

Abstract
Ligand-gated channels, in which a substrate transport pathway is formed as a result of the binding of a small-molecule chemical messenger, constitute a diverse class of membrane proteins with important functions in prokaryotic and eukaryotic organisms. Despite their widespread nature, no ligand-gated channels have yet been found within the outer membrane (OM) of Gram-negative bacteria. Here we show, using in vivo transport assays, intrinsic tryptophan fluorescence and X-ray crystallography, that high-affinity (submicromolar) substrate binding to the OM long-chain fatty acid transporter FadL from Escherichia coli causes conformational changes in the N terminus that open up a channel for substrate diffusion. The OM long-chain fatty acid transporter FadL from E. coli is a unique paradigm for OM diffusion-driven transport, in which ligand gating within a β-barrel membrane protein is a prerequisite for channel formation.
The outer membrane (OM) of Gram-negative bacteria is a very efficient permeability barrier and therefore contains proteins that form channels for the uptake of small molecules required for cell growth and function. Virtually all OM channels form β-barrels that are stable under the harsh conditions frequently encountered on the outside of the cells. Most OM channels, such as general porins and substrate-specific channels, mediate small-molecule substrate uptake via passive diffusion (1). These proteins either do not bind their substrates (e.g., porins) or, in the case of substrate-specific channels, they bind their substrates with only low affinities (submillimolar to millimolar). Originally, members of these two classes of channels were thought not to undergo conformational changes during transport (1). Recent studies have shown, however, that OM diffusion channels can mediate complex transport processes in which the channels are not necessarily static pores (2–4). Despite these advances, OM ligand-gated channels, in which binding of a small molecule opens up a transport channel, have not yet been described. The closest known examples of ligand-gated channels in the OM are the TonB-dependent receptors (TBDRs). These are active transporters that mediate the uptake of relatively large (> 600 Da) hydrophilic solutes such as vitamin B12 and iron siderophores. Members of this class of channels, exemplified by Escherichia coli FhuA and FepA, have been characterized in detail both biochemically and structurally. TBDRs form large, 22-stranded β-barrels with an N-terminal plug domain that occludes the lumen of the barrel. Upon high-affinity ligand (nanomolar) binding on the extracellular side of the cell, conformational changes occur that are transmitted to the TonB box, a short N-terminal sequence within the TBDR that becomes exposed to the periplasmic space. Thus, ligand binding in TBDRs generates conformational changes within the receptor, but these do not lead to an open channel. Only after interaction with the inner membrane protein TonB and utilizing the proton motive force via a mechanism that is still unclear, a transport channel is formed in TBDRs (5, 6). All of the above-mentioned OM proteins mediate the transport of hydrophilic molecules.
The long-chain fatty acid (LCFA) transporter FadL from E. coli (EcFadL) is the archetypal member of a large family of OM proteins mediating the passive diffusion of hydrophobic molecules across the OM, including xenobiotics destined for biodegradation (7, 8). EcFadL has an N-terminal “plug” domain in the lumen of the barrel, and an opening in the barrel wall due to an inward-pointing kink in one of the β-strands (9). We have recently shown that substrate uptake by EcFadL occurs via lateral diffusion (10). The defining feature of this mechanism is that the transport pathway is not across the membrane like in other membrane transporters; instead, the hydrophobic substrates diffuse from the barrel lumen laterally into the outer leaflet of the OM via the opening in the barrel wall (10). Crystal structures suggest that EcFadL has a low-affinity binding site between two loops on the extracellular surface of the protein, and a high-affinity site in the lumen of the barrel, close to the N terminus (9). The substrate binding sites are part of a long hydrophobic channel that leads from the extracellular surface to the opening in the barrel wall. However, the N-terminal three residues of the plug domain, most notably F3, interrupt the channel and block access of the substrate to the lateral opening (Fig. 1). The closed channel is stabilized by an interaction between the α-amino group and the carboxyl group of the conserved residue D348 (9) (Fig. 1). The structural data therefore suggest that the N terminus has to move in order to open the channel.
Fig. 1.

