073/pnas.0605457103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gasser H. Hallstrom S. Redl H. Schlag G. Oxidatively modified plasma phospholipids containing reactive carbonyl functions measured by HPLC: Evidence for phosphatidylcholine-bound aldehydes in plasma of burn patients. Free Radic Res. 1995;22:327–336. doi: 10.3109/10715769509145645. [DOI] [PubMed] [Google Scholar]
- 110.Geiger B. Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol. 2001;13:584–592. doi: 10.1016/s0955-0674(00)00255-6. [DOI] [PubMed] [Google Scholar]
- 111.George J. Afek A. Gilburd B. Levkovitz H. Shaish A. Goldberg I. Kopolovic Y. Wick G. Shoenfeld Y. Harats D. Hyperimmunization of apo-E-deficient mice with homologous malondialdehyde low-density lipoprotein suppresses early atherogenesis. Atherosclerosis. 1998;138:147–152. doi: 10.1016/s0021-9150(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 112.Gershov D. Kim S. Brot N. Elkon KB. C-reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: Implications for systemic autoimmunity. J Exp Med. 2000;192:1353–1364. doi: 10.1084/jem.192.9.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gharavi NM. Alva JA. Mouillesseaux KP. Lai C. Yeh M. Yeung W. Johnson J. Szeto WL. Hong L. Fishbein M. Wei L. Pfeffer LM. Berliner JA. Role of the Jak/STAT pathway in the regulation of interleukin-8 transcription by oxidized phospholipids in vitro and in atherosclerosis in vivo. J Biol Chem. 2007;282:31460–31468. doi: 10.1074/jbc.M704267200. [DOI] [PubMed] [Google Scholar]
- 114.Gharavi NM. Baker NA. Mouillesseaux KP. Yeung W. Honda HM. Hsieh X. Yeh M. Smart EJ. Berliner JA. Role of endothelial nitric oxide synthase in the regulation of SREBP activation by oxidized phospholipids. Circ Res. 2006;98:768–776. doi: 10.1161/01.RES.0000215343.89308.93. [DOI] [PubMed] [Google Scholar]
- 115.Gharavi NM. Gargalovic PS. Chang I. Araujo JA. Clark MJ. Szeto WL. Watson AD. Lusis AJ. Berliner JA. High-density lipoprotein modulates oxidized phospholipid signaling in human endothelial cells from proinflammatory to anti-inflammatory. Arterioscler Thromb Vasc Biol. 2007;27:1346–1353. doi: 10.1161/ATVBAHA.107.141283. [DOI] [PubMed] [Google Scholar]
- 116.Gillotte-Taylor K. Boullier A. Witztum JL. Steinberg D. Quehenberger O. Scavenger receptor class B type I as a receptor for oxidized low density lipoprotein. J Lipid Res. 2001;42:1474–1482. [PubMed] [Google Scholar]
- 117.Girotti AW. Kriska T. Role of lipid hydroperoxides in photo-oxidative stress signaling. Antioxid Redox Signal. 2004;6:301–310. doi: 10.1089/152308604322899369. [DOI] [PubMed] [Google Scholar]
- 118.Gniwotta C. Morrow JD. Roberts LJ. Kuhn H. Prostaglandin F2-like compounds, F2-isoprostanes, are present in increased amounts in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1997;17:3236–3241. doi: 10.1161/01.atv.17.11.3236. [DOI] [PubMed] [Google Scholar]
- 119.Gopfert MS. Siedler F. Siess W. Sellmayer A. Structural identification of oxidized acyl-phosphatidylcholines that induce platelet activation. J Vasc Res. 2005;42:120–132. doi: 10.1159/000083461. [DOI] [PubMed] [Google Scholar]
- 120.Govindarajan B. Laird J. Salomon RG. Bhattacharya SK. Isolevuglandin-modified proteins, including elevated levels of inactive calpain-1, accumulate in glaucomatous trabecular meshwork. Biochemistry. 2008;47:817–825. doi: 10.1021/bi701517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Goyal J. Wang K. Liu M. Subbaiah PV. Novel function of lecithin-cholesterol acyltransferase. Hydrolysis of oxidized polar phospholipids generated during lipoprotein oxidation. J Biol Chem. 1997;272:16231–16239. doi: 10.1074/jbc.272.26.16231. [DOI] [PubMed] [Google Scholar]
- 122.Greenberg ME. Li XM. Gugiu BG. Gu X. Qin J. Salomon RG. Hazen SL. The lipid whisker model of the structure of oxidized cell membranes. J Biol Chem. 2008;283:2385–2396. doi: 10.1074/jbc.M707348200. [DOI] [PubMed] [Google Scholar]
- 123.Greenberg ME. Sun M. Zhang R. Febbraio M. Silverstein R. Hazen SL. Oxidized phosphatidylserine–CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J Exp Med. 2006;203:2613–2625. doi: 10.1084/jem.20060370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Groeneweg M. Vergouwe MN. Scheffer PG. Vermue HP. Sollewijn G. Sijbers AM. Leitinger N. Hofker MH. de Winther MP. Modification of LDL with oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) results in a novel form of minimally modified LDL that modulates gene expression in macrophages. Biochim Biophys Acta. 2008;1781:336–343. doi: 10.1016/j.bbalip.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 125.Gruber F. Oskolkova O. Leitner A. Mildner M. Mlitz V. Lengauer B. Kadl A. Mrass P. Kronke G. Binder BR. Bochkov VN. Leitinger N. Tschachler E. Photooxidation generates biologically active phospholipids that induce heme oxygenase-1 in skin cells. J Biol Chem. 2007;282:16934–16941. doi: 10.1074/jbc.M702523200. [DOI] [PubMed] [Google Scholar]
- 126.Gu X. Sun M. Gugiu B. Hazen S. Crabb JW. Salomon RG. Oxidatively truncated docosahexaenoate phospholipids: Total synthesis, generation, and peptide adduction chemistry. J Org Chem. 2003;68:3749–3761. doi: 10.1021/jo026721t. [DOI] [PubMed] [Google Scholar]
- 127.Gu X. Zhang W. Salomon RG. Fe2+ catalyzes vitamin E-induced fragmentation of hydroperoxy and hydroxy endoperoxides that generates gamma-hydroxy alkenals. J Am Chem Soc. 2007;129:6088–6089. doi: 10.1021/ja0689785. [DOI] [PubMed] [Google Scholar]
- 128.Gugiu BG. Mesaros CA. Sun M. Gu X. Crabb JW. Salomon RG. Identification of oxidatively truncated ethanolamine phospholipids in retina and their generation from polyunsaturated phosphatidylethanolamines. Chem Res Toxicol. 2006;19:262–271. doi: 10.1021/tx050247f. [DOI] [PubMed] [Google Scholar]
- 129.Gugiu BG. Mouillesseaux K. Duong V. Herzog T. Hekimian A. Koroniak L. Vondriska TM. Watson AD. Protein targets of oxidized phospholipids in endothelial cells. J Lipid Res. 2008;49:510–520. doi: 10.1194/jlr.M700264-JLR200. [DOI] [PubMed] [Google Scholar]
- 130.Han KH. Chang MK. Boullier A. Green SR. Li A. Glass CK. Quehenberger O. Oxidized LDL reduces monocyte CCR2 expression through pathways involving peroxisome proliferator-activated receptor gamma. J Clin Invest. 2000;106:793–802. doi: 10.1172/JCI10052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hartwich J. Dembinska–Kiec A. Gruca A. Motyka M. Partyka L. Skrzeczynska J. Bzowska M. Pryjma J. Huber J. Leitinger N. Schmitz G. Regulation of platelet adhesion by oxidized lipoproteins and oxidized phospholipids. Platelets. 2002;13:141–151. doi: 10.1080/09533710022149368. [DOI] [PubMed] [Google Scholar]
- 132.Haseruck N. Erl W. Pandey D. Tigyi G. Ohlmann P. Ravanat C. Gachet C. Siess W. The plaque lipid lysophosphatidic acid stimulates platelet activation and platelet-monocyte aggregate formation in whole blood: Involvement of P2Y1 and P2Y12 receptors. Blood. 2004;103:2585–2592. doi: 10.1182/blood-2003-04-1127. [DOI] [PubMed] [Google Scholar]
- 133.Hattori M. Arai H. Inoue K. Purification and characterization of bovine brain platelet-activating factor acetylhydrolase. J Biol Chem. 1993;268:18748–18753. [PubMed] [Google Scholar]
- 134.Hazen SL. Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J Biol Chem. 2008;283:15527–15531. doi: 10.1074/jbc.R700054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Herbrand U. Ahmadian MR. p190-RhoGAP as an integral component of the Tiam1/Rac1-induced downregulation of Rho. Biol Chem. 2006;387:311–317. doi: 10.1515/BC.2006.041. [DOI] [PubMed] [Google Scholar]
- 136.Hoebe K. Georgel P. Rutschmann S. Du X. Mudd S. Crozat K. Sovath S. Shamel L. Hartung T. Zahringer U. Beutler B. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 137.Hoff HF. O'Neil J. Wu Z. Hoppe G. Salomon RL. Phospholipid hydroxyalkenals: Biological and chemical properties of specific oxidized lipids present in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2003;23:275–282. doi: 10.1161/01.atv.0000051407.42536.73. [DOI] [PubMed] [Google Scholar]
- 138.Holland R. Hawkins AE. Eggler AL. Mesecar AD. Fabris D. Fishbein JC. Prospective type 1 and type 2 disulfides of Keap1 protein. Chem Res Toxicol. 2008;21:2051–2060. doi: 10.1021/tx800226m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Honda HM. Leitinger N. Frankel M. Goldhaber JI. Natarajan R. Nadler JL. Weiss JN. Berliner JA. Induction of monocyte binding to endothelial cells by MM-LDL: role of lipoxygenase metabolites. Arterioscler Thromb Vasc Biol. 1999;19:680–686. doi: 10.1161/01.atv.19.3.680. [DOI] [PubMed] [Google Scholar]
- 140.Horkko S. Binder CJ. Shaw PX. Chang MK. Silverman G. Palinski W. Witztum JL. Immunological responses to oxidized LDL. Free Radic Biol Med. 2000;28:1771–1779. doi: 10.1016/s0891-5849(00)00333-6. [DOI] [PubMed] [Google Scholar]
