

Abstract
Mitochondria are organelles of eukaryotic cells that contain their own genetic material and evolved from prokaryotic ancestors some 2 billion years ago. They are the main source of the cell's energy supply and are involved in such important processes as apoptosis, mitochondrial diseases, and aging. During recent years it also became apparent that mitochondria display a complex dynamical behavior of fission and fusion, the function of which is as yet unknown. In this paper we develop a concise theory that explains why fusion and fission have evolved, how these processes are related to the accumulation of mitochondrial mutants during aging, why the mitochondrial DNA has to be located close to the respiration complexes where most radicals are generated, and what selection pressures shaped the slightly different structure of animal and plant mitochondria. We believe that this “organelle control” theory will help in understanding key processes involved in the evolution of the mitochondrial genome and the aging process.
Keywords: mechanistic aging theory, mathematical model
Mitochondria are cellular organelles present in all higher eukaryotes, responsible for bioenergy (ATP) production. Mitochondria have their own genetic material, mtDNA, and evolved from bacterial endosymbionts, raising many broad issues in evolutionary biology (1–4). However, during the course of evolution, it appears that large parts of the genomes of the originally autonomous mitochondrial ancestors have become transferred to the host cell nucleus, leaving only a small fraction of the genes required for mitochondrial functions encoded within mtDNA. Nevertheless, mitochondria undergo their own cycles of replication and removal within the cell; thus, there is turnover of mitochondria in both mitotic and postmitotic cells, which provides opportunities for selection at the level of the organelle.
In recent decades it became clear that mtDNA mutations are involved in many diseases (5) and also in the aging process (6–10). As well as producing ATP, mitochondria are the major site of production of damaging reactive oxygen species (ROS), which are formed as by-products of the respiratory chain reactions. Thus, mitochondria are believed to be both the major source and a primary target of ROS-induced damage, resulting in high levels of mtDNA mutation. Intriguingly, it appears that generally when cells exhibit severe mitochondrial deficiency, a single mitochondrial mutation has overtaken the population of wild-type mitochondria in the cell (11, 12). This is surprising, because, first, it is not obvious how a functionally impaired mutant can outcompete the healthy organelles and, second, within aged tissues there is a considerable spectrum of different mtDNA mutations such that individual mitochondrially defective cells are taken over by different mtDNA mutations.
Another important finding of recent years is that individual mitochondria do not exist as permanently distinct entities, as has long been believed, but instead form a dynamic network within which the mitochondria regularly exchange proteins, mtDNA, and lipids by rapid fusion and fission processes (13, 14).
Why mitochondria fuse and divide is not yet understood. It has been suggested that filamentous mitochondria might be used as intracellular power cables (15), but that theory does not explain fission and the high fusion/fission rates. Another idea is that mitochondrial fusion is a mechanism to rescue damaged mitochondria by enabling transcomplementation of missing proteins. However, recent experimental results show that an intact proton gradient is required for fusion, indicating that it is instead much more likely that fission is a process to separate dysfunctional organelles from the network (14). However, if fusion is not a rescue process, what else could be the function of this elaborate cellular mechanism? In this paper we argue that
i) mitochondrial fusion is a necessary consequence of the fact that most mitochondrial genes have, over the course of evolution, migrated to the nucleus;
ii) this fusion process makes possible the accumulation of mitochondrial mutants;
iii) mitochondrial fission is then necessary to combat this accumulation; and
iv) point iii requires that the mtDNA is attached to OXPHOS complexes in the mitochondrial membrane, exactly where most ROS are produced.
Origin of Mitochondrial Fusion
Mitochondria are endosymbionts that derived from α-proteobacteria that formed a symbiosis with archaebacteria ∼2 billion years ago. During the course of evolution almost all of the mitochondrial genes have been transferred to the nucleus. The proteins encoded by these genes are synthesized in the cytoplasm and then imported into the mitochondria using complex molecular machinery. This transfer was necessary to reduce the potential for conflict between the host cell and symbiont (16) and it also avoided the metabolic burden to maintain hundreds of full-sized genomes in a single host cell.
Despite the advantages of a central control of all organelles, this arrangement runs into problems if the mitochondria remain always as separate entities. The problem is that central control cannot take account of the different protein requirements of the individual mitochondria. Although mitochondria perform in principle the same functions, their requirements for the different mitochondrial proteins quickly begin to diverge, because mitochondria at different cellular locations (close to the nucleus or close to actin/myosin filaments) have different metabolic activities. When only a single set of mitochondrial genes exists (in the nucleus), regulation at the level of transcription or translation can lead to only a single cytoplasmic concentration for a given protein and is thus not suitable to respond in an optimal way to different requirements of different individual mitochondria. A possible solution would be a cytoplasmic excess of mitochondrial proteins whereby each organelle could control the import rate of each protein according to its needs. However, to develop and maintain such a protein-specific import control would be difficult and would also waste precious membrane space that is needed for OXPHOS complexes. For a subset of mitochondrial proteins, namely the proteins of the respiratory chain itself, a somewhat similar mechanism has actually been proposed. Allen proposed that cells regulate the synthesis rate of OXPHOS proteins by sensing the redox state of individual mitochondria, followed by transcriptional regulation of the local mtDNA molecules (17). This would explain why not all organelle genes can be transferred into the nucleus. OXPHOS complexes consist of proteins encoded by the mtDNA as well as proteins encoded by the nucleus. According to the redox sensing mechanism the nuclear components are always synthesized in excess and become degraded if there are no mitochondrial components available with which to combine to form an OXPHOS complex. In this special case, the complex-assembly process functions as protein-specific import control. Although this mechanism works, there would be less waste of nuclear components and thereby greater metabolic efficiency, if all mitochondria had the same demand.