The N terminus of FadL is connected to the high-affinity binding site. (A) Stereo cartoon viewed from the side, showing the locations of F3, W298, and D348 within the wild-type EcFadL structure (Protein Data Bank ID 1T16). The N terminus (residues 1–3) is colored red, with the rest of the plug domain colored pink. The LCFA-mimicking LDAO detergent molecule bound in the high-affinity binding site is colored dark green (nitrogens blue, oxygens red). The lateral opening in the barrel wall is indicated with an orange asterisk. Residues 405–411 in strand S14 have been removed to show the LDAO detergent molecule more clearly. The extracellular side is at the top of the figure. (B) Overview showing the complete EcFadL structure for reference, with the hydrophobic core of the OM indicated as horizontal lines. This and the following figures were made with PyMOL (14).
Results and Discussion
The N terminus Is Important for LCFA Transport.
To test the role of the N terminus in LCFA transport we made two N-terminal EcFadL plug truncation mutants, in which three (ΔN3) and eight (ΔN8) N-terminal residues are removed (SI Text). Surprisingly, the in vivo oleate transport activities of both N-terminal plug truncation mutant proteins show drastically impaired LCFA transport (Fig. 2). Because removal of the N terminus should generate an open channel for LCFA diffusion, the low transport rates imply that LCFA binding is impaired in the truncation mutants. This conclusion makes sense considering that F3, absent in both truncation mutants, is part of the high-affinity LCFA binding site (9) (Fig. 1). The three N-terminal (plug) residues are conserved as [small]-[small]-[large hydrophobic] in FadL channels, suggesting that the presence of a large hydrophobic residue at position 3 is functionally important. We tested this notion with the less drastic, N-terminal point mutants F3E and F3G, and found that these proteins are also characterized by very low oleate uptake activities (Fig. 2). Together these results show that the N terminus, especially F3, plays an important role in LCFA transport.
Fig. 2.

Oleate transport and binding by FadL proteins. (A) In vivo [3H]-oleate transport activities of wild-type (gray bar) and mutant EcFadL proteins, grouped by color (N-terminal mutants, red; 0Trp and single-Trp mutants, cyan). The weighted average and sigma values of moles oleate transported/(nanogram × min ) are reported as % wild-type, and obtained as previously reported (7). The zero-Trp mutant is labeled 0Trp-F0W for consistency. The gray vertical line marks 100% wild-type activity. Palmitate plate growth is graded in order “-” (no growth), “+,” “++,” and “++ +” (wild-type). ND, not determined. For more information, see Figs. S6–S9. (B and C) In vitro oleate and LDAO binding to EcFadL mutants monitored by intrinsic fluorescence. Green, 0Trp-F298W (oleate); magenta, 0Trp-F298W (LDAO); blue, 0Trp-F298W-ΔN3 (oleate); red, 0Trp-F0W (oleate). (B) Normalized fluorescence emission intensity at 319 nm vs. oleate concentration. (C) Peak emission wavelength vs. oleate/LDAO concentration. A quadratic fit is plotted for oleate titration to 0Trp-F298W in B and C. Note the extended concentration range for the LDAO titration. For oleate it is not possible to perform titrations > 10 μM due to Rayleigh scattering effects.
Structural Characterization of N-Terminal EcFadL Mutants.
As the next step, we determined structures of the N-terminal mutant proteins by X-ray crystallography (Fig. 3 and Table S1). Strikingly, although the membrane-embedded parts of the ΔN3 and ΔN8 (Fig. S1) mutant structures are very similar to wild-type EcFadL (Fig. 3), the mutants lack electron density for residues 235–267 in loop L4 and residues 299–324 in loop L5. In addition, no detergent molecules are visible in the low- and high-affinity binding sites (Fig. 3), which makes sense considering that a number of hydrophobic residues in loops L4 and L5 are part of the LCFA binding sites (9). Our interpretation of the structural data is that the removal of F3 destabilizes the LCFA binding site, leading to increased flexibility of loops L4 and L5. Besides having disordered substrate binding sites, the ΔN3 and ΔN8 mutant structures also have open channels due to the truncation of the N terminus (Fig. 4). Thus, dramatically decreased LCFA binding is the likely reason for the low transport activities of the N-terminal truncation mutants. The crystal structure of the F3E point mutant shows that its LCFA binding sites are also disordered, indicating that substitution of F3 for a negatively charged amino acid is also detrimental to LCFA binding. It should be noted that electron density for the L4/L5 loops is absent/partially present only in mutants that are severely affected in the N terminus (ΔN3, ΔN8, F3E). In addition, the crystal packing in the N-terminal mutants suggests that neighboring molecules are close enough to provide lattice contacts with the L4/L5 loops (Fig. S2). These two arguments suggest that the observed disorder in the loops is not due to a lack of crystal lattice contacts, but a direct consequence of the mutations within the N terminus. The structure of the final, most conservative point mutant F3G is similar to wild-type EcFadL, with density present for all loops and a detergent molecule bound in the high-affinity site (Fig. 3). However, this detergent molecule has moved compared to that bound in the wild-type protein, occupying a position where the side chain of F3 would be (Fig. 3). Thus, the absence of the F3 side chain has resulted in the partial release of the detergent from its binding site, in accordance with our notion based on the more severe mutants. The N terminus in the F3G mutant, like that in wild-type FadL, still prevents access of a substrate to the lateral opening (Fig. 4), demonstrating that the removal of the F3 side chain alone is not sufficient to alleviate the blockage of the channel by the N terminus.