- 141.Horkko S. Bird DA. Miller E. Itabe H. Leitinger N. Subbanagounder G. Berliner JA. Friedman P. Dennis EA. Curtiss LK. Palinski W. Witztum JL. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid–protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103:117–128. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Horkko S. Miller E. Branch DW. Palinski W. Witztum JL. The epitopes for some antiphospholipid antibodies are adducts of oxidized phospholipid and beta2 glycoprotein 1 (and other proteins) Proc Nat! Acad Sci USA. 1997;94:10356–10361. doi: 10.1073/pnas.94.19.10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Horkko S. Miller E. Dudl E. Reaven P. Curtiss LK. Zvaifler NJ. Terkeltaub R. Pierangeli SS. Branch DW. Palinski W. Witztum JL. Antiphospholipid antibodies are directed against epitopes of oxidized phospholipids. Recognition of cardiolipin by monoclonal antibodies to epitopes of oxidized low density lipoprotein. J Clin Invest. 1996;98:815–825. doi: 10.1172/JCI118854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hosoya T. Maruyama A. Kang MI. Kawatani Y. Shibata T. Uchida K. Warabi E. Noguchi N. Itoh K. Yamamoto M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem. 2005;280:27244–27250. doi: 10.1074/jbc.M502551200. [DOI] [PubMed] [Google Scholar]
- 145.Hsiai TK. Cho SK. Reddy S. Hama S. Navab M. Demer LL. Honda HM. Ho CM. Pulsatile flow regulates monocyte adhesion to oxidized lipid-induced endothelial cells. Arterioscler Thromb Vasc Bio! 2001;21:1770–1776. doi: 10.1161/hq1001.097104. [DOI] [PubMed] [Google Scholar]
- 146.Huang LS. Kang JS. Kim MR. Sok DE. Oxygenation of arachidonoyl lysophospholipids by lipoxygenases from soybean, porcine leukocyte, or rabbit reticulocyte. J Agric Food Chem. 2008;56:1224–1232. doi: 10.1021/jf073016i. [DOI] [PubMed] [Google Scholar]
- 147.Huang MS. Lu J. Ivanov Y. Sage AP. Tseng W. Demer LL. Tintut Y. Hyperlipidemia impairs osteoanabolic effects of PTH. J Bone Miner Res. 2008;23:1672–1679. doi: 10.1359/JBMR.080513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huang MS. Morony S. Lu J. Zhang Z. Bezouglaia O. Tseng W. Tetradis S. Demer LL. Tintut Y. Atherogenic phospholipids attenuate osteogenic signaling by BMP-2 and parathyroid hormone in osteoblasts. J Biol Chem. 2007;282:21237–21243. doi: 10.1074/jbc.M701341200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Huang Z. Jiang J. Tyurin VA. Zhao Q. Mnuskin A. Ren J. Belikova NA. Feng W. Kurnikov IV. Kagan VE. Cardiolipin deficiency leads to decreased cardiolipin peroxidation and increased resistance of cells to apoptosis. Free Radic Biol Med. 2008;44:1935–1944. doi: 10.1016/j.freeradbiomed.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huber J. Furnkranz A. Bochkov VN. Patricia MK. Lee H. Hedrick CC. Berliner JA. Binder BR. Leitinger N. Specific monocyte adhesion to endothelial cells induced by oxidized phospholipids involves activation of cPLA2 and lipoxygenase. J Lipid Res. 2006;47:1054–1062. doi: 10.1194/jlr.M500555-JLR200. [DOI] [PubMed] [Google Scholar]
- 151.Huber J. Vales A. Mitulovic G. Blumer M. Schmid R. Witztum JL. Binder BR. Leitinger N. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler Thromb Vasc Biol. 2002;22:101–107. doi: 10.1161/hq0102.101525. [DOI] [PubMed] [Google Scholar]
- 152.Hulthe J. Antibodies to oxidized LDL in atherosclerosis development. Clinical and animal studies. Clin Chim Acta. 2004;348:1–8. doi: 10.1016/j.cccn.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 153.Imai Y. Kuba K. Neely GG. Yaghubian-Malhami R. Perkmann T. van LG. Ermolaeva M. Veldhuizen R. Leung YH. Wang H. Liu H. Sun Y. Pasparakis M. Kopf M. Mech C. Bavari S. Peiris JS. Slutsky AS. Akira S. Hultqvist M. Holmdahl R. Nicholls J. Jiang C. Binder CJ. Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Imbusch R. Mueller MJ. Formation of isoprostane F(2)-like compounds (phytoprostanes F(1)) from alpha-linolenic acid in plants. Free Radic Biol Med. 2000;28:720–726. doi: 10.1016/s0891-5849(00)00154-4. [DOI] [PubMed] [Google Scholar]
- 155.Isakson BE. Kronke G. Kadl A. Leitinger N. Duling BR. Oxidized phospholipids alter vascular connexin expression, phosphorylation, and heterocellular communication. Arterioscler Thromb Vasc Biol. 2006;26:2216–2221. doi: 10.1161/01.ATV.0000237608.19055.53. [DOI] [PubMed] [Google Scholar]
- 156.Ishii H. Tezuka T. Ishikawa H. Takada K. Oida K. Horie S. Oxidized phospholipids in oxidized low-density lipoprotein down-regulate thrombomodulin transcription in vascular endothelial cells through a decrease in the binding of RARbeta-RXRalpha heterodimers and Sp1 and Sp3 to their binding sequences in the TM promoter. Blood. 2003;101:4765–4774. doi: 10.1182/blood-2002-08-2428. [DOI] [PubMed] [Google Scholar]
- 157.Ishikawa K. Navab M. Leitinger N. Fogelman AM. Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest. 1997;100:1209–1216. doi: 10.1172/JCI119634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jerlich A. Pitt AR. Schaur RJ. Spickett CM. Pathways of phospholipid oxidation by HOCl in human LDL detected by LC-MS. Free Radic Biol Med. 2000;28:673–682. doi: 10.1016/s0891-5849(99)00273-7. [DOI] [PubMed] [Google Scholar]
- 159.Jerlich A. Schaur RJ. Pitt AR. Spickett CM. The formation of phosphatidylcholine oxidation products by stimulated phagocytes. Free Radic Res. 2003;37:645–653. doi: 10.1080/1071576031000091720. [DOI] [PubMed] [Google Scholar]
- 160.Jin Y. Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol. 2007;47:263–292. doi: 10.1146/annurev.pharmtox.47.120505.105337. [DOI] [PubMed] [Google Scholar]
- 161.Jyrkkanen HK. Kansanen E. Inkala M. Kivela AM. Hurttila H. Heinonen SE. Goldsteins G. Jauhiainen S. Tiainen S. Makkonen H. Oskolkova O. Afonyushkin T. Koistinaho J. Yamamoto M. Bochkov VN. Yla-Herttuala S. Levonen AL. Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ Res. 2008;103:e1–e9. doi: 10.1161/CIRCRESAHA.108.176883. [DOI] [PubMed] [Google Scholar]
- 162.Kadl A. Galkina E. Leitinger N. Induction of CCR2-dependent macrophage accumulation by oxidized phospholipids in the air-pouch model of inflammation. Arthritis Rheum. 2009;60:1362–1371. doi: 10.1002/art.24448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kadl A. Huber J. Gruber F. Bochkov VN. Binder BR. Leitinger N. Analysis of inflammatory gene induction by oxidized phospholipids in vivo by quantitative real-time RT-PCR in comparison with effects of LPS. Vascul Pharmacol. 2002;38:219–227. doi: 10.1016/s1537-1891(02)00172-6. [DOI] [PubMed] [Google Scholar]
- 164.Kagan VE. Bayir HA. Belikova NA. Kapralov O. Tyurina YY. Tyurin VA. Jiang J. Stoyanovsky DA. Wipf P. Kochanek PM. Greenberger JS. Pitt B. Shvedova AA. Borisenko G. Cytochrome c/cardiolipin relations in mitochondria: A kiss of death. Free Radic Biol Med. 2009;46:1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kagan VE. Borisenko GG. Tyurina YY. Tyurin VA. Jiang J. Potapovich AI. Kini V. Amoscato AA. Fujii Y. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med. 2004;37:1963–1985. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 166.Kagan VE. Tyurin VA. Jiang J. Tyurina YY. Ritov VB. Amoscato AA. Osipov AN. Belikova NA. Kapralov AA. Kini V. Vlasova II. Zhao Q. Zou M. Di P. Svistunenko DA. Kurnikov IV. Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 167.Kanter JL. Narayana S. Ho PP. Catz I. Warren KG. Sobel RA. Steinman L. Robinson WH. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138–143. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- 168.Kar NS. Ashraf MZ. Valiyaveettil M. Podrez EA. Mapping and characterization of the binding site for specific oxidized phospholipids and oxidized low density lipoprotein of scavenger receptor CD36. J Biol Chem. 2008;283:8765–8771. doi: 10.1074/jbc.M709195200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Karara A. Dishman E. Falck JR. Capdevila JH. Endogenous epoxyeicosatrienoyl-phospholipids. A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. J Biol Chem. 1991;266:7561–7569. [PubMed] [Google Scholar]
- 170.Karvonen J. Paivansalo M. Kesaniemi YA. Horkko S. Immunoglobulin M type of autoantibodies to oxidized low-density lipoprotein has an inverse relation to carotid artery atherosclerosis. Circulation. 2003;108:2107–2112. doi: 10.1161/01.CIR.0000092891.55157.A7. [DOI] [PubMed] [Google Scholar]
- 171.Katsumi A. Orr AW. Tzima E. Schwartz MA. Integrins in mechano-transduction. J Biol Chem. 2004;279:12001–12004. doi: 10.1074/jbc.R300038200. [DOI] [PubMed] [Google Scholar]
- 172.Kawai Y. Kiyokawa H. Kimura Y. Kato Y. Tsuchiya K. Terao J. Hypochlorous acid-derived modification of phospholipids: Characterization of amino phospholipids as regulatory molecules for lipid peroxidation. Biochemistry. 2006;45:14201–14211. doi: 10.1021/bi0610909. [DOI] [PubMed] [Google Scholar]
- 173.Kensler TW. Wakabayashi N. Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 174.Khallou-Laschet J. Tupin E. Caligiuri G. Poirier B. Thieblemont N. Gaston AT. Vandaele M. Bleton J. Tchapla A. Kaveri SV. Rudling M. Nicoletti A. Atheroprotective effect of adjuvants in apolipoprotein E knockout mice. Atherosclerosis. 2006;184:330–341. doi: 10.1016/j.atherosclerosis.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 175.Kiechl S. Willeit J. Mayr M. Viehweider B. Oberhollenzer M. Kronenberg F. Wiedermann CJ. Oberthaler S. Xu Q. Witztum JL. Tsimikas S. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: Prospective results from the Bruneck study. Arterioscler Thromb Vasc Biol. 2007;27:1788–1795. doi: 10.1161/ATVBAHA.107.145805. [DOI] [PubMed] [Google Scholar]
- 176.Kim JA. Territo MC. Wayner E. Carlos TM. Parhami F. Smith CW. Haberland ME. Fogelman AM. Berliner JA. Partial characterization of leukocyte binding molecules on endothelial cells induced by minimally oxidized LDL. Arterioscler Thromb. 1994;14:427–433. doi: 10.1161/01.atv.14.3.427. [DOI] [PubMed] [Google Scholar]
- 177.Kinnunen PK. Lipid bilayers as osmotic response elements. Cell Physiol Biochem. 2000;10:243–250. doi: 10.1159/000016360. [DOI] [PubMed] [Google Scholar]