Mitochondrial fusion represents a general solution to these problems by equilibrating the concentrations of all nuclear-encoded mitochondrial proteins over all organelles, effectively forming a single mitochondrial compartment in the cell. Transcriptional control of a single set of mitochondrial genes (in the nucleus) is then sufficient to respond in an optimal way to a single set of protein concentrations in the mitochondrial matrix. No new protein-specific import control needs to be evolved and the fraction of wasted nuclear OXPHOS components can be minimized.
Modeling Equilibration via Fusion.
Can fusion of mitochondria equilibrate the concentration of the matrix proteins, despite the continuous tendency of the mitochondria to develop diverging concentrations? A mathematical model helps to illustrate these processes. For simplicity we assume a system of just three mitochondria that have different requirements for the same protein, C. To implement these differences mathematically we use identical import rates for the protein, but different degradation rates. The following set of ordinary differential equations describes this system including import, degradation, and diffusion between the three mitochondria:
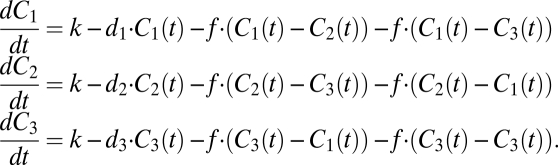 |
The parameter f controls the diffusion between the mitochondria and with no diffusion (f = 0) the steady-state concentrations of C inside the mitochondria are given by Ciss = k/di. Fig. 1 shows how the steady-state concentrations converge with increasing diffusion rate f. Without fusion, however, the mitochondrion with the lowest demand (represented by the lowest degradation rate) ends up with the highest steady-state level and the mitochondrion with the highest demand (highest degradation rate) has the lowest steady-state level.
Fig. 1.

Steady-state levels of protein concentration C inside the three different mitochondria depending on the diffusion rate f. Parameter values are k = 10, d1 = 1, d2 = 2, d3 = 3.
In summary we propose that mitochondrial fusion developed as a consequence of the migration of mitochondrial genes to the nucleus and the concomitant problem of controlling the demands of hundreds of organelles with a single set of mitochondrial genes.
Accumulation of Mitochondrial Mutants
The mitochondrial theory of aging (also known as the free radical theory of aging) states that ROS, which are by-products of respiration, continuously damage membranes, proteins, and mtDNA and thus cause the accumulation of molecular and cellular damage that is responsible for aging. It has been suggested that mtDNA damage leads to increased ROS production, which in turn leads to even more mtDNA damage. This idea has been called “the vicious cycle” (18, 19). However, the introduction of single-cell studies has revealed that affected cells tend to contain only a single type of mtDNA mutation that has displaced the wild type (WT) mitochondria to a large degree. Furthermore, neighboring cells are taken over by different mutants. This result suggests that the observed mutations have a selection advantage over the WT, leading to a clonal expansion of the single mutant, which is inconsistent with the vicious cycle idea that predicts a multitude of different mutants in the same cell.
One proposed mechanism to account for clonal expansion is “survival of the slowest” (SOS) (20). According to this idea mitochondrial mutants generate less of the membrane-damaging superoxide anion radical and therefore accumulate membrane damage at a slower rate. If mitochondrial degradation is then linked to the amount of membrane damage, it is possible to show that mitochondrial mutants would have a selection advantage (21). However, this mechanism works only if mitochondria are separate entities. If fusion occurs, the necessary link between genotype and phenotype is broken; i.e., there is no longer the necessary connection between the mutated mtDNA and the mutant organelle's membrane that suffers less damage. The ubiquitous and frequent occurrence of mitochondrial fusions therefore speaks clearly against the SOS hypothesis.