Fig. 3.

Effect of N-terminal mutations on the structure of EcFadL. Cartoons viewed from the side, shown in identical orientations, for closed wild-type EcFadL [Protein Data Bank (PDB) ID 1T16; ref. 6] and the mutants ΔN3, F3E, F3G, and D348R. In addition, the structure of a putative open wild-type channel is shown (PDB ID 1T1L; ref. 6). Loops L3–L5 are indicated in wild-type FadL. Detergent (green) bound in the low-affinity binding site is indicated with “L,” and that in the high-affinity binding site by “H.” Residue R348 in the D348R mutant is indicated with magenta sticks, with two conformations for the guanidinium group. The N terminus (residues G1–F3) is colored red, and W298 is colored blue. The dotted line in the wild-type protein corresponds to the center of the slabs through surfaces shown in Fig. 4.
Fig. 4.

The LCFA diffusion pathway in wild-type EcFadL and mutant proteins. Shown are surface slabs viewed from the extracellular side, showing the diffusion pathway from the high-affinity binding site to the lateral opening as a dark tube. In the closed wild-type channel and in the F3G mutant, the N terminus (red) blocks the diffusion pathway. In the ΔN3 mutant and in the open wild-type channel, the diffusion pathway (shown as a yellow arrow) is uninterrupted because of the removal (ΔN3) or conformational change of the N terminus. Detergent molecules bound in the high-affinity binding sites are shown as green stick models.
Residue D348 Constrains the N Terminus of EcFadL.
In order to obtain more information about the effect of the conformation and dynamics of the N terminus on LCFA transport, we made the D348A EcFadL mutant. D348 is conserved in FadL channels that have high LCFA transport activity, but not in FadL channels dedicated to monoaromatic hydrocarbon transport (11), suggestive of an LCFA-specific role. Within the closed channel, the D348 carboxylate interacts with the A1 α-amino group (Fig. 1), presumably stabilizing its conformation. Strikingly, the D348A EcFadL mutant has very low oleate transport activity and does not grow on palmitate plates (Fig. 2), supporting the importance of an interaction between D348 and the N terminus. Surprisingly, the crystal structure of D348A, including the conformation of the N terminus, is virtually identical to wild-type EcFadL (Fig. S3, Cα rmsd 0.35 Å). Apparently the conformation of the “unconstrained” N terminus favored under the crystallization conditions is similar to the “D348-constrained” N terminus in the wild-type protein. We therefore made a D348R mutant, with the goal of physically forcing the N terminus to move via electrostatic and steric clashes of the introduced arginine side chain with the α-amino group. As expected, LCFA transport in this mutant is abolished (Fig. 2). The crystal structure of the D348R mutant shows the same dramatic changes in loops L4/L5 as the ΔN3 and F3E mutants compared to wild-type EcFadL (Fig. 3). In all these mutants, the N terminus (including F3) is either absent (ΔN3) or has moved (F3E, D348R) as a consequence of the introduced changes. Although it is unlikely that the structures of these mutants represent intermediates in the transport cycle, we do infer that a movement of the N terminus will cause a disruption of the substrate binding sites.
High-Affinity Substrate Binding Triggers an N-Terminal Conformational Change.