- 178.Knapp S. Matt U. Leitinger N. van der PT. Oxidized phospholipids inhibit phagocytosis and impair outcome in gram-negative sepsis in vivo. J Immunol. 2007;178:993–1001. doi: 10.4049/jimmunol.178.2.993. [DOI] [PubMed] [Google Scholar]
- 179.Kobayashi M. Li L. Iwamoto N. Nakajima-Takagi Y. Kaneko H. Nakayama Y. Eguchi M. Wada Y. Kumagai Y. Yamamoto M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol. 2009;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kono N. Inoue T. Yoshida Y. Sato H. Matsusue T. Itabe H. Niki E. Aoki J. Arai H. Protection against oxidative stress-induced hepatic injury by intracellular type II platelet-activating factor acetylhydrolase by metabolism of oxidized phospholipids in vivo. J Biol Chem. 2008;283:1628–1636. doi: 10.1074/jbc.M708622200. [DOI] [PubMed] [Google Scholar]
- 181.Kriska T. Marathe GK. Schmidt JC. McIntyre TM. Girotti AW. Phospholipase action of platelet-activating factor acetylhydrolase, but not paraoxonase-1, on long fatty acyl chain phospholipid hydroperoxides. J Biol Chem. 2007;282:100–108. doi: 10.1074/jbc.M608135200. [DOI] [PubMed] [Google Scholar]
- 182.Kronke G. Bochkov VN. Huber J. Gruber F. Bluml S. Furnkranz A. Kadl A. Binder BR. Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J Biol Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- 183.Kuhn H. Belkner J. Wiesner R. Brash AR. Oxygenation of biological membranes by the pure reticulocyte lipoxygenase. J Biol Chem. 1990;265:18351–18361. [PubMed] [Google Scholar]
- 184.Kuhn H. Belkner J. Zaiss S. Fahrenklemper T. Wohlfeil S. Involvement of 15-lipoxygenase in early stages of atherogenesis. J Exp Med. 1994;179:1903–1911. doi: 10.1084/jem.179.6.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kuzmenko AI. Wu H. Bridges JP. McCormack FX. Surfactant lipid peroxidation damages surfactant protein A and inhibits interactions with phospholipid vesicles. J Lipid Res. 2004;45:1061–1068. doi: 10.1194/jlr.M300360-JLR200. [DOI] [PubMed] [Google Scholar]
- 186.Ky B. Burke A. Tsimikas S. Wolfe ML. Tadesse MG. Szapary PO. Witztum JL. FitzGerald GA. Rader DJ. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol. 2008;51:1653–1662. doi: 10.1016/j.jacc.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 187.Lang JD. McArdle PJ. O'Reilly PJ. Matalon S. Oxidant–antioxidant balance in acute lung injury. Chest. 2002;122:314S–320S. doi: 10.1378/chest.122.6_suppl.314s. [DOI] [PubMed] [Google Scholar]
- 188.Lee CY. Seet RC. Huang SH. Long LH. Halliwell B. Different patterns of oxidized lipid products in plasma and urine of dengue fever, stroke and Parkinsons disease patients. Cautions in the use of biomarkers of oxidative stress. Antioxid Redox Signal. 2009;11:407–420. doi: 10.1089/ars.2008.2179. [DOI] [PubMed] [Google Scholar]
- 189.Lee H. Shi W. Tontonoz P. Wang S. Subbanagounder G. Hedrick CC. Hama S. Borromeo C. Evans RM. Berliner JA. Nagy L. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ Res. 2000;87:516–521. doi: 10.1161/01.res.87.6.516. [DOI] [PubMed] [Google Scholar]
- 190.Lee S. Gharavi NM. Honda H. Chang I. Kim B. Jen N. Li R. Zimman A. Berliner JA. A role for NADPH oxidase 4 in the activation of vascular endothelial cells by oxidized phospholipids. Free Radic Biol Med. 2009;47:145–151. doi: 10.1016/j.freeradbiomed.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Leitinger N. Oxidized phospholipids as modulators of inflammation in atherosclerosis. Curr Opin Lipidol. 2003;14:421–430. doi: 10.1097/00041433-200310000-00002. [DOI] [PubMed] [Google Scholar]
- 192.Leitinger N. Tyner TR. Oslund L. Rizza C. Subbanagounder G. Lee H. Shih PT. Mackman N. Tigyi G. Territo MC. Berliner JA. Vora DK. Structurally similar oxidized phospholipids differentially regulate endothelial binding of monocytes and neutrophils. Proc Natl Acad Sci USA. 1999;96:12010–12015. doi: 10.1073/pnas.96.21.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Leroyer AS. Isobe H. Leseche G. Castier Y. Wassef M. Mallat Z. Binder BR. Tedgui A. Boulanger CM. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol. 2007;49:772–777. doi: 10.1016/j.jacc.2006.10.053. [DOI] [PubMed] [Google Scholar]
- 194.Lesnefsky EJ. Hoppel CL. Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochim Biophys Acta. 2008;1777:1020–1027. doi: 10.1016/j.bbabio.2008.05.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Levonen AL. Landar A. Ramachandran A. Ceaser EK. Dickinson DA. Zanoni G. Morrow JD. Darley-Usmar VM. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defenses in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li R. Chen W. Yanes R. Lee S. Berliner JA. OKL38 is an oxidative stress response gene stimulated by oxidized phospholipids. J Lipid Res. 2007;48:709–715. doi: 10.1194/jlr.M600501-JLR200. [DOI] [PubMed] [Google Scholar]
- 197.Li R. Mouillesseaux KP. Montoya D. Cruz D. Gharavi N. Dun M. Koroniak L. Berliner JA. Identification of prostaglandin E2 receptor subtype 2 as a receptor activated by OxPAPC. Circ Res. 2006;98:642–650. doi: 10.1161/01.RES.0000207394.39249.fc. [DOI] [PubMed] [Google Scholar]
- 198.Li Y. Schwabe RF. Vries-Seimon T. Yao PM. Gerbod-Giannone MC. Tall AR. Davis RJ. Flavell R. Brenner DA. Tabas I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: Model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280:21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 199.Libby P. Ridker PM. Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 200.Lloberas N. Torras J. Herrero-Fresneda I. Cruzado JM. Riera M. Hurtado I. Grinyo JM. Postischemic renal oxidative stress induces inflammatory response through PAF and oxidized phospholipids. Prevention by antioxidant treatment. FASEB J. 2002;16:908–910. doi: 10.1096/fj.01-0880fje. [DOI] [PubMed] [Google Scholar]
- 201.Loidl A. Sevcsik E. Riesenhuber G. Deigner HP. Hermetter A. Oxidized phospholipids in minimally modified low density lipoprotein induce apoptotic signaling via activation of acid sphingomyelinase in arterial smooth muscle cells. J Biol Chem. 2003;278:32921–32928. doi: 10.1074/jbc.M306088200. [DOI] [PubMed] [Google Scholar]
- 202.Lucerna M. Zernecke A. de NR. de Jager SC. Bot I. van der LC. Kholova I. Liehn EA. van Berkel TJ. Yla-Herttuala S. Weber C. Biessen EA. Vascular endothelial growth factor-A induces plaque expansion in ApoE knock-out mice by promoting de novo leukocyte recruitment. Blood. 2007;109:122–129. doi: 10.1182/blood-2006-07-031773. [DOI] [PubMed] [Google Scholar]
- 203.Ma Y. Malbon CC. Williams DL. Thorngate FE. Altered gene expression in early atherosclerosis is blocked by low level apolipoprotein E. PLoS One. 2008;3:e2503. doi: 10.1371/journal.pone.0002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ma Z. Li J. Yang L. Mu Y. Xie W. Pitt B. Li S. Inhibition of LPS- and CpG DNA-induced TN F-alpha response by oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2004;286:L808–L816. doi: 10.1152/ajplung.00220.2003. [DOI] [PubMed] [Google Scholar]
- 205.Malle E. Marsche G. Arnhold J. Davies MJ. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim Biophys Acta. 2006;1761:392–415. doi: 10.1016/j.bbalip.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 206.Malleier JM. Oskolkova O. Bochkov V. Jerabek I. Sokolikova B. Perkmann T. Breuss J. Binder BR. Geiger M. Regulation of protein C inhibitor (PCI) activity by specific oxidized and negatively charged phospholipids. Blood. 2007;109:4769–4776. doi: 10.1182/blood-2006-09-046953. [DOI] [PubMed] [Google Scholar]
- 207.Malliri A. van ES. Huveneers S. Collard JG. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. J Biol Chem. 2004;279:30092–30098. doi: 10.1074/jbc.M401192200. [DOI] [PubMed] [Google Scholar]
- 208.Manevich Y. Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med. 2005;38:1422–1432. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 209.Manser E. Loo TH. Koh CG. Zhao ZS. Chen XQ. Tan L. Tan I. Leung T. Lim L. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol Cell. 1998;1:183–192. doi: 10.1016/s1097-2765(00)80019-2. [DOI] [PubMed] [Google Scholar]
- 210.Marathe GK. Davies SS. Harrison KA. Silva AR. Murphy RC. Castro-Faria Neto H. Prescott SM. Zimmerman GA. McIntyre TM. Inflammatory platelet-activating factor-like phospholipids in oxidized low density lipoproteins are fragmented alkyl phosphatidylcholines. J Biol Chem. 1999;274:28395–28404. doi: 10.1074/jbc.274.40.28395. [DOI] [PubMed] [Google Scholar]
- 211.Marathe GK. Prescott SM. Zimmerman GA. McIntyre TM. Oxidized LDL contains inflammatory PAF-like phospholipids. Trends Cardiovasc Med. 2001;11:139–142. doi: 10.1016/s1050-1738(01)00100-1. [DOI] [PubMed] [Google Scholar]
- 212.Marathe GK. Zimmerman GA. McIntyre TM. Platelet-activating factor acetylhydrolase, and not paraoxonase-1, is the oxidized phospholipid hydrolase of high density lipoprotein particles. J Biol Chem. 2003;278:3937–3947. doi: 10.1074/jbc.M211126200. [DOI] [PubMed] [Google Scholar]
- 213.Marathe GK. Zimmerman GA. Prescott SM. McIntyre TM. Activation of vascular cells by PAF-like lipids in oxidized LDL. Vascul Pharmacol. 2002;38:193–200. doi: 10.1016/s1537-1891(02)00169-6. [DOI] [PubMed] [Google Scholar]