The fact that mitochondrial fusions do occur revives an earlier idea that the selection advantage of deletion mutants is their reduced size, which allows them to replicate faster than WT (22–24). This idea had fallen into disfavor because even though the mutant might have a replication advantage it should also suffer from a severe lack of ATP and a reduced proton gradient, both of which are important for fast mitochondrial growth. However, most important for the dismissal of the replication-rate idea was the fact that the time required for the replication of the mtDNA is only 1–2 h (25), whereas the division time of mitochondria is ∼1–3 wk (26–28). Thus, it has been argued that it is very difficult to imagine how mtDNA replication could be the rate-limiting step in mitochondrial multiplication (29, 30). However, when fusion occurs, all of the mitochondria of a cell will effectively form one large compartment with constant mixing of mtDNA as well as matrix and membrane components. Under these conditions, mtDNA deletions no longer lead to mitochondrion-specific energy and proton gradient deficiencies, and, most importantly, the competing entities are no longer mitochondria, but individual mtDNA molecules. For the outcome of this competition only the mtDNA replication times are important, and the longer mitochondrial division time becomes irrelevant. Indeed, using computer simulations it could be shown that this “survival of the tiny” idea can very well reproduce experimental findings regarding the distribution of deletion sizes in rat muscle fibers (31).
Thus, we propose that mitochondrial fusion is the underlying mechanism that opens the door for the clonal expansion of mitochondrial deletion mutants.
Combating the Accumulation of Mitochondrial Mutants
The replacement of WT mitochondria by mutants does sooner or later impair the functional status of the cell, because important functions, such as ATP production, are diminished. It is therefore of great importance for the cell to develop mechanisms to combat this accumulation. Recent results show that mitochondrial fission might just be this mechanism. Twig et al. (14) demonstrated that mitochondrial fragments often show unequal membrane potential after fission and that fragments with lowered potential are more likely to undergo mitophagy. The authors propose that a random segregation of mtDNAs during the fission process can lead to a separation of defective mtDNAs, which are then preferentially degraded (32). The efficiency of this mechanism would be increased by the observation that mitochondrial fragments can fuse with the network only if they have a normal membrane potential (14).
This is an interesting idea, but it works only with certain additional assumptions. As mentioned earlier, selection can act only if there is a link between genotype and phenotype. If mtDNAs freely diffuse within the mitochondrial network without connection to their gene products, the proposed mechanism cannot work because mitochondrial fragments with the selected phenotype (diminished proton gradient) will not necessarily contain the corresponding genotype (mutant mtDNA).
How can such a link be created despite the mixing caused by the fusion events? It is known that the mtDNA is attached to the inner mitochondrial membrane and we propose that it is actually connected to respiration complexes that are encoded by this specific molecule of mtDNA.
In prokaryotes, transcription and translation are closely linked, so that the proteins are synthesized in close proximity to the encoding genes. Mitochondria are derived from bacteria and also have a close connection between transcription and translation. Proteins of the OXPHOS complexes might therefore be synthesized and integrated into the membrane close to the encoding mtDNA. In prokaryotes, such a connection between translation and membrane insertion is known as transertion (33) and studies show that the same type of coupling probably also occurs in yeast mitochondria (34). This view is also supported by the observation that mtRNA remains localized at the nucleoids where it was synthesized (35). Furthermore, recent evidence indicates that the respiration complexes are not found as monomers, but are organized in supercomplexes (36–38) and even respiratory strings (39). Such an arrangement not only speeds up the enzyme kinetics by substrate channeling, but also connects mtDNA to its gene products. It has been shown that supercomplexes contain members of all respiratory complexes encoded by mtDNA, i.e., I, III, and IV (37, 38). Thus, if mtDNA is attached to one of the complexes, a link to all of them exists. And indeed, there is experimental evidence that proteins of respiration complex I are connected to nucleoids (40). Furthermore, studies using fluorescence recovery after photobleaching (FRAP) of mitochondrial protein mobility show not only that OXPHOS complexes have a very low diffusion constant, but also that ∼30% of these complexes are not mobile at all (41).
The proposed link between genotype and phenotype is, of course, not as tight as the classical connection (DNA inside cell), because proteins might diffuse away before or after they are inserted into the membrane. To investigate whether selection might also work for a leaky link between genotype and phenotype we developed a mathematical model.
Modeling of Leaky Link Between Genotype and Phenotype.
We study the competition between wild-type DNA (wt) and mutant DNA (mt) by setting up a system of two coupled differential equations. We assume that wild-type and mutant DNA grow exponentially (limited by the carrying capacity K) and are degraded depending on their degradation rates and leakiness L. For L = 0, there is no leak and the DNAs are linked only to their own gene products, resulting in pure degradation rates d1 and d2. A leakiness of L = 0.5 means that there is random mixing of proteins and DNA and consequently mutant and wild type have the same effective degradation rate given by the mean of d1 and d2. The idea of a carrying capacity is derived from population models. It reflects limiting resources and leads to a logistic growth pattern. We assume that the growth rate of mutant is higher than that of the wild type, c2 > c1, and that the cell counteracts this by also keeping the degradation rate of mutant higher than that of wild type, d2 > d1:
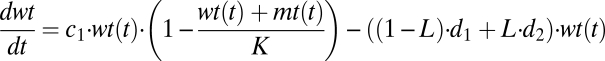 |
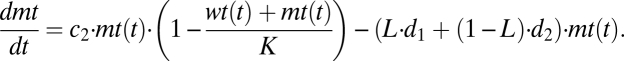 |
With the substitutions
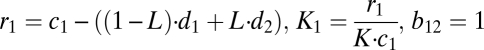 |
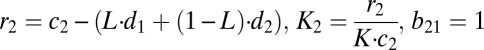 |
we obtain the expressions
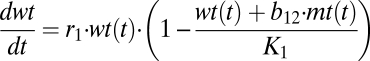 |
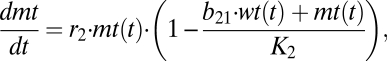 |
which correspond exactly to equations 3.29 and 3.30 of the book Mathematical Biology by J. D. Murray (42); i.e., the analysis given by Murray holds one-to-one for our case. Because in our case b12 = b21 = 1, the system always has only one globally stable steady state. That single steady state means either the wild-type or the mutant DNA survives, irrespective of the starting concentrations. If K2/K1 < 1, then the wild type prevails and if K1/K2 < 1, then the mutant form dominates.