The structural and transport data suggest an important functional link between the N terminus and the high-affinity LCFA binding site, mediated by the side chain of F3. More specifically, the data suggest that a conformational change in the N terminus leads to LCFA substrate release from the binding site. We hypothesized that binding of the substrate in the high-affinity site might trigger the N-terminal conformational change, resulting in the subsequent release of the substrate and further transport. In order to test this model, we developed an in vitro LCFA binding assay based on intrinsic tryptophan fluorescence (SI Text). We removed the 11 endogenous Trp residues in EcFadL (0Trp-F0W) and reintroduced a single-Trp residue as a reporter at position 298 (0Trp-F298W). Residue W298, which is endogenous to EcFadL and highly conserved in FadL channels (9), is close to the N terminus and contacts the aromatic ring of F3 (Fig. 1). Because W298 is not close to the detergent in the high-affinity binding site (closest distance is approximately 8 Å; Fig. 1), this residue is ideally positioned to report on a conformational change of the N terminus. The 0Trp-F0W and the 0Trp-F298W mutants are active in oleate uptake (Fig. 2A and SI Text), indicating that none of the tryptophan residues in EcFadL are required for transport. For the Trp-less control mutant (0Trp-F0W), titration with oleate generated a gradual, linear decrease in fluorescence intensity, with λmax remaining constant at approximately 326 nm (Fig. 2 and Fig. S4). Similar titrations were obtained for several other single-Trp mutants (SI Text). These results indicate that the 23 tyrosine residues, as well as Trp residues at various sites in EcFadL, are “inert” to oleate addition under the experimental conditions. By contrast, titration of the 0Trp-F298W mutant with oleate caused a clear, saturable increase in fluorescence intensity together with a blue shift for λmax (Fig. 2 an Fig. S4), indicating that W298 experiences a significant, specific change in local environment (12) and is dequenched with increasing oleate concentration (Fig. 2). A quadratic model function fit to the normalized intensity data produces an apparent Kd of 0.2 ± 0.1 μM for oleate binding by EcFadL. This affinity is very high for a diffusion-driven channel, confirming our previous prediction based on structural arguments (9). The fluorescence data for 0Trp-F298W support our hypothesis that substrate binding in the high-affinity site causes a conformational shift of the N terminus, and we propose that this shift opens the channel. The fluorescence data for 0Trp-F298W-ΔN3 support this notion. The removal of the N-terminal three residues in the 0Trp-F298W mutant protein abolishes oleate binding and therefore the substrate-induced conformational changes in the N terminus (Fig. 2). Together, the data suggest that EcFadL forms a ligand-gated channel, with an N-terminal gate that signals the presence of a ligand in the binding site to open the channel.
An interesting question is why lauryldimethylamine-oxide (LDAO) detergent is observed in the high-affinity binding site of wild-type FadL, given the fact that LDAO is only used at the initial stages of purification (Methods; ref. 9), and considering our notion that substrate binding at this site leads to a decreased affinity. One possible explanation is that LDAO binding does not trigger conformational changes—i.e., LDAO is not a transport substrate. It is not possible to test directly if detergents are transported by FadL for two reasons: (i) they are relatively polar and will enter E. coli via porins, and (ii) unlike LCFAs, detergents are not metabolized by E. coli and consequently there is no sink that drives transport to measurable levels (SI Results and Discussion). In order to ascertain whether LDAO functions like an LCFA substrate, we have measured LDAO binding in our in vitro binding assay using the 0Trp-F298W reporter protein. Interestingly, LDAO titration only causes small and gradual changes in the fluorescence spectra (Fig. 2), indicating that LDAO, unlike oleate, does not cause conformational changes in the N terminus. Thus, LDAO is likely to be an inhibitor.
The observed submicromolar affinity of EcFadL for oleate may have some interesting implications for substrate acquisition in vivo. For E. coli, the physiological fatty acid substrates within the intestine are most likely derived from dietary triglycerides. Those triglycerides are present in mixed micelles with bile salts and converted into glycerol and LCFAs by pancreatic lipase. The LCFA concentration in the mixed micelles is unknown, but undoubtedly much higher than the concentrations of free fatty acids, which are essentially insoluble in water (the aqueous solubility of palmitate is approximately 10 nM; ref. 13). Thus, substrate acquisition by EcFadL is likely to be very efficient within the gut due to the high affinity of the channel for the substrate.
A Model for Ligand-Gating in EcFadL.