- 214.MartIn–Fontecha A. Sebastiani S. Hopken UE. Uguccioni M. Lipp M. Lanzavecchia A. Sallusto F. Regulation of dendritic cell migration to the draining lymph node: Impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615–621. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mastorikou M. Mackness M. Mackness B. Defective metabolism of oxidized phospholipid by HDL from people with type 2 diabetes. Diabetes. 2006;55:3099–3103. doi: 10.2337/db06-0723. [DOI] [PubMed] [Google Scholar]
- 216.McEver RP. Cummings RD. Perspectives series: Cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100:485–491. doi: 10.1172/JCI119556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.McIntyre TM. Prescott SM. Stafforini DM. The emerging roles of PAF acetylhydrolase. J Lipid Res. 2009;50:S255–S259. doi: 10.1194/jlr.R800024-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Meg li FM. Sabatini K. Respiration state IV-generated ROS destroy the mitochondrial bilayer packing order in vitro. An EPR study. FEBS Lett. 2003;550:185–189. doi: 10.1016/s0014-5793(03)00861-5. [DOI] [PubMed] [Google Scholar]
- 219.Megli FM. Sabatini K. Mitochondrial phospholipid bilayer structure is ruined after liver oxidative injury in vivo. FEBS Lett. 2004;573:68–72. doi: 10.1016/j.febslet.2004.07.057. [DOI] [PubMed] [Google Scholar]
- 220.Mehta D. Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 221.Memon RA. Staprans I. Noor M. Holleran WM. Uchida Y. Moser AH. Feingold KR. Grunfeld C. Infection and inflammation induce LDL oxidation in vivo. Arterioscler Thromb Vasc Biol. 2000;20:1536–1542. doi: 10.1161/01.atv.20.6.1536. [DOI] [PubMed] [Google Scholar]
- 222.Milne GL. Yin H. Morrow JD. Human biochemistry of the isoprostane pathway. J Biol Chem. 2008;283:15533–15537. doi: 10.1074/jbc.R700047200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Milne GL. Zanoni G. Porta A. Sasi S. Vidari G. Musiek ES. Freeman ML. Morrow JD. The cyclopentenone product of lipid peroxidation, 15-A2t-isoprostane, is efficiently metabolized by HepG2 cells via conjugation with glutathione. Chem Res Toxicol. 2004;17:17–25. doi: 10.1021/tx034213o. [DOI] [PubMed] [Google Scholar]
- 224.Miyamoto S. Martinez GR. Rettori D. Augusto O. Medeiros MH. Di MP. Linoleic acid hydroperoxide reacts with hypochlorous acid, generating peroxyl radical intermediates and singlet molecular oxygen. Proc Natl Acad Sci USA. 2006;103:293–298. doi: 10.1073/pnas.0508170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Montuschi P. Barnes P. Roberts LJ. Insights into oxidative stress: The isoprostanes. Curr Med Chem. 2007;14:703–717. doi: 10.2174/092986707780059607. [DOI] [PubMed] [Google Scholar]
- 226.Moore KJ. Kunjathoor VV. Koehn SL. Manning JJ. Tseng AA. Silver JM. McKee M. Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Morrow JD. Awad JA. Boss HJ. Blair IA. Roberts LJ. Noncyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc Natl Acad Sci USA. 1992;89:10721–10725. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Morrow JD. Awad JA. Wu A. Zackert WE. Daniel VC. Roberts LJ. Nonenzymatic free radical-catalyzed generation of thromboxane-like compounds (isothromboxanes) in vivo. J Biol Chem. 1996;271:23185–23190. doi: 10.1074/jbc.271.38.23185. [DOI] [PubMed] [Google Scholar]
- 229.Moumtzi A. Trenker M. Flicker K. Zenzmaier E. Saf R. Hermetter A. Import and fate of fluorescent analogs of oxidized phospholipids in vascular smooth muscle cells. J Lipid Res. 2007;48:565–582. doi: 10.1194/jlr.M600394-JLR200. [DOI] [PubMed] [Google Scholar]
- 230.Mullins RD. How WASP-family proteins and the Arp2/3 complex convert intracellular signals into cytoskeletal structures. Curr Opin Cell Biol. 2000;12:91–96. doi: 10.1016/s0955-0674(99)00061-7. [DOI] [PubMed] [Google Scholar]
- 231.Musiek ES. Breeding RS. Milne GL. Zanoni G. Morrow JD. McLaughlin B. Cyclopentenone isoprostanes are novel bioactive products of lipid oxidation which enhance neurodegeneration. J Neurochem. 2006;97:1301–1313. doi: 10.1111/j.1471-4159.2006.03797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Mwaikambo BR. Sennlaub F. Ong H. Chemtob S. Hardy P. Activation of CD36 inhibits and induces regression of inflammatory corneal neovascularization. Invest Ophthalmol Vis Sci. 2006;47:4356–4364. doi: 10.1167/iovs.05-1656. [DOI] [PubMed] [Google Scholar]
- 233.Nagata Y. Yamamoto Y. Niki E. Reaction of phosphatidylcholine hydroperoxide in human plasma: the role of peroxidase and lecithin:cholesterol acyltransferase. Arch Biochem Biophys. 1996;329:24–30. doi: 10.1006/abbi.1996.0187. [DOI] [PubMed] [Google Scholar]
- 234.Nagy L. Tontonoz P. Alvarez JG. Chen H. Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 235.Nakanishi H. Iida Y. Shimizu T. Taguchi R. Analysis of oxidized phosphatidylcholines as markers for oxidative stress, using multiple reaction monitoring with theoretically expanded data sets with reversed-phase liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1366–1374. doi: 10.1016/j.jchromb.2008.09.041. [DOI] [PubMed] [Google Scholar]
- 236.Navab M. Anantharamaiah GM. Fogelman AM. The effect of apolipoprotein mimetic peptides in inflammatory disorders other than atherosclerosis. Trends Cardiovasc Med. 2008;18:61–66. doi: 10.1016/j.tcm.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Navab M. Anantharamaiah GM. Hama S. Hough G. Reddy ST. Frank JS. Garber DW. Handattu S. Fogelman AM. D-4F and statins synergize to render HDL antiinflammatory in mice and monkeys and cause lesion regression in old apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1426–1432. doi: 10.1161/01.ATV.0000167412.98221.1a. [DOI] [PubMed] [Google Scholar]
- 238.Navab M. Anantharamaiah GM. Reddy ST. Fogelman AM. Apolipoprotein A-I mimetic peptides and their role in atherosclerosis prevention. Nat Clin Pract Cardiovasc Med. 2006;3:540–547. doi: 10.1038/ncpcardio0661. [DOI] [PubMed] [Google Scholar]
- 239.Navab M. Hama SY. Hough GP. Subbanagounder G. Reddy ST. Fogelman AM. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J Lipid Res. 2001;42:1308–1317. [PubMed] [Google Scholar]
- 240.Nguyen T. Sherratt PJ. Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 241.Nigam S. Schewe T. Phospholipase A(2)s and lipid peroxidation. Biochim Biophys Acta. 2000;1488:167–181. doi: 10.1016/s1388-1981(00)00119-0. [DOI] [PubMed] [Google Scholar]
- 242.Niki E. Yoshida Y. Saito Y. Noguchi N. Lipid peroxidation: Mechanisms, inhibition, and biological effects. Biochem Biophys Res Commun. 2005;338:668–676. doi: 10.1016/j.bbrc.2005.08.072. [DOI] [PubMed] [Google Scholar]
- 243.Nilsson J. Calara F. Regnstrom J. Hultgardh-Nilsson A. Ameli S. Cercek B. Shah PK. Immunization with homologous oxidized low density lipoprotein reduces neointimal formation after balloon injury in hypercholesterolemic rabbits. J Am Coll Cardiol. 1997;30:1886–1891. doi: 10.1016/s0735-1097(97)00366-5. [DOI] [PubMed] [Google Scholar]
- 244.Nonas S. Birukova AA. Fu P. Xing J. Chatchavalvanich S. Bochkov VN. Leitinger N. Garcia JG. Birukov KG. Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit Care. 2008;12:R27. doi: 10.1186/cc6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Nonas S. Miller I. Kawkitinarong K. Chatchavalvanich S. Gorshkova I. Bochkov VN. Leitinger N. Natarajan V. Garcia JG. Birukov KG. Oxidized phospholipids reduce vascular leak and inflammation in rat model of acute lung injury. Am J Respir Crit Care Med. 2006;173:1130–1138. doi: 10.1164/rccm.200511-1737OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.O'Donnell VB. Eiserich JP. Chumley PH. Jablonsky MJ. Krishna NR. Kirk M. Barnes S. Darley–Usmar VM. Freeman BA. Nitration of unsaturated fatty acids by nitric oxide-derived reactive nitrogen species peroxynitrite, nitrous acid, nitrogen dioxide, and nitronium ion. Chem Res Toxicol. 1999;12:83–92. doi: 10.1021/tx980207u. [DOI] [PubMed] [Google Scholar]
- 247.Ohkura N. Hiraishi S. Itabe H. Hamuro T. Kamikubo Y. Takano T. Matsuda J. Horie S. Oxidized phospholipids in oxidized low-density lipoprotein reduce the activity of tissue factor pathway inhibitor through association with its carboxy-terminal region. Antioxid Redox Signal. 2004;6:705–712. doi: 10.1089/1523086041361686. [DOI] [PubMed] [Google Scholar]
- 248.Oskolkova OV. Afonyushkin T. Leitner A. von SE. Gargalovic PS. Lusis AJ. Binder BR. Bochkov VN. ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: Critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood. 2008;112:330–339. doi: 10.1182/blood-2007-09-112870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Owens GK. Kumar MS. Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 250.Pagano RE. Longmuir KJ. Phosphorylation, transbilayer movement, and facilitated intracellular transport of diacylglycerol are involved in the uptake of a fluorescent analog of phosphatidic acid by cultured fibroblasts. J Biol Chem. 1985;260:1909–1916. [PubMed] [Google Scholar]
- 251.Palinski W. Horkko S. Miller E. Steinbrecher UP. Powell HC. Curtiss LK. Witztum JL. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98:800–814. doi: 10.1172/JCI118853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Palinski W. Miller E. Witztum JL. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci USA. 1995;92:821–825. doi: 10.1073/pnas.92.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Papadopoulou C. Corrigall V. Taylor PR. Poston RN. The role of the chemokines MCP-1, GRO-alpha, IL-8 and their receptors in the adhesion of monocytic cells to human atherosclerotic plaques. Cytokine. 2008;43:181–186. doi: 10.1016/j.cyto.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Papaiahgari S. Kleeberger SR. Cho HY. Kalvakolanu DV. Reddy SP. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J Biol Chem. 2004;279:42302–42312. doi: 10.1074/jbc.M408275200. [DOI] [PubMed] [Google Scholar]