Increasing leakiness L increases the quotient K2/K1 and eventually leads to the domination of the mutant form. In principle, the cell can compensate for the detrimental effects of leakiness by selectively increasing the degradation rate, d2, of the mutant. Fig. 2 shows which combinations of leakiness, L, and degradation ratio, d2/d1, lead to the survival of wild type or mutant. Even for a leaky genotype/phenotype link (L > 0) it is possible to achieve survival of the wild type. However, the larger the degree of leakiness is, the stronger has to be the selection pressure (in the form of the degradation ratio, d2/d1) until finally at a critical value of L = c1/(c1 + c2), compensation is no longer possible.
Fig. 2.

Stability diagram showing for which combinations of leakiness, L, and the ratio of mutant versus wild-type degradation rate, d2/d1, wild-type respectively mutant DNA survives. Parameter values are c1 = 1.5, c2 = 2.
Selection against mtDNA mutants absolutely requires a connection between genotype and phenotype and our proposed mechanism provides such a link. At the same time it explains why mtDNA has to be attached to the mitochondrial membrane, a place of intensive ROS production and hence a place most dangerous for mtDNA.
Random Drift
Whereas we propose that the accumulation of mitochondrial deletion mutants is driven by a replicative selection advantage, it has been proposed that a pure random drift can also explain the clonal expansion of mitochondrial DNA mutations (29, 43, 44). The authors performed computer simulations in which they followed the fate of mtDNA molecules for 120 in silico years assuming a half-life of 10 d and different mutation rates (29). They found that after 80 y the fraction of COX-negative cells was comparable with what is seen experimentally in humans (assuming that cells with >60% mutant DNA are COX-negative and a mutation rate of 10−5 per replication). They also showed that most of these cells harbor only one type of mutant if the mutation rate is small enough. Higher mutation rates, however, led to a mixture of different mutants per cell, which would not be in agreement with observations. This result is an important feature of the drift hypothesis that can be used to differentiate it from theories that assume a selection advantage for mtDNA mutants.
Drift requires a long time from the emergence of a mutation until it reaches the threshold to cause COX deficiency. Applied to short-lived animals (e.g., rats) it would require a much higher mutation rate to achieve the same fraction of COX-negative cells as in humans after 3 instead of 80 y. However, as the simulations have shown, this outcome would also mean a much higher degree of heteroplasmy. This result, however, is inconsistent with studies showing that, as in humans, aged rat cells tended to contain only a single mitochondrial deletion mutant (12).
Whereas it seems that the drift hypothesis is not applicable for deletion mutants, the situation might be different for point mutations. Having the same size as wild type, the latter would not enjoy a replicative advantage. Any progressive increase in their frequency is therefore more likely to follow a random drift. The accumulation of point mutations and deletions is therefore most likely be caused by two separate mechanisms, as has also been noted by Durham et al. (44). The drift hypothesis predicts that the degree of heteroplasmy is inversely proportional to species life span. It would therefore be interesting to determine the heteroplasmy of point mutations in such different species as mouse and human.
Plant, Fungi, and Protist Mitochondria
Once damage to the mitochondrial DNA has happened, there are two principal ways to deal with it. The first is to remove the damage and this seems to be the option that is used in the animal kingdom. Mitochondrial fission creates small fragments. If by chance such a fragment is stochastically enriched in a defective membrane potential, it can be preferentially degraded. Because of the link between genotype and phenotype, this also removes preferentially the damaged mtDNA. However, an alternative option is to repair the damage. This option could be possible because there are many mtDNA molecules that all carry the same information. A section of mtDNA sequence that is lost by deletion could in principle be repaired using the information of other, intact mtDNA molecules. We propose that this option is used by plants, fungi, and protists. It is known that the DNA of plant mitochondria consists of a complex mixture of circular molecules of different sizes that seem to undergo frequent recombination events (45, 46). If this is true, it has important consequences. Because deletion mutants are repaired, there is probably no danger that smaller, but functionally deficient mtDNA molecules will accumulate with age. However, a definitive answer might depend on a detailed understanding of the system of recombination events. Because there is no need for preferential degradation, it also means that no connection between genotype and phenotype is required. In summary, this result means that although the mitochondria of these species also need fusion to equilibrate their proteins, they have found a very different way to prevent the accumulation of deletion mutants with age. Thus, large parts of the above-developed theory apply only to animal mitochondria.