Why does FadL-mediated LCFA transport require gating? At least in the case of EcFadL, and likely for other FadL channels as well, substrate binding of substrates occurs with high affinity. Tight binding requires a substrate release mechanism that, in the case of FadL channels, does not depend on external energy input. A conformational change in a channel-blocking N terminus that is physically linked (via F3) to the substrate binding site could provide such a mechanism. Within the closed state of the channel, the α-amino group interacts with D348 and the N-terminal gate blocks substrate access to the lateral opening (Fig. 5). Binding of LCFAs to the high-affinity site perturbs the side chain of F3 and causes a conformational change in the N terminus, promoting substrate release and generating an open channel that now allows diffusion of the substrate from the high-affinity binding site to the lateral opening in the β-barrel wall (10) (Fig. 5). The open state of the channel may resemble that present in the structure of wild-type EcFadL crystallized previously in a hexagonal space group (9) (Figs. 3 and 4). Within this latter structure, W298 is in a very different environment compared to the closed channel due to a conformational change in the N terminus (Fig. S5), supporting our intrinsic tryptophan fluorescence data.
Fig. 5.

Model for ligand-gated transport in EcFadL. In the closed state of the channel (Left), the α-amino group of the N terminus interacts with the carboxyl group of D348 and the side chain of F3 forms part of the high-affinity binding site. Substrate (green square) binding in the high-affinity site causes a conformational change of the N terminus. The resulting displacement of F3 from the binding site leads to substrate release and opens the channel (Right). The substrate subsequently moves through the lateral gap into the outer leaflet of the OM (7).
The structural similarity of FadL channels with widely different substrate specificities (11) suggests that the ligand-gating mechanism observed for EcFadL may be common to FadL channels. In the case of EcFadL, the ligand that opens the N-terminal gate is also the transported substrate. This feature is unusual for ligand-gated channels but not unprecedented, as exemplified by the Ca2+ release-activated Ca2+ channels of human T cells (14). EcFadL is an example of a ligand-gated diffusion channel within the bacterial OM, demonstrating that channels formed by β-barrels can display mechanistic intricacies that are more commonly observed in α-helical membrane proteins.
Methods
FadL Mutagenesis.
The Escherichia coli fadL gene, including the signal sequence and a C-terminal hexahistidine tag, was cloned as previously described (10) into the pBAD22 vector, which is under the control of the arabinose-inducible promoter (15). Mutations were introduced into the fadL gene by using the QuikChange® Site-Directed Mutagenesis kit (Stratagene), and the mutations were verified by nucleotide sequencing. The ΔN3 and ΔN8 mutant proteins have deletions of residues F3–L5 and F3–S10, respectively, in order to preserve the original cleavage site of EcFadL for signal peptidase, which cleaves after the serine in the sequence AWSAGF.
Protein Purification, Crystallization and Structure Determination of FadL Mutants.
For crystallization, the FadL mutant proteins were expressed from the pBAD22 plasmids (15) in E. coli C43 (DE3) cells grown in 2× yeast extract and tryptone medium by induction with 0.2% (wt/vol) arabinose for 6 h at 30 °C. Cells were harvested, and the proteins were purified from the total membrane fraction as previously described (10) by Ni affinity chromatography in LDAO, gel filtration chromatography in 2 mM LDAO (Anatrace), and a final gel filtration chromatography step in 13 mM C8E4 (Anatrace). The Trp mutants used in the fluorescence assay were purified in a similar way, but with the following modifications: The detergent was β-decyl-maltoside (DM; Anatrace), and ion exchange chromatography was performed after the second gel filtration column, either Source 15Q (Pharmacia) at pH 8 or Source 15S (Pharmacia) at pH 5, followed by dialysis in 10 mM Tris, 10–100 mM NaCl, 10% glycerol, 0.1–0.2% (2–4 mM) DM, pH 8. DM was chosen due to its large, polar maltose headgroup, which we reasoned might prevent binding to the low- and high-affinity binding sites in FadL. The D348A mutant was purified in a different way, in an (unsuccessful) attempt to obtain a crystal structure with bound oleate. Total membrane extraction was performed in 1% DM and 1 mM Na-oleate, followed by purification by gel filtration chromatography in DM (first column) and C8E4 (second column). Oleate was omitted from the gel filtration buffers because solutions containing > 0.1 mM oleate in DM/C8E4 are turbid. All proteins were dialyzed overnight against C8E4 gel filtration buffer, concentrated to 5–10 mg/mL and flash frozen in liquid nitrogen.
