

Abstract
The transfer free energies of the twenty natural amino acid side chains from water to phospholipid bilayers make a major contribution to the assembly and function of membrane proteins. Measurements of those transfer free energies will facilitate the identification of membrane protein sequences and aid in the understanding of how proteins interact with membranes during key biological events. We report the first water-to-bilayer transfer free energy scale (i.e., a “hydrophobicity scale”) for the twenty natural amino acid side chains measured in the context of a native transmembrane protein and a phospholipid bilayer. Our measurements reveal parity for apolar side-chain contributions between soluble and membrane proteins and further demonstrate that an arginine side-chain placed near the middle of a lipid bilayer is accommodated with much less energetic cost than predicted by molecular dynamics simulations.
Keywords: solvation, thermodynamics
The transfer of amino acid side chains from water into phospholipid lipid bilayers is a fundamental energetic contribution to the total thermodynamic stability of transmembrane proteins (1, 2). A hydrophobicity scale that ranks the water-to-bilayer transfer energies of the twenty natural amino acid side chains could allow the identification of genes that code for membrane proteins (3–6) and aid the understanding of how proteins interact with membranes during key biological events, such as cell signaling, drug binding, and the gating of ion-channels. However, despite the abundance of available hydrophobicity scales, there have been no experimental measurements of water-to-bilayer transfer free energies of amino acid side chains that derive from the partitioning of a native transmembrane protein from water to the interior of a phospholipid bilayer. Existing estimations of water-to-bilayer partitioning of side chains were obtained from systems comprising small compounds to represent proteins and/or organic solvents to mimic bilayers. The only experimental hydrophobicity scale derived from proteins and bilayers (the “translocon scale”) described the translocon-to-bilayer transition (7, 8). Mediated membrane insertion by the translocon complex does not explain the stability of membrane proteins in lipid bilayers relative to a water-solvated state.
Here we achieve thermodynamic measurements of amino acid side-chain transfer into lipid bilayers in the context of a natively folded transmembrane protein spanning a phospholipid bilayer. Our measurements reveal the hydrophobic effect contributes equivalently to the stability of both membrane and soluble proteins (9). Our experiments further demonstrate that a membrane protein can accommodate an arginine side chain placed near the apolar middle of a lipid bilayer with much less cost in energy than has been previously predicted by