Observations and Predictions
An interesting feature of mitochondrial genome organization is that mtDNA molecules are organized in nucleoids within the mitochondria, with ∼5–10 mtDNAs per nucleoid (47). It has been shown that nucleoids hardly ever exchange mtDNAs (48), which means that accumulating mutant mtDNA molecules cluster in nucleoids. If mitochondrial fission is accompanied by a random segregation of mtDNAs, the chance of producing a fragment with mainly damaged (or WT) mtDNAs will be greater if the genomes are already organized in clusters that do not exchange members. This outcome is simply because the clustering of mutant molecules means that the variance between fission fragments in their burdens of mutant mtDNA molecules will be greater than if the mtDNA molecules segregated individually. Margineantu et al. (49) used FISH to study mitochondrial morphology and found that nucleoids are distributed at more or less regular spatial intervals within the mitochondria and similar results have also been found by others (35, 50, 51). If, as proposed, translation and membrane insertion are coupled in mitochondria, then this naturally leads to a uniform distribution of mtDNAs. Around each nucleoid there is a patch of membrane into which new proteins are incorporated that are encoded by the mtDNAs of the nucleoids. This process automatically pushes the individual nucleoids away from each other, resulting in a regular distribution.
A central point of our hypothesis is the proposed attachment of nucleoids to their own gene products (respiration complexes) in the inner mitochondrial membrane. It should be possible to test this prediction experimentally by using cells that contain two different types of mtDNA that code for protein variants that can be differentiated via monoclonal antibodies. Imaging experiments using those antibodies together with FISH should reveal whether the gene products of the different mtDNAs colocalize, as we predict, with the corresponding mtDNA.
The (leaky) link between genotype and phenotype also has implications for the structure of mtDNA. This link can be established only between the mtDNA and membrane proteins, but not with diffusible matrix proteins. In agreement with this requirement the human mitochondrial genome (and other animal genomes) codes only for membrane proteins. We suggest that several converging selection forces have acted on the evolving mitochondrial genome. First, there was the need to transfer control from the mitochondria to the nucleus, as discussed earlier. Second, there was the need for the (leaky) link that required removal of genes encoding nonmembrane proteins from the mtDNA. Third, there will have been selection to reduce mtDNA size by competition between functional mtDNA molecules. As a consequence animal mtDNA is extremely compact as exemplified by the lack of introns, noncoding regions between genes, the small size of tRNA and rRNA genes, and several other features. The advantage might be small, but this selection pressure will have acted in the same direction for the last 2000 million years. This size competition of mitochondrial genomes on an evolutionary timescale is different from the size competition of deletion mutants during the life span of an organism, but it supports the idea that a reduced size does indeed confer a selection advantage to mitochondrial genomes.
In the previous section we proposed that plants, fungi, and protists repair mitochondrial deletion mutants instead of removing them. This repair leads to different consequences for the mitochondrial structure than in the case of animals. As explained earlier, no link between genotype and phenotype is required and hence it is no problem if matrix proteins are encoded by the mtDNA. Also, deletions in the germ line do not generate the same selection pressure as in animals, because they can be reversed by recombination events. Thus, among the three sources of selection pressure toward a smaller genome that we identified for animal mtDNA, only one applies to plants, namely, the advantage that is gained from transferring organelle genes to the nucleus, thus minimizing the potential for conflict. Consequently, we would expect that the mitochondrial genomes of plants, fungi, and protists will have lost most of their genes during evolution, but that they might be larger than the mitochondrial genome of animals, which is exactly what is seen. The mitochondrial genome of Arabidopsis thaliana, for instance, is 367 kbp long, contains 23 introns, and codes for several nonmembrane proteins in the form of ribosomal proteins (52). Similarly, also the genomes of Tetrahymena and Paramecium are quite large (>40 kbp) and contain nonmembrane proteins (53).
Our proposal for how the cell combats the accumulation of mtDNA mutants integrates the suggestion of Twig et al. (14, 32) with the leaky link idea. Fission generates fragments, some of which are enriched in mtDNA mutants that are then preferentially degraded. Fission effectively breaks up the mitochondrial network into patches of membrane (containing nucleoids, proteins, and lipids) . This process has the potential to separate proteins from the nucleoids, where they have been synthesized. This separation would be no problem as long as the resulting fragments are always degraded. However, this is not the case. If the fragments are energetically intact or too large, they are not degraded, but will fuse again with the network. This outcome means proteins can be separated from their nucleoids and transported to another place in the network, thus increasing the leakiness of the proposed link between genotype and phenotype. Is there a way for the cell to avoid this mixing? This process seems to be a difficult feat, but another surprising observation made by Twig et al. (14) could provide the solution. They showed that fusion and fission are connected, in the sense that fission occurs at the same location where an earlier fusion event happened. Although the mechanism is as yet unclear, it seems that fragments can “remember” to a certain degree the borderline of the membrane patch that was inserted during fusion and then remove the membrane patch exactly along this border during fission. This is exactly the kind of mechanism needed to avoid the mixing that might otherwise be caused by fusion and fission.