Crystals were obtained by the hanging drop method at 22 °C using commercially available screens (The Classics and MB Class II, Qiagen, MemGold Plus) or inhouse screens. The mutant FadL proteins crystallized under the following conditions: ΔN3, 0.1 M Hepes (pH 7.5), 20% (wt/vol) PEG 10,000, 8% (vol/vol) ethylene glycol; ΔN8, 0.05 M magnesium acetate, 0.05 M N-(2-acetamido) iminoacetic acid (pH 7), 25% (wt/vol) PEG 4,000; D348A, 15–17% PEG 4,000, 0.2M KCl, pH 6.3 with protein was dialyzed in 10 mM NaOAc, 50 mM NaCl, 10% glycerol, 0.4% C8E4, pH 5.5; F3E: 15 mM tricine pH 8.5 24% wt/vol PEG 4,000; F3G: 0.1 M NaOAc pH 5.5 8.8% wt/vol PEG 2,000 monomethyl ether; D348R: 360 mM NaCl/0.1% wt/vol NaN3 15 mM NaPi 9.9% wt/vol PEG 4,000, pH 7.0. The crystals were flash frozen (100 K) in reservoir solution containing C8E4 and 15–25% (vol/vol) glycerol by plunging in liquid nitrogen.
Diffraction data were obtained on beamline X6A at the National Synchrotron Light Source (Brookhaven National Laboratory). Datasets were integrated and scaled using HKL2000 (16). During model building of the F3E mutant protein, density consistent with bound metal ions was observed in the electron density maps. We reasoned that this density might originate from bound nickel ions introduced during the metal-affinity chromatography step. For this reason, we reprocessed (and refined) the F3E dataset as “anomalous” within HKL2000. The structures of the mutant proteins were solved by molecular replacement using Phaser within CCP4 (17); the monoclinic FadL structure (Protein Data Bank ID 1T16) without the N-terminal 40 amino acids and the S3 kink residues 99–108 was used in all cases as the search model. Model building was done using COOT (18), and refinement was done with CNS (19) or PHENIX (20). Refinement of the mutant data was extensive and included a first round of high-temperature simulated annealing (5,000–10,000 K starting temperature). Figures were made using PYMOL (21). Data collection and refinement statistics for the mutant FadL proteins are summarized in Table S1.
In Vivo LCFA Transport Assays.
For transport assays, E. coli LS6164 (ΔfadR ΔfadL) was transformed with the pBAD22 plasmids carrying the mutant fadL genes. As a negative control, the pBAD22 plasmid carrying the OM nucleoside transporter Tsx was introduced into E. coli LS6164 (ΔfadR ΔfadL). The ability of the mutant proteins to support growth on LCFAs was measured by plating cells (5 × 106 cfu mL-1) on agar plates containing 5 mM sodium palmitate (Sigma), 0.5% (wt/vol) Brij 58, M9 minimal medium, and 1.5% (wt/vol) Noble agar (Difco). Growth was scored after 96 h incubation at 37 °C. No arabinose was used in the plate growth experiments because the resulting higher expression levels of FadL proteins are toxic for the cells and lead to inhibition of colony growth. Toxicity effects of (outer) membrane protein expression are commonly observed; in the case of liquid cultures, the toxicity effects manifest themselves by decreasing culture densities after induction of expression.
Transport assays with radiolabeled oleic acid were performed with modifications to the method described (10, 22). E. coli LS6164 (ΔfadR ΔfadL) cells with the FadL mutant pBAD22 plasmids were grown in LB medium to midlog phase, and FadL expression was induced with 0.001% (wt/vol) arabinose at 37 °C for 1 h. Cells were harvested, washed in EB1 buffer (10 mM citric acid, 0.8 mM magnesium sulfate, 20 mM sodium ammonium phosphate, 60 mM potassium phosphate, 0.02 mM thiamine), and resuspended to an OD600 of 1 in EB1 buffer with 0.5% (wt/vol) Brij 58. After 30 min starvation at 37 °C, samples were taken for Western immunoblotting analysis and to measure the transport activity for oleic acid. The transport assay was performed by diluting the cells in prewarmed EB1 buffer containing 20 mM glucose, 0.008% (wt/vol) Brij 58, 0.2 mM oleic acid, and 0.02 μCi [3H-9,10]-oleic acid (1 Ci = 37 GBq) (Sigma; specific activity 40 Ci mmol-1). At specified time intervals, samples were removed and filtered through 0.45-mm membrane filters (Metricel GN-6, Pall Life Sciences). Filters were washed with EB1 buffer containing 0.5% Brij 58, and the radioactivity retained on the filters was counted. To determine the amount of FadL protein expressed in the outer membrane, cells were incubated with BugBuster (Pierce) while shaking for 20 min at 25 °C, and centrifuged for 10 min at 20,800 × g. Proteins were quantified by Western immunoblotting using an antiHis antibody (Qiagen), and specific transport rates were calculated as picomole oleic acid per minute per nanogram FadL protein. The relative quantities of wild-type, W298F, 0Trp-F339W, 0Trp-F298W, and 0Trp-F0W proteins on Western blots were confirmed by SDS-PAGE of partially purified samples (i.e., after nickel chromatography) from E. coli D10.