- 255.Parhami F. Fang ZT. Yang B. Fogelman AM. Berliner JA. Stimulation of Gs and inhibition of Gi protein functions by minimally oxidized LDL. Arterioscler Thromb Vasc Biol. 1995;15:2019–2024. doi: 10.1161/01.atv.15.11.2019. [DOI] [PubMed] [Google Scholar]
- 256.Parhami F. Morrow AD. Balucan J. Leitinger N. Watson AD. Tintut Y. Berliner JA. Demer LL. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. 1997;17:680–687. doi: 10.1161/01.atv.17.4.680. [DOI] [PubMed] [Google Scholar]
- 257.Parry GC. Mackman N. Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol. 1997;159:5450–5456. [PubMed] [Google Scholar]
- 258.Parthasarathy S. Rankin SM. Role of oxidized low density lipoprotein in atherogenesis. Prog Lipid Res. 1992;31:127–143. doi: 10.1016/0163-7827(92)90006-5. [DOI] [PubMed] [Google Scholar]
- 259.Pegorier S. Stengel D. Durand H. Croset M. Ninio E. Oxidized phospholipid: POVPC binds to platelet-activating-factor receptor on human macrophages. Implications in atherosclerosis. Atherosclerosis. 2006;188:433–443. doi: 10.1016/j.atherosclerosis.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 260.Pidkovka NA. Cherepanova OA. Yoshida T. Alexander MR. Deaton RA. Thomas JA. Leitinger N. Owens GK. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101:792–801. doi: 10.1161/CIRCRESAHA.107.152736. [DOI] [PubMed] [Google Scholar]
- 261.Pinderski Oslund LJ. Hedrick CC. Olvera T. Hagenbaugh A. Territo M. Berliner JA. Fyfe AI. Interleukin-10 blocks atherosclerotic events in vitro and in vivo. Arterioscler Thromb Vasc Biol. 1999;19:2847–2853. doi: 10.1161/01.atv.19.12.2847. [DOI] [PubMed] [Google Scholar]
- 262.Pirog KA. Stabinsky Y. Goldman R. Peprotech. Cytokine index. In: Stabinsky Y, editor; Goldman R, editor. Rocky Hill, New Jersey USA: PeproTech Inc.; 2006. [Google Scholar]
- 263.Podrez EA. Byzova TV. Febbraio M. Salomon RG. Ma Y. Valiyaveettil M. Poliakov E. Sun M. Finton PJ. Curtis BR. Chen J. Zhang R. Silverstein RL. Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Podrez EA. Poliakov E. Shen Z. Zhang R. Deng Y. Sun M. Finton PJ. Shan L. Febbraio M. Hajjar DP. Silverstein RL. Hoff HF. Salomon RG. Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- 265.Podrez EA. Poliakov E. Shen Z. Zhang R. Deng Y. Sun M. Finton PJ. Shan L. Gugiu B. Fox PL. Hoff HF. Salomon RG. Hazen SL. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem. 2002;277:38503–38516. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- 266.Poliakov E. Brennan ML. Macpherson J. Zhang R. Sha W. Narine L. Salomon RG. Hazen SL. Isolevuglandins, a novel class of isoprostenoid derivatives, function as integrated sensors of oxidant stress and are generated by myeloperoxidase in vivo. FASEB J. 2003;17:2209–2220. doi: 10.1096/fj.03-0086com. [DOI] [PubMed] [Google Scholar]
- 267.Poliakov E. Meer SG. Roy SC. Mesaros C. Salomon RG. Iso[7]LGD2- protein adducts are abundant in vivo and free radical-induced oxidation of an arachidonyl phospholipid generates this D series isolevuglandin in vitro. Chem Res Toxicol. 2004;17:613–622. doi: 10.1021/tx034185+. [DOI] [PubMed] [Google Scholar]
- 268.Pontsler AV. St. Hilaire A. Marathe GK. Zimmerman GA. McIntyre TM. Cyclooxygenase-2 is induced in monocytes by peroxisome proliferator activated receptor gamma and oxidized alkyl phospholipids from oxidized low density lipoprotein. J Biol Chem. 2002;277:13029–13036. doi: 10.1074/jbc.M109546200. [DOI] [PubMed] [Google Scholar]
- 269.Pratico D. Iuliano L. Mauriello A. Spagnoli L. Lawson JA. Rokach J. Maclouf J. Violi F. FitzGerald GA. Localization of distinct F2-isoprostanes in human atherosclerotic lesions. J Clin Invest. 1997;100:2028–2034. doi: 10.1172/JCI119735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Pratico D. Tangirala RK. Horkko S. Witztum JL. Palinski W. FitzGerald GA. Circulating autoantibodies to oxidized cardiolipin correlate with isoprostane F(2alpha)-VI levels and the extent of atherosclerosis in ApoE-deficient mice: Modulation by vitamin E. Blood. 2001;97:459–464. doi: 10.1182/blood.v97.2.459. [DOI] [PubMed] [Google Scholar]
- 271.Qiao J. Huang F. Lum H. PKA inhibits RhoA activation: A protection mechanism against endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2003;284:L972–L980. doi: 10.1152/ajplung.00429.2002. [DOI] [PubMed] [Google Scholar]
- 272.Qiao J. Huang F. Naikawadi RP. Kim KS. Said T. Lum H. Lysophosphatidylcholine impairs endothelial barrier function through the G protein-coupled receptor GPR4. Am J Physiol Lung Cell Mol Physiol. 2006;291:L91–101. doi: 10.1152/ajplung.00508.2005. [DOI] [PubMed] [Google Scholar]
- 273.Qin J. Goswami R. Balabanov R. Dawson G. Oxidized phosphatidylcholine is a marker for neuroinflammation in multiple sclerosis brain. J Neurosci Res. 2007;85:977–984. doi: 10.1002/jnr.21206. [DOI] [PubMed] [Google Scholar]
- 274.Radi R. Beckman JS. Bush KM. Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- 275.Rahaman SO. Lennon DJ. Febbraio M. Podrez EA. Hazen SL. Silverstein RL. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4:211–221. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Ravandi A. Babaei S. Leung R. Monge JC. Hoppe G. Hoff H. Kamido H. Kuksis A. Phospholipids and oxophospholipids in atherosclerotic plaques at different stages of plaque development. Lipids. 2004;39:97–109. doi: 10.1007/s11745-004-1207-5. [DOI] [PubMed] [Google Scholar]
- 277.Reaven PD. Mechanisms of atherosclerosis: role of LDL oxidation. Adv Exp Med Biol. 1994;366:113–128. doi: 10.1007/978-1-4615-1833-4_9. [DOI] [PubMed] [Google Scholar]
- 278.Reddy S. Hama S. Grijalva V. Hassan K. Mottahedeh R. Hough G. Wadleigh DJ. Navab M. Fogelman AM. Mitogen-activated protein kinase phosphatase 1 activity is necessary for oxidized phospholipids to induce monocyte chemotactic activity in human aortic endothelial cells. J Biol Chem. 2001;276:17030–17035. doi: 10.1074/jbc.M011663200. [DOI] [PubMed] [Google Scholar]
- 279.Reddy ST. Nguyen JT. Grijalva V. Hough G. Hama S. Navab M. Fogelman AM. Potential role for mitogen-activated protein kinase phosphatase-1 in the development of atherosclerotic lesions in mouse models. Arterioscler Thromb Vasc Biol. 2004;24:1676–1681. doi: 10.1161/01.ATV.0000138342.94314.64. [DOI] [PubMed] [Google Scholar]
- 280.Roberts II LJ. Montine TJ. Markesbery WR. Tapper AR. Hardy P. Chemtob S. Dettbarn WD. Morrow JD. Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. J Biol Chem. 1998;273:13605–13612. doi: 10.1074/jbc.273.22.13605. [DOI] [PubMed] [Google Scholar]
- 281.Roberts LJ. Fessel JP. The biochemistry of the isoprostane, neuroprostane, and isofuran pathways of lipid peroxidation. Chem Phys Lipids. 2004;128:173–186. doi: 10.1016/j.chemphyslip.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 282.Roberts LJ. Milne GL. Isoprostanes. J Lipid Res. 2009;50 Suppl:S219–S223. doi: 10.1194/jlr.R800037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Roberts LJ. Morrow JD. Products of the isoprostane pathway: Unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808–820. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Rodenburg J. Vissers MN. Wiegman A. Miller ER. Ridker PM. Witztum JL. Kastelein JJ. Tsimikas S. Oxidized low-density lipoprotein in children with familial hypercholesterolemia and unaffected siblings: Effect of pravastatin. J Am Coll Cardiol. 2006;47:1803–1810. doi: 10.1016/j.jacc.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 285.Ron D. Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 286.Rouhanizadeh M. Hwang J. Clempus RE. Marcu L. Lassegue B. Sevanian A. Hsiai TK. Oxidized-1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine induces vascular endothelial superoxide production: Implication of NADPH oxidase. Free Radic Biol Med. 2005;39:1512–1522. doi: 10.1016/j.freeradbiomed.2005.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Ryter SW. Alam J. Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 288.Sabatini K. Mattila JP. Megli FM. Kinnunen PK. Characterization of two oxidatively modified phospholipids in mixed monolayers with DPPC. Biophys J. 2006;90:4488–4499. doi: 10.1529/biophysj.105.080176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Safa O. Hensley K. Smirnov MD. Esmon CT. Esmon NL. Lipid oxidation enhances the function of activated protein C. J Biol Chem. 2001;276:1829–1836. doi: 10.1074/jbc.M005931200. [DOI] [PubMed] [Google Scholar]
- 290.Salomon RG. Isolevuglandins, oxidatively truncated phospholipids, and atherosclerosis. Ann NY Acad Sci. 2005;1043:327–342. doi: 10.1196/annals.1333.040. [DOI] [PubMed] [Google Scholar]
- 291.Salomon RG. Levuglandins and isolevuglandins: Stealthy toxins of oxidative injury. Antioxid Redox Signal. 2005;7:185–201. doi: 10.1089/ars.2005.7.185. [DOI] [PubMed] [Google Scholar]
- 292.Salomon RG. Batyreva E. Kaur K. Sprecher DL. Schreiber MJ. Crabb JW. Penn MS. DiCorletoe AM. Hazen SL. Podrez EA. Isolevuglandin–protein adducts in humans: Products of free radical-induced lipid oxidation through the isoprostane pathway. Biochim Biophys Acta. 2000;1485:225–235. doi: 10.1016/s1388-1981(00)00038-x. [DOI] [PubMed] [Google Scholar]