Finally, the arguments that we put forward, to explain why it is necessary for mitochondria to equilibrate the concentrations of all nuclear-encoded mitochondrial proteins, are equally true for chloroplasts. Our hypothesis thus predicts that chloroplasts also have to exchange proteins to improve cellular efficiency and control. Although no fusion of chloroplasts has been observed to date, it has been known for a long time that chloroplasts form long thin tubules, called stromules, that can connect to other chloroplasts (54, 55). In 1997 Koehler et al. (56) labeled plastid stroma of tobacco and petunia plants using green fluorescent protein (GFP) and observed a flow of GFP between connected plastids after photobleaching. These findings were later also observed by others (56, 57). Thus, as predicted and required by our hypothesis, chloroplasts equilibrate their proteins exactly like mitochondria, but using a different mechanism.
Summary
Mitochondria are organelles that provide the cell with energy and that are causally involved in several diseases and probably also the aging process. Important mitochondrial processes that emerged over the last years are the clonal accumulation of mitochondrial mutants and the dynamic character of these organelles, involving frequent fusion and fission events. The mechanistic basis of the first and the origin and function of the second process are as yet poorly understood. Here we present a hypothesis that provides an evolutionary explanation for mitochondrial fusion and suggests that it is causally involved in the accumulation of mitochondrial mutants during animal aging. Specifically we propose that
i) mitochondrial fusion is necessary to control hundreds of mitochondria in an optimal way with only a single set of mitochondrial genes that are located in the nucleus;
ii) the fusion process opens the door for the accumulation of mitochondrial deletion mutants in animal cells, which have a selection advantage on the basis of their small size;
iii) mitochondrial fission together with selective degradation or delayed fusion of damaged mitochondria is then necessary to counteract this accumulation;
iv) a link between genotype and phenotype is necessary for this strategy to work and this link is provided by nucleoids being attached to OXPHOS complexes located in the inner mitochondrial membrane; and
v) plants, fungi, and protists repair mitochondrial deletion mutants instead of removing them and thus points ii–iv apply only to animal cells.
Surprisingly few assumptions are necessary for our hypothesis and these are supported either by mathematical analysis (equilibrating the effect of fusion events, selection efficiency even with leaky genotype/phenotype link) or by recent experimental findings (OXPHOS supercomplexes, transertion in bacteria and yeast). Our idea also provides an explanation for the extremely compact size of mitochondrial DNA and for the structural differences seen between plant and animal mtDNA.
The strongest prediction from our hypothesis is that exchange of nuclear-encoded proteins should occur not only in mitochondria, but also in chloroplasts. And indeed such a phenomenon has already been observed experimentally. Chloroplasts do not fuse like mitochondria, but they do produce long tubules through which proteins are quickly exchanged. Because these tubules appear not to allow the exchange of chloroplast DNA (at least none was observed), this solution for protein exchange prevents the spread of cpDNA mutants. As already mentioned by others (56), the tubules resemble bacterial pili and thus it is possible that the machinery for the exchange of genetic material in bacteria served as an evolutionary basis for the machinery to equilibrate protein levels between chloroplasts and maybe also mitochondria. Thus, although prokaryotes are not known to fuse, they might have provided the molecular mechanisms that evolved into the fusion machinery.
Our “organelle control” theory connects many important mitochondrial processes within a common mechanistic framework and provides an explanation for the evolution of these processes. Further critical testing of the hypothesis will and has to be aided by the development of detailed spatially explicit models, in conjunction with experimental tests. We strongly believe that the presented idea will help in understanding key processes involved in the evolution of the mitochondrial genome and the aging process.
Acknowledgments
We thank Neil Blackstone for interesting discussions and Heinz Osiewacz for pointing us to relevant literature. T.B.L.K. is supported by the Biotechnology and Biological Sciences Research Council Centre for Integrated Systems Biology of Aging and Nutrition and by the United Kingdom National Institute for Health Research (NIHR) Biomedical Research Centre for Aging and Age-Related Disease award to the Newcastle upon Tyne Foundation Hospitals National Health Service (NHS) Trust. This work was partly funded by a Glenn Foundation award to T.B.L.K.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.W.G. is a guest editor invited by the Editorial Board.