In Vitro and in Vivo LCFA Binding Assays.
The in vitro ADIFAB (acrylodan-labeled fatty acid binding protein; Molecular Probes, Inc.) binding assay was performed and data were analyzed essentially as described previously (23). Briefly, 0.4–2 mM protein purified in LDAO and DM was added to buffer (10 mM Hepes, 50 mM NaCl, 10% glycerol, 0.05% LDAO) containing 0.2–0.5 mM ADIFAB. Na-oleate (30 mM stock) was added stepwise to 10–20 mM final concentration and fluorescence emission spectra were recorded from 420–550 nm with excitation at 386 nm. Control experiments were performed without added membrane proteins (micelles only). Fatty acid binding to ADIFAB causes a decrease in fluorescence at 432 nm with a concomitant increase in fluorescence at 505 nm, and from these changes specific oleate binding to empty micelles, FadL and Tsx were determined as described. In vivo 3H oleate binding was performed as described (24). For this experiment, LS6929 cells expressing FadL or OmpG from pB22 plasmids were grown to OD600 0.5–0.6, followed by induction with 0.001% arabinose for 1 h at 37 °C. The low concentration of arabinose was chosen to prevent cell lysis, which is severe at higher arabinose concentrations. Cells were harvested by centrifugation, washed in 10 mM phosphate buffer containing 10 mM NaCl and adjusted to OD600 approximately 1.0. Following starvation for 30 min at 37 °C (24), 3H-oleate/BSA were added to 500 mL of cells and incubated for 15 min at 37 °C. Aliquots of cells were filtered through GN-6 membranes, washed with phosphate/NaCl buffer followed by scintillation counting of the filters. Total oleate concentrations of 17 and 170 nM and different molar ratios of oleate:BSA were used, with similar results. Dipyrromethene boron difluoride (BODIPY)-C16 binding to LS6929 cells was done in a similar way as described above for 3H-oleate. BODIPY-C16 and BSA were preincubated at different molar ratios (with the fatty acid concentration kept constant at 0.25 or 2.5 mM) for 1 h at 37 °C. Fluorescence was measured with a TECAN™ Safire™ fluorimeter by excitation at 488 nm and measuring fluorescence emission at 514 nm.
Fluorescence Spectroscopy.
Fluorescence spectra were recorded from an ISS™ PC1 Photon Counting Spectrofluorometer with ISIS™ Vinci™ data collection software version BETA.1.5 (2002–2004, ISS, Inc.). Excitation wavelength was 295 nm. Lamp set to 17 A dc (ILC Technology™). Emission and excitation bandwidth was set to 4 nm using 1.0-mm slits. Δλ/step = 1.0 nm; Approximately 90-s scan time. Assay buffer: 10 mM Tris•HCl, pH 7.95 (pH 8.0), 0.05–0.10% DM, 1.0 μM protein, 10% glycerol. A single step binding quadratic model was fit to the intensity data at 319 nm for 0Trp-F298W, with protein concentration P, binding constant K, zeroF0 shift dF, and sodium oleate concentration x (25):
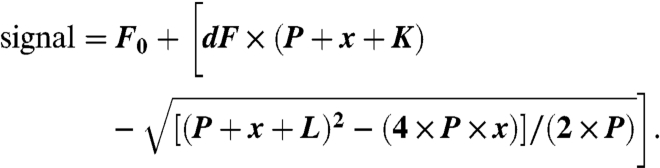 |
Plots were generated with Gnuplot 4.4.
Supplementary Material
Acknowledgments.
We thank David Lambright for helpful discussions. We are furthermore indebted to the personnel of beamlines 23-ID B and D of the Advanced Photon Source and beamlines X25 and X6A of the National Synchrotron Light Source for generous beamtime and beamline support, and Shyamasri Biswas and Komi Akpalu for help with construction of the 0Trp mutants. This work was supported by National Institutes of Health research Grant 5R01GM074824 to B.v.d.B.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3PGR, 2R89, 2R8A, 3PF1, 3PGU, 3PGS).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1018532108/-/DCSupplemental.