- 293.Salomon RG. Sha W. Brame C. Kaur K. Subbanagounder G. O'Neil J. Hoff HF. Roberts LJ. Protein adducts of iso[4]levuglandin E2, a product of the isoprostane pathway, in oxidized low density lipoprotein. J Biol Chem. 1999;274:20271–20280. doi: 10.1074/jbc.274.29.20271. [DOI] [PubMed] [Google Scholar]
- 294.Santrock J. Gorski RA. O'Gara JF. Products and mechanism of the reaction of ozone with phospholipids in unilamellar phospholipid vesicles. Chem Res Toxicol. 1992;5:134–141. doi: 10.1021/tx00025a023. [DOI] [PubMed] [Google Scholar]
- 295.Savaskan NE. Ufer C. Kuhn H. Borchert A. Molecular biology of glutathione peroxidase 4: From genomic structure to developmental expression and neural function. Biol Chem. 2007;388:1007–1017. doi: 10.1515/BC.2007.126. [DOI] [PubMed] [Google Scholar]
- 296.Schenkein HA. Berry CR. Purkall D. Burmeister JA. Brooks CN. Tew JG. Phosphorylcholine-dependent cross-reactivity between dental plaque bacteria and oxidized low-density lipoproteins. Infect Immun. 2001;69:6612–6617. doi: 10.1128/IAI.69.11.6612-6617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Schneider C. Porter NA. Brash AR. Routes to 4-hydroxynonenal: fundamental issues in the mechanisms of lipid peroxidation. J Biol Chem. 2008;283:15539–15543. doi: 10.1074/jbc.R800001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Schnurr K. Belkner J. Ursini F. Schewe T. Kuhn H. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase controls the activity of the 15-lipoxygenase with complex substrates and preserves the specificity of the oxygenation products. J Biol Chem. 1996;271:4653–4658. doi: 10.1074/jbc.271.9.4653. [DOI] [PubMed] [Google Scholar]
- 299.Schroder M. Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 300.Sekhar KR. Crooks PA. Sonar VN. Friedman DB. Chan JY. Meredith MJ. Starnes JH. Kelton KR. Summar SR. Sasi S. Freeman ML. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-ylmethylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–5645. [PubMed] [Google Scholar]
- 301.Servitja JM. Marinissen MJ. Sodhi A. Bustelo XR. Gutkind JS. Rac1 function is required for Src-induced transformation. Evidence of a role for Tiam1 and Vav2 in Rac activation by Src. J Biol Chem. 2003;278:34339–34346. doi: 10.1074/jbc.M302960200. [DOI] [PubMed] [Google Scholar]
- 302.Seyerl M. Bluml S. Kirchberger S. Bochkov VN. Oskolkova O. Majdic O. Stockl J. Oxidized phospholipids induce anergy in human peripheral blood T cells. Eur J Immunol. 2008;38:778–787. doi: 10.1002/eji.200737619. [DOI] [PubMed] [Google Scholar]
- 303.Shaw PX. Horkko S. Chang MK. Curtiss LK. Palinski W. Silverman GJ. Witztum JL. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. 2000;105:1731–1740. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Shaw PX. Horkko S. Tsimikas S. Chang MK. Palinski W. Silverman GJ. Chen PP. Witztum JL. Human-derived antioxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler Thromb Vasc Biol. 2001;21:1333–1339. doi: 10.1161/hq0801.093587. [DOI] [PubMed] [Google Scholar]
- 305.Shih PT. Brennan ML. Vora DK. Territo MC. Strahl D. Elices MJ. Lusis AJ. Berliner JA. Blocking very late antigen-4 integrin decreases leukocyte entry and fatty streak formation in mice fed an atherogenic diet. Circ Res. 1999;84:345–351. doi: 10.1161/01.res.84.3.345. [DOI] [PubMed] [Google Scholar]
- 306.Shih PT. Elices MJ. Fang ZT. Ugarova TP. Strahl D. Territo MC. Frank JS. Kovach NL. Cabanas C. Berliner JA. Vora DK. Minimally modified low-density lipoprotein induces monocyte adhesion to endothelial connecting segment-1 by activating beta1 integrin. J Clin Invest. 1999;103:613–625. doi: 10.1172/JCI5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Sigal NH. Dumont FJ. Cyclosporin A, FK-506, and rapamycin: Pharmacologic probes of lymphocyte signal transduction. Annu Rev Immunol. 1992;10:519–560. doi: 10.1146/annurev.iy.10.040192.002511. [DOI] [PubMed] [Google Scholar]
- 308.Silaste ML. Rantala M. Alfthan G. Aro A. Witztum JL. Kesaniemi YA. Horkko S. Changes in dietary fat intake alter plasma levels of oxidized low-density lipoprotein and lipoprotein(a) Arterioscler Thromb Vasc Biol. 2004;24:498–503. doi: 10.1161/01.ATV.0000118012.64932.f4. [DOI] [PubMed] [Google Scholar]
- 309.Silva AR. de Assis EF. Caiado LF. Marathe GK. Bozza MT. McIntyre TM. Zimmerman GA. Prescott SM. Bozza PT. Castro-Faria-Neto HC. Monocyte chemoattractant protein-1 and 5-lipoxygenase products recruit leukocytes in response to platelet-activating factor-like lipids in oxidized low-density lipoprotein. J Immunol. 2002;168:4112–4120. doi: 10.4049/jimmunol.168.8.4112. [DOI] [PubMed] [Google Scholar]
- 310.Singleton PA. Chatchavalvanich S. Fu P. Xing J. Birukova AA. Fortune JA. Klibanov AM. Garcia JG. Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104:978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Singleton PA. Dudek SM. Chiang ET. Garcia JG. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005;19:1646–1656. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- 312.Smiley PL. Stremler KE. Prescott SM. Zimmerman GA. McIntyre TM. Oxidatively fragmented phosphatidylcholines activate human neutrophils through the receptor for platelet-activating factor. J Biol Chem. 1991;266:11104–11110. [PubMed] [Google Scholar]
- 313.Smyth EM. Grosser T. Wang M. Yu Y. FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50:5423–5428. doi: 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Song WL. Lawson JA. Reilly D. Rokach J. Chang CT. Giasson B. FitzGerald GA. Neurofurans, novel indices of oxidant stress derived from docosahexaenoic acid. J Biol Chem. 2008;283:6–16. doi: 10.1074/jbc.M706124200. [DOI] [PubMed] [Google Scholar]
- 315.Spector AA. Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 316.Spickett CM. Chlorinated lipids and fatty acids: An emerging role in pathology. Pharmacol Ther. 2007;115:400–409. doi: 10.1016/j.pharmthera.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 317.Spickett CM. Rennie N. Winter H. Zambonin L. Landi L. Jerlich A. Schaur RJ. Pitt AR. Detection of phospholipid oxidation in oxidatively stressed cells by reversed-phase HPLC coupled with positive-ionization electrospray [correction of electroscopy] MS. Biochem J. 2001;355:449–457. doi: 10.1042/0264-6021:3550449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Spite M. Baba SP. Ahmed Y. Barski OA. Nijhawan K. Petrash JM. Bhatnagar A. Srivastava S. Substrate specificity and catalytic efficiency of aldo-keto reductases with phospholipid aldehydes. Biochem J. 2007;405:95–105. doi: 10.1042/BJ20061743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Spiteller P. Spiteller G. 9-Hydroxy-10,12-octadecadienoic acid (9-HODE) and 13-hydroxy-9,11-octadecadienoic acid (13-HODE): Excellent markers for lipid peroxidation. Chemistry and Physics of Lipids. 1997;89:131–139. [Google Scholar]
- 320.Srivastava S. Spite M. Trent JO. West MB. Ahmed Y. Bhatnagar A. Aldose reductase-catalyzed reduction of aldehyde phospholipids. J Biol Chem. 2004;279:53395–53406. doi: 10.1074/jbc.M403416200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Stafforini DM. Sheller JR. Blackwell TS. Sapirstein A. Yull FE. McIntyre TM. Bonventre JV. Prescott SM. Roberts LJ. Release of free F2-isoprostanes from esterified phospholipids is catalyzed by intracellular and plasma platelet-activating factor acetylhydrolases. J Biol Chem. 2006;281:4616–4623. doi: 10.1074/jbc.M507340200. [DOI] [PubMed] [Google Scholar]
- 322.Steinbrecher UP. Fisher M. Witztum JL. Curtiss LK. Immunogenicity of homologous low density lipoprotein after methylation, ethylation, acetylation, or carbamylation: Generation of antibodies specific for derivatized lysine. J Lipid Res. 1984;25:1109–1116. [PubMed] [Google Scholar]
- 323.Steinbrecher UP. Zhang HF. Lougheed M. Role of oxidatively modified LDL in atherosclerosis. Free Radic Biol Med. 1990;9:155–168. doi: 10.1016/0891-5849(90)90119-4. [DOI] [PubMed] [Google Scholar]
- 324.Stenson WF. Parker CW. Metabolism of arachidonic acid in ionophore-stimulated neutrophils. Esterification of a hydroxylated metabolite into phospholipids. J Clin Invest. 1979;64:1457–1465. doi: 10.1172/JCI109604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Strauss G. Osen W. Debatin KM. Induction of apoptosis and modulation of activation and effector function in T cells by immunosuppressive drugs. Clin Exp Immunol. 2002;128:255–266. doi: 10.1046/j.1365-2249.2002.01777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Stremler KE. Stafforini DM. Prescott SM. McIntyre TM. Human plasma platelet-activating factor acetylhydrolase. Oxidatively fragmented phospholipids as substrates. J Biol Chem. 1991;266:11095–11103. [PubMed] [Google Scholar]
- 327.Subbanagounder G. Deng Y. Borromeo C. Dooley AN. Berliner JA. Salomon RG. Hydroxy alkenal phospholipids regulate inflammatory functions of endothelial cells. Vascul Pharmacol. 2002;38:201–209. doi: 10.1016/s1537-1891(02)00170-2. [DOI] [PubMed] [Google Scholar]
- 328.Subbanagounder G. Leitinger N. Schwenke DC. Wong JW. Lee H. Rizza C. Watson AD. Faull KF. Fogelman AM. Berliner JA. Determinants of bioactivity of oxidized phospholipids. Specific oxidized fatty acyl groups at the sn-2 position. Arterioscler Thromb Vasc Biol. 2000;20:2248–2254. doi: 10.1161/01.atv.20.10.2248. [DOI] [PubMed] [Google Scholar]
- 329.Subbanagounder G. Wong JW. Lee H. Faull KF. Miller E. Witztum JL. Berliner JA. Epoxyisoprostane and epoxycyclopentenone phospholipids regulate monocyte chemotactic protein-1 and interleukin-8 synthesis. Formation of these oxidized phospholipids in response to interleukin-1 beta. J Biol Chem. 2002;277:7271–7281. doi: 10.1074/jbc.M107602200. [DOI] [PubMed] [Google Scholar]