References
- 1.Margulis L. Symbiosis in Cell Evolution: Life and Its Environment on the Early Earth. San Francisco: Freeman; 1981. p. 419. [Google Scholar]
- 2.Schwartz RM, Dayhoff MO. Origins of prokaryotes, eukaryotes, mitochondria, and chloroplasts. Science. 1978;199:395–403. doi: 10.1126/science.202030. [DOI] [PubMed] [Google Scholar]
- 3.Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 4.von Heijne G. Why mitochondria need a genome. FEBS Lett. 1986;198:1–4. doi: 10.1016/0014-5793(86)81172-3. [DOI] [PubMed] [Google Scholar]
- 5.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 6.Harman D. The biologic clock: The mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 7.Harman D. Free radical theory of aging: Consequences of mitochondrial aging. Age (Omaha) 1983;6:86–94. [Google Scholar]
- 8.Miquel J. An integrated theory of aging as the result of mitochondrial-DNA mutation in differentiated cells. Arch Gerontol Geriatr. 1991;12:99–117. doi: 10.1016/0167-4943(91)90022-i. [DOI] [PubMed] [Google Scholar]
- 9.Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 10.Miquel J, Economos AC, Fleming J, Johnson JE., Jr Mitochondrial role in cell aging. Exp Gerontol. 1980;15:575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- 11.Khrapko K, et al. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–2441. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao Z, Wanagat J, McKiernan SH, Aiken JM. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: Analysis by laser-capture microdissection. Nucleic Acids Res. 2001;29:4502–4508. doi: 10.1093/nar/29.21.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duvezin-Caubet S, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- 14.Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skulachev VP. Power transmission along biological membranes. J Membr Biol. 1990;114:97–112. doi: 10.1007/BF01869092. [DOI] [PubMed] [Google Scholar]
- 16.Blackstone NW, Kirkwood TBL. In: Mitochondria and Programmed Cell Death. “Slave Revolt” or Community Homeostasis? Genetic and Cultural Evolution of Cooperation. Hammerstein P, editor. Cambridge, MA: MIT Press; 2003. [Google Scholar]
- 17.Allen JF. Control of gene expression by redox potential and the requirement for chloroplast and mitochondrial genomes. J Theor Biol. 1993;165:609–631. doi: 10.1006/jtbi.1993.1210. [DOI] [PubMed] [Google Scholar]
- 18.Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: Implications for carcinogenesis and aging? Free Radic Biol Med. 1990;8:523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- 19.Arnheim N, Cortopassi G. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutat Res. 1992;275:157–167. doi: 10.1016/0921-8734(92)90020-p. [DOI] [PubMed] [Google Scholar]
- 20.de Grey ADNJ. A proposed refinement of the mitochondrial free radical theory of aging. BioEssays. 1997;19:161–166. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- 21.Kowald A, Kirkwood TB. Accumulation of defective mitochondria through delayed degradation of damaged organelles and its possible role in the ageing of post-mitotic and dividing cells. J Theor Biol. 2000;202:145–160. doi: 10.1006/jtbi.1999.1046. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi J, et al. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc Natl Acad Sci USA. 1991;88:10614–10618. doi: 10.1073/pnas.88.23.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallace DC. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science. 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 24.Takai D, Isobe K, Hayashi J-I. Transcomplementation between different types of respiration-deficient mitochondria with different pathogenic mutant mitochondrial DNAs. J Biol Chem. 1999;274:11199–11202. doi: 10.1074/jbc.274.16.11199. [DOI] [PubMed] [Google Scholar]
- 25.Davis AF, Clayton DA. In situ localization of mitochondrial DNA replication in intact mammalian cells. J Cell Biol. 1996;135:883–893. doi: 10.1083/jcb.135.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korr H, Kurz C, Seidler TO, Sommer D, Schmitz C. Mitochondrial DNA synthesis studied autoradiographically in various cell types in vivo. Braz J Med Biol Res. 1998;31:289–298. doi: 10.1590/s0100-879x1998000200012. [DOI] [PubMed] [Google Scholar]
- 27.Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–2429. [PubMed] [Google Scholar]
- 28.Huemer RP, Lee KD, Reeves AE, Bickert C. Mitochondrial studies in senescent mice. II. Specific activity, buoyant density, and turnover of mitochondrial DNA. Exp Gerontol. 1971;6:327–334. doi: 10.1016/0531-5565(71)90001-5. [DOI] [PubMed] [Google Scholar]
- 29.Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68:802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Grey ADNJ. The Mitochondrial Free Radical Theory of Aging. Austin, TX: R. G. Landes; 1999. [Google Scholar]
- 31.Kowald A, Jendrach M, Pohl S, Bereiter-Hahn J, Hammerstein P. On the relevance of mitochondrial fusions for the accumulation of mitochondrial deletion mutants: A modelling study. Aging Cell. 2005;4:273–283. doi: 10.1111/j.1474-9726.2005.00169.x. [DOI] [PubMed] [Google Scholar]