References
- 1.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanabe M, Nimigean CM, Iverson TM. Structural basis for solute transport, nucleotide regulation, and immunological recognition of Neisseria meningitidis PorB. Proc Natl Acad Sci USA. 2010;107:6811–6816. doi: 10.1073/pnas.0912115107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hong H, Szabo G, Tamm LK. Electrostatic couplings in OmpA ion-channel gating suggest a mechanism for pore opening. Nat Chem Biol. 2006;2:627–635. doi: 10.1038/nchembio827. [DOI] [PubMed] [Google Scholar]
- 4.Henderson NS, Ng TW, Talukder I, Thanassi DG. Function of the usher N-terminus in catalysing pilus assembly. Mol Microbiol. 2011;79:954–967. doi: 10.1111/j.1365-2958.2010.07505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferguson AD, Deisenhofer J. TonB-dependent receptors: Structural perspectives. Biochim Biophys Acta. 2002;1565:318–332. doi: 10.1016/s0005-2736(02)00578-3. [DOI] [PubMed] [Google Scholar]
- 6.Noinaj N, Guillier M, Barnard TJ, Buchanan SK. TonB-dependent transporters: regulation, structure, and function. Annu Rev Microbiol. 2010;64:43–60. doi: 10.1146/annurev.micro.112408.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van den Berg B. The FadL family: Unusual transporters for unusual substrates. Curr Opin Struct Biol. 2005;15:401–407. doi: 10.1016/j.sbi.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 8.van den Berg B. Going forward laterally: Transmembrane passage of hydrophobic molecules through protein channel walls. ChemBioChem. 2010;11:1339–1343. doi: 10.1002/cbic.201000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Berg B, Black PN, Clemons WM, Jr, Rapoport TA. Crystal structure of the long-chain fatty acid transporter FadL. Science. 2004;304:1506–1509. doi: 10.1126/science.1097524. [DOI] [PubMed] [Google Scholar]
- 10.Hearn EM, Patel DR, Lepore BW, Indic M, van den Berg B. Transmembrane passage of hydrophobic compounds through a protein channel wall. Nature. 2009;458:367–370. doi: 10.1038/nature07678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hearn EM, Patel DR, van den Berg B. Outer-membrane transport of aromatic hydrocarbons as a first step in biodegradation. Proc Natl Acad Sci USA. 2008;105:8601–18606. doi: 10.1073/pnas.0801264105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd Ed. New York: Kluwer; 1999. p. 453. [Google Scholar]
- 13.Vorum H, Brodersen R, Kragh-Hansen U, Pedersen AO. Solubility of long-chain fatty acids in phosphate buffer at pH 7.4. Biochim Biophys Acta. 1992;1126:135–142. doi: 10.1016/0005-2760(92)90283-2. [DOI] [PubMed] [Google Scholar]
- 14.Cahalan MD, et al. Molecular basis of the CRAC channel. Cell Calcium. 2007;42:133–144. doi: 10.1016/j.ceca.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guzman L, Belin D, Carson M, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor D. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 17.Collaborative computational project N. The CCP4 suite—programs for protein crystallography. Acta Cryst D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 18.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of COOT. Acta Cryst D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunger AT. Version 1.2 of the crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 20.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.PyMOL Molecular Graphics System. Cambridge, MA: Schrödinger, LLC; 2011. v1.4. [Google Scholar]
- 22.Kumar G, Black P. Bacterial long-chain fatty acid transport. Identification of amino acid residues within the outer membrane protein FadL required for activity. J Biol Chem. 1993;268:15469–15476. [PubMed] [Google Scholar]
- 23.Richieri G, Ogata R, Kleinfeld A. The measurement of free fatty acid concentration with the fluorescent probe ADIFAB: A practical guide for the use of the ADIFAB probe. Mol Cell Biochem. 1999;192:87–94. [PubMed] [Google Scholar]
- 24.Black PN, Zhang Q. Evidence that His110 of the protein FadL in the outer membrane of Escherichia coli is involved in the binding and uptake of long-chain fatty acids: possible role of this residue in carboxylate binding. Biochem J. 1995;310:389–394. doi: 10.1042/bj3100389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Copeland RA. Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis. New York: Wiley-VCH; 2000. p. 94. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.