- 330.Subramanian VS. Goyal J. Miwa M. Sugatami J. Akiyama M. Liu M. Subbaiah PV. Role of lecithin-cholesterol acyltransferase in the metabolism of oxidized phospholipids in plasma: Studies with platelet-activating factor-acetyl hydrolase-deficient plasma. Biochim Biophys Acta. 1999;1439:95–109. doi: 10.1016/s1388-1981(99)00072-4. [DOI] [PubMed] [Google Scholar]
- 331.Sun M. Finnemann SC. Febbraio M. Shan L. Annangudi SP. Podrez EA. Hoppe G. Darrow R. Organisciak DT. Salomon RG. Silverstein RL. Hazen SL. Light- induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: A potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J Biol Chem. 2006;281:4222–4230. doi: 10.1074/jbc.M509769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Takai Y. Sasaki T. Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 333.Takayama F. Egashira T. Yamanaka Y. Effect of diclofenac, a non-steroidal anti-inflammatory drug, on lipid peroxidation caused by ischemia-reperfusion in rat liver. Jpn J Pharmacol. 1994;64:71–78. doi: 10.1254/jjp.64.71. [DOI] [PubMed] [Google Scholar]
- 334.Tallman KA. Kim HY. Ji JX. Szapacs ME. Yin H. McIntosh TJ. Liebler DC. Porter NA. Phospholipid–protein adducts of lipid peroxidation: Synthesis and study of new biotinylated phosphatidylcholines. Chem Res Toxicol. 2007;20:227–234. doi: 10.1021/tx600331s. [DOI] [PubMed] [Google Scholar]
- 335.Tapon N. Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- 336.Tokumura A. Toujima M. Yoshioka Y. Fukuzawa K. Lipid peroxidation in low density lipoproteins from human plasma and egg yolk promotes accumulation of 1-acyl analogues of platelet-activating factor-like lipids. Lipids. 1996;31:1251–1258. doi: 10.1007/BF02587909. [DOI] [PubMed] [Google Scholar]
- 337.Tokumura A. Tsutsumi T. Tsukatani H. Transbilayer movement and metabolic fate of ether-linked phosphatidic acid (1-O-octadecyl-2-acetyl-sn-glycerol-3-phosphate) in guinea pig peritoneal polymorphonuclear leukocytes. J Biol Chem. 1992;267:7275–7283. [PubMed] [Google Scholar]
- 337a.Tsimikas S. In vivo markers of oxidative stress and therapeutic interventions. Am J Cardiol. 2008;101:34D–42D. doi: 10.1016/j.amjcard.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 338.Tsimikas S. Bergmark C. Beyer RW. Patel R. Pattison J. Miller E. Juliano J. Witztum JL. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. J Am Coll Cardiol. 2003;41:360–370. doi: 10.1016/s0735-1097(02)02769-9. [DOI] [PubMed] [Google Scholar]
- 339.Tsimikas S. Brilakis ES. Lennon RJ. Miller ER. Witztum JL. McConnell JP. Kornman KS. Berger PB. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J Lipid Res. 2007;48:425–433. doi: 10.1194/jlr.M600361-JLR200. [DOI] [PubMed] [Google Scholar]
- 340.Tsimikas S. Kiechl S. Willeit J. Mayr M. Miller ER. Kronenberg F. Xu Q. Bergmark C. Weger S. Oberhollenzer F. Witztum JL. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: Five-year prospective results from the Bruneck study. J Am Coll Cardiol. 2006;47:2219–2228. doi: 10.1016/j.jacc.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 341.Tsimikas S. Lau HK. Han KR. Shortal B. Miller ER. Segev A. Curtiss LK. Witztum JL. Strauss BH. Percutaneous coronary intervention results in acute increases in oxidized phospholipids and lipoprotein(a): Short-term and long-term immunologic responses to oxidized low-density lipoprotein. Circulation. 2004;109:3164–3170. doi: 10.1161/01.CIR.0000130844.01174.55. [DOI] [PubMed] [Google Scholar]
- 342.Tsimikas S. Palinski W. Halpern SE. Yeung DW. Curtiss LK. Witztum JL. Radiolabeled MDA2, an oxidation-specific, monoclonal antibody, identifies native atherosclerotic lesions in vivo. J Nucl Cardiol. 1999;6:41–53. doi: 10.1016/s1071-3581(99)90064-8. [DOI] [PubMed] [Google Scholar]
- 343.Tsimikas S. Witztum JL. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr Opin Lipidol. 2008;19:369–377. doi: 10.1097/MOL.0b013e328308b622. [DOI] [PubMed] [Google Scholar]
- 344.Tuominen A. Miller YI. Hansen LF. Kesaniemi YA. Witztum JL. Horkko S. A natural antibody to oxidized cardiolipin binds to oxidized low-density lipoprotein, apoptotic cells, and atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2006;26:2096–2102. doi: 10.1161/01.ATV.0000233333.07991.4a. [DOI] [PubMed] [Google Scholar]
- 345.Tyurin VA. Tyurina YY. Feng W. Mnuskin A. Jiang J. Tang M. Zhang X. Zhao Q. Kochanek PM. Clark RS. Bayir H. Kagan VE. Mass-spectrometric characterization of phospholipids and their primary peroxidation products in rat cortical neurons during staurosporine-induced apoptosis. J Neurochem. 2008;107:1614–1633. doi: 10.1111/j.1471-4159.2008.05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Tyurina YY. Serinkan FB. Tyurin VA. Kini V. Yalowich JC. Schroit AJ. Fadeel B. Kagan VE. Lipid antioxidant, etoposide, inhibits phosphatidylserine externalization and macrophage clearance of apoptotic cells by preventing phosphatidylserine oxidation. J Biol Chem. 2004;279:6056–6064. doi: 10.1074/jbc.M309929200. [DOI] [PubMed] [Google Scholar]
- 347.Tyurina YY. Tyurin VA. Epperly MW. Greenberger JS. Kagan VE. Oxidative lipidomics of gamma-irradiation-induced intestinal injury. Free Radic Biol Med. 2008;44:299–314. doi: 10.1016/j.freeradbiomed.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 348.Tyurina YY. Tyurin VA. Zhao Q. Djukic M. Quinn PJ. Pitt BR. Kagan VE. Oxidation of phosphatidylserine: A mechanism for plasma membrane phospholipid scrambling during apoptosis? Biochem Biophys Res Commun. 2004;324:1059–1064. doi: 10.1016/j.bbrc.2004.09.102. [DOI] [PubMed] [Google Scholar]
- 349.Uhlson C. Harrison K. Allen CB. Ahmad S. White CW. Murphy RC. Oxidized phospholipids derived from ozone-treated lung surfactant extract reduce macrophage and epithelial cell viability. Chem Res Toxicol. 2002;15:896–906. doi: 10.1021/tx010183i. [DOI] [PubMed] [Google Scholar]
- 350.Vaarala O. Puurunen M. Lukka M. Alfthan G. Leirisalo-Repo M. Aho K. Palosuo T. Affinity-purified cardiolipin-binding antibodies show heterogeneity in their binding to oxidized low-density lipoprotein. Clin Exp Immunol. 1996;104:269–274. doi: 10.1046/j.1365-2249.1996.21728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Van Lenten BJ. Wagner AC. Jung CL. Ruchala P. Waring AJ. Lehrer RI. Watson AD. Hama S. Navab M. Anantharamaiah GM. Fogelman AM. Anti-inflammatory apoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human apoA-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Van Lenten BJ. Wagner AC. Navab M. Anantharamaiah GM. Hui EK. Nayak DP. Fogelman AM. D-4F, an apolipoprotein A-I mimetic peptide, inhibits the inflammatory response induced by influenza A infection of human type II pneumocytes. Circulation. 2004;110:3252–3258. doi: 10.1161/01.CIR.0000147232.75456.B3. [DOI] [PubMed] [Google Scholar]
- 353.van Nieuw Amerongen GP. van DS. Vermeer MA. Collard JG. van Hinsbergh V. Activation of RhoA by thrombin in endothelial hyperpermeability: Role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 354.van Nieuw Amerongen GP. van Hinsbergh V. Cytoskeletal effects of rho-like small guanine nucleotide-binding proteins in the vascular system. Arterioscler Thromb Vasc Biol. 2001;21:300–311. doi: 10.1161/01.atv.21.3.300. [DOI] [PubMed] [Google Scholar]
- 355.Vaneker M. Joosten LA. Heunks LM. Snijdelaar DG. Halbertsma FJ. van EJ. Netea MG. van der Hoeven JG. Scheffer GJ. Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology. 2008;109:465–472. doi: 10.1097/ALN.0b013e318182aef1. [DOI] [PubMed] [Google Scholar]
- 356.VanRollins M. Woltjer RL. Yin H. Morrow JD. Montine TJ. F2-dihomoisoprostanes arise from free radical attack on adrenic acid. J Lipid Res. 2008;49:995–1005. doi: 10.1194/jlr.M700503-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Vila A. Tallman KA. Jacobs AT. Liebler DC. Porter NA. Marnett LJ. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem Res Toxicol. 2008;21:432–444. doi: 10.1021/tx700347w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Vohl MC. Neville TA. Kumarathasan R. Braschi S. Sparks DL. A novel lecithin-cholesterol acyltransferase antioxidant activity prevents the formation of oxidized lipids during lipoprotein oxidation. Biochemistry. 1999;38:5976–5981. doi: 10.1021/bi982258w. [DOI] [PubMed] [Google Scholar]
- 359.Volanakis JE. Human C-reactive protein: Expression, structure, and function. Mol Immunol. 2001;38:189–197. doi: 10.1016/s0161-5890(01)00042-6. [DOI] [PubMed] [Google Scholar]
- 360.von Schlieffen E. Oskolkova OV. Schabbauer G. Gruber F. Bluml S. Genest M. Kadl A. Marsik C. Knapp S. Chow J. Leitinger N. Binder BR. Bochkov VN. Multi-hit inhibition of circulating and cell-associated components of the Toll-like receptor 4 pathway by oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2008;29:356–362. doi: 10.1161/ATVBAHA.108.173799. [DOI] [PubMed] [Google Scholar]
- 361.Vora DK. Fang ZT. Liva SM. Tyner TR. Parhami F. Watson AD. Drake TA. Territo MC. Berliner JA. Induction of P-selectin by oxidized lipoproteins. Separate effects on synthesis and surface expression. Circ Res. 1997;80:810–818. doi: 10.1161/01.res.80.6.810. [DOI] [PubMed] [Google Scholar]
- 362.Waddington E. Sienuarine K. Puddey I. Croft K. Identification and quantitation of unique fatty acid oxidation products in human atherosclerotic plaque using high- performance liquid chromatography. Anal Biochem. 2001;292:234–244. doi: 10.1006/abio.2001.5075. [DOI] [PubMed] [Google Scholar]
- 363.Wakabayashi N. Dinkova-Kostova AT. Holtzclaw WD. Kang MI. Kobayashi A. Yamamoto M. Kensler TW. Talalay P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 2004;101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Walton KA. Cole AL. Yeh M. Subbanagounder G. Krutzik SR. Modlin RL. Lucas RM. Nakai J. Smart EJ. Vora DK. Berliner JA. Specific phospholipid oxidation products inhibit ligand activation of toll-like receptors 4 and 2. Arterioscler Thromb Vasc Biol. 2003;23:1197–1203. doi: 10.1161/01.ATV.0000079340.80744.B8. [DOI] [PubMed] [Google Scholar]
- 365.Walton KA. Gugiu BG. Thomas M. Basseri RJ. Eliav DR. Salomon RG. Berliner JA. A role for neutral sphingomyelinase activation in the inhibition of LPS action by phospholipid oxidation products. J Lipid Res. 2006;47:1967–1974. doi: 10.1194/jlr.M600060-JLR200. [DOI] [PubMed] [Google Scholar]
- 366.Walton KA. Hsieh X. Gharavi N. Wang S. Wang G. Yeh M. Cole AL. Berliner JA. Receptors involved in the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine-mediated synthesis of interleukin-8. A role for Toll-like receptor 4 and a glycosylphosphatidylinositol-anchored protein. J Biol Chem. 2003;278:29661–29666. doi: 10.1074/jbc.M300738200. [DOI] [PubMed] [Google Scholar]
- 367.Warabi E. Takabe W. Minami T. Inoue K. Itoh K. Yamamoto M. Ishii T. Kodama T. Noguchi N. Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: Role of reactive oxygen/nitrogen species. Free Radic Biol Med. 2007;42:260–269. doi: 10.1016/j.freeradbiomed.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 368.Watson AD. Leitinger N. Navab M. Faull KF. Horkko S. Witztum JL. Palinski W. Schwenke D. Salomon RG. Sha W. Subbanagounder G. Fogelman AM. Berliner JA. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 369.Watson AD. Navab M. Hama SY. Sevanian A. Prescott SM. Stafforini DM. McIntyre TM. Du BN. Fogelman AM. Berliner JA. Effect of platelet activating factoracetylhydrolase on the formation and action of minimally oxidized low density lipoprotein. J Clin Invest. 1995;95:774–782. doi: 10.1172/JCI117726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Watson AD. Subbanagounder G. Welsbie DS. Faull KF. Navab M. Jung ME. Fogelman AM. Berliner JA. Structural identification of a novel pro-inflammatory epoxyisoprostane phospholipid in mildly oxidized low density lipoprotein. J Biol Chem. 1999;274:24787–24798. doi: 10.1074/jbc.274.35.24787. [DOI] [PubMed] [Google Scholar]
- 371.Weinstein EA. Li H. Lawson JA. Rokach J. FitzGerald GA. Axelsen PH. Prothrombinase acceleration by oxidatively damaged phospholipids. J Biol Chem. 2000;275:22925–22930. doi: 10.1074/jbc.M002438200. [DOI] [PubMed] [Google Scholar]
- 372.Wenk J. Schuller J. Hinrichs C. Syrovets T. Azoitei N. Podda M. Wlaschek M. Brenneisen P. Schneider LA. Sabiwalsky A. Peters T. Sulyok S. Dissemond J. Schauen M. Krieg T. Wirth T. Simmet T. Scharffetter-Kochanek K. Overexpression of phospholipid-hydroperoxide glutathione peroxidase in human dermal fibroblasts abrogates UVA irradiation-induced expression of interstitial collagenase/matrix metalloproteinase-1 by suppression of phosphatidylcholine hydroperoxide-mediated NFkappaB activation and interleukin-6 release. J Biol Chem. 2004;279:45634–45642. doi: 10.1074/jbc.M408893200. [DOI] [PubMed] [Google Scholar]
- 373.White CR. Datta G. Zhang Z. Gupta H. Garber DW. Mishra VK. Palgunachari MN. Handattu SP. Chaddha M. Anantharamaiah GM. HDL therapy for cardiovascular diseases: The road to HDL mimetics. Curr Atheroscler Rep. 2008;10:405–412. doi: 10.1007/s11883-008-0063-6. [DOI] [PubMed] [Google Scholar]
- 374.Winterbourn CC. van den Berg JJ. Roitman E. Kuypers FA. Chlorohydrin formation from unsaturated fatty acids reacted with hypochlorous acid. Arch Biochem Biophys. 1992;296:547–555. doi: 10.1016/0003-9861(92)90609-z. [DOI] [PubMed] [Google Scholar]
- 375.Wittchen ES. Worthylake RA. Kelly P. Casey PJ. Quilliam LA. Burridge K. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J Biol Chem. 2005;280:11675–11682. doi: 10.1074/jbc.M412595200. [DOI] [PubMed] [Google Scholar]
- 376.Wittwer J. Hersberger M. The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot Essent Fatty Acids. 2007;77:67–77. doi: 10.1016/j.plefa.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 377.Wondrak GT. Jacobson MK. Jacobson EL. Endogenous UVA photosensitizers: Mediators of skin photodamage and novel targets for skin photoprotection. Photochem Photobiol Sci. 2006;5:215–237. doi: 10.1039/b504573h. [DOI] [PubMed] [Google Scholar]
- 378.Yamamoto T. Suzuki T. Kobayashi A. Wakabayashi J. Maher J. Motohashi H. Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Yang Y. Cheng JZ. Singhal SS. Saini M. Pandya U. Awasthi S. Awasthi YC. Role of glutathione S-transferases in protection against lipid peroxidation. Overexpression of hGSTA2-2 in K562 cells protects against hydrogen peroxide-induced apoptosis and inhibits JNK and caspase 3 activation. J Biol Chem. 2001;276:19220–19230. doi: 10.1074/jbc.M100551200. [DOI] [PubMed] [Google Scholar]
- 380.Yeh M. Cole AL. Choi J. Liu Y. Tulchinsky D. Qiao JH. Fishbein MC. Dooley AN. Hovnanian T. Mouilleseaux K. Vora DK. Yang WP. Gargalovic P. Kirchgessner T. Shyy JY. Berliner JA. Role for sterol regulatory element-binding protein in activation of endothelial cells by phospholipid oxidation products. Circ Res. 2004;95:780–788. doi: 10.1161/01.RES.0000146030.53089.18. [DOI] [PubMed] [Google Scholar]
- 381.Yeh M. Gharavi NM. Choi J. Hsieh X. Reed E. Mouillesseaux KP. Cole AL. Reddy ST. Berliner JA. Oxidized phospholipids increase interleukin 8 (IL-8) synthesis by activation of the c-src/signal transducers and activators of transcription (STAT)3 pathway. J Biol Chem. 2004;279:30175–30181. doi: 10.1074/jbc.M312198200. [DOI] [PubMed] [Google Scholar]
- 382.Yeh M. Leitinger N. de MR. Onai N. Matsushima K. Vora DK. Berliner JA. Reddy ST. Increased transcription of IL-8 in endothelial cells is differentially regulated by TNFalpha and oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2001;21:1585–1591. doi: 10.1161/hq1001.097027. [DOI] [PubMed] [Google Scholar]
- 383.Yin H. Brooks JD. Gao L. Porter NA. Morrow JD. Identification of novel autoxidation products of the omega-3 fatty acid eicosapentaenoic acid in vitro and in vivo. J Biol Chem. 2007;282:29890–29901. doi: 10.1074/jbc.M703108200. [DOI] [PubMed] [Google Scholar]
- 384.Yoshida T. Gan Q. Owens GK. Kruppel-like factor 4, Elk-1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol. 2008;295:C1175–C1182. doi: 10.1152/ajpcell.00288.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Yoshimi N. Ikura Y. Sugama Y. Kayo S. Ohsawa M. Yamamoto S. Inoue Y. Hirata K. Itabe H. Yoshikawa J. Ueda M. Oxidized phosphatidylcholine in alveolar macrophages in idiopathic interstitial pneumonias. Lung. 2005;183:109–121. doi: 10.1007/s00408-004-2525-0. [DOI] [PubMed] [Google Scholar]
- 386.Yurkova I. Huster D. Arnhold J. Free radical fragmentation of cardiolipin by cytochrome c. Chem Phys Lipids. 2009;158:16–21. doi: 10.1016/j.chemphyslip.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 387.Zagol-Ikapitte I. Masterson TS. Amarnath V. Montine TJ. Andreasson KI. Boutaud O. Oates JA. Prostaglandin H(2)-derived adducts of proteins correlate with Alzheimer's disease severity. J Neurochem. 2005;94:1140–1145. doi: 10.1111/j.1471-4159.2005.03264.x. [DOI] [PubMed] [Google Scholar]
- 388.Zhan X. Brown B. Slobod KS. Hurwitz JL. Inhibition of ex vivo-expanded cytotoxic T-lymphocyte function by high-dose cyclosporine. Transplantation. 2003;76:739–740. doi: 10.1097/01.TP.0000078623.64968.E5. [DOI] [PubMed] [Google Scholar]
- 389.Zhang DD. Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Zhang Q. Sitzman LA. Al-Hassani M. Cai S. Pollok KE. Travers JB. Hingtgen CM. Involvement of platelet-activating factor in ultraviolet B-induced hyperalgesia. J Invest Dermatol. 2009;129:167–174. doi: 10.1038/jid.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Zhang Q. Southall MD. Mezsick SM. Johnson C. Murphy RC. Konger RL. Travers JB. Epidermal peroxisome proliferator-activated receptor gamma as a target for ultraviolet B radiation. J Biol Chem. 2005;280:73–79. doi: 10.1074/jbc.M409795200. [DOI] [PubMed] [Google Scholar]
- 392.Zhang W. Salomon RG. Oxidized phospholipids, isolevuglandins, and atherosclerosis. Mol Nutr Food Res. 2005;49:1050–1062. doi: 10.1002/mnfr.200500056. [DOI] [PubMed] [Google Scholar]
- 393.Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci. 2001;26:724–732. doi: 10.1016/s0968-0004(01)01973-9. [DOI] [PubMed] [Google Scholar]
- 394.Zhou J. Lhotak S. Hilditch BA. Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 395.Zhou X. Caligiuri G. Hamsten A. Lefvert AK. Hansson GK. LDL immunization induces T-cell-dependent antibody formation and protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:108–114. doi: 10.1161/01.atv.21.1.108. [DOI] [PubMed] [Google Scholar]
- 396.Zimman A. Mouillesseaux KP. Le T. Gharavi NM. Ryvkin A. Graeber TG. Chen TT. Watson AD. Berliner JA. Vascular endothelial growth factor receptor 2 plays a role in the activation of aortic endothelial cells by oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2007;27:332–338. doi: 10.1161/01.ATV.0000252842.57585.df. [DOI] [PubMed] [Google Scholar]