- 32.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woldringh CL. The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Mol Microbiol. 2002;45:17–29. doi: 10.1046/j.1365-2958.2002.02993.x. [DOI] [PubMed] [Google Scholar]
- 34.Bryan AC, Rodeheffer MS, Wearn CM, Shadel GS. Sls1p is a membrane-bound regulator of transcription-coupled processes involved in Saccharomyces cerevisiae mitochondrial gene expression. Genetics. 2002;160:75–82. doi: 10.1093/genetics/160.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iborra FJ, Kimura H, Cook PR. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004;2:9. doi: 10.1186/1741-7007-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schäfer E, et al. Architecture of active mammalian respiratory chain supercomplexes. J Biol Chem. 2006;281:15370–15375. doi: 10.1074/jbc.M513525200. [DOI] [PubMed] [Google Scholar]
- 37.Seelert H, et al. From protons to OXPHOS supercomplexes and Alzheimer's disease: Structure-dynamics-function relationships of energy-transducing membranes. Biochim Biophys Acta. 2009;1787:657–671. doi: 10.1016/j.bbabio.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 38.Frenzel M, Rommelspacher H, Sugawa MD, Dencher NA. Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex. Exp Gerontol. 2010;45:563–572. doi: 10.1016/j.exger.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Wittig I, Schägger H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta. 2009;1787:672–680. doi: 10.1016/j.bbabio.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Bogenhagen DF. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J Biol Chem. 2006;281:25791–25802. doi: 10.1074/jbc.M604501200. [DOI] [PubMed] [Google Scholar]
- 41.Sukhorukov VM, et al. Determination of protein mobility in mitochondrial membranes of living cells. Biochim Biophys Acta. 2010;1798:2022–2032. doi: 10.1016/j.bbamem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 42.Murray JD. Mathematical Biology. 2nd Ed. Heidelberg: Springer; 1993. [Google Scholar]
- 43.Chinnery PF, Samuels DC. Relaxed replication of mtDNA: A model with implications for the expression of disease. Am J Hum Genet. 1999;64:1158–1165. doi: 10.1086/302311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durham SE, Samuels DC, Chinnery PF. Is selection required for the accumulation of somatic mitochondrial DNA mutations in post-mitotic cells? Neuromuscul Disord. 2006;16:381–386. doi: 10.1016/j.nmd.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Atlan A, Couvet D. A model simulating the dynamics of plant mitochondrial genomes. Genetics. 1993;135:213–222. doi: 10.1093/genetics/135.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Albert B, Godelle B, Atlan A, De Paepe R, Gouyon PH. Dynamics of plant mitochondrial genome: Model of a three-level selection process. Genetics. 1996;144:369–382. doi: 10.1093/genetics/144.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clay Montier LL, Deng JJ, Bai Y. Number matters: Control of mammalian mitochondrial DNA copy number. J Genet Genomics. 2009;36:125–131. doi: 10.1016/S1673-8527(08)60099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilkerson RW, Schon EA, Hernandez E, Davidson MM. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J Cell Biol. 2008;181:1117–1128. doi: 10.1083/jcb.200712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Margineantu DH, et al. Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion. 2002;1:425–435. doi: 10.1016/s1567-7249(02)00006-5. [DOI] [PubMed] [Google Scholar]
- 50.Kang D, Hamasaki N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: Overview of its multiple roles. Ann N Y Acad Sci. 2005;1042:101–108. doi: 10.1196/annals.1338.010. [DOI] [PubMed] [Google Scholar]
- 51.Legros F, Malka F, Frachon P, Lombès A, Rojo M. Organization and dynamics of human mitochondrial DNA. J Cell Sci. 2004;117:2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- 52.Unseld M, Marienfeld JR, Brandt P, Brennicke A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat Genet. 1997;15:57–61. doi: 10.1038/ng0197-57. [DOI] [PubMed] [Google Scholar]
- 53.Burger G, et al. Complete sequence of the mitochondrial genome of Tetrahymena pyriformis and comparison with Paramecium aurelia mitochondrial DNA. J Mol Biol. 2000;297:365–380. doi: 10.1006/jmbi.2000.3529. [DOI] [PubMed] [Google Scholar]
- 54.Wildman SG, Hongladarom T, Honda SI. Chloroplasts and mitochondria in living plant cells: Cinephotomicrographic studies. Science. 1962;138:434–436. doi: 10.1126/science.138.3538.434. [DOI] [PubMed] [Google Scholar]
- 55.Weier TE, Thomson WW. The grana of starch-free chloroplasts of Nicotiana rustica. J Cell Biol. 1962;13:89–108. doi: 10.1083/jcb.13.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Köhler RH, Cao J, Zipfel WR, Webb WW, Hanson MR. Exchange of protein molecules through connections between higher plant plastids. Science. 1997;276:2039–2042. doi: 10.1126/science.276.5321.2039. [DOI] [PubMed] [Google Scholar]
- 57.Kwok EY, Hanson MR. In vivo analysis of interactions between GFP-labeled microfilaments and plastid stromules. BMC Plant Biol. 2004;4:2. doi: 10.1186/1471-2229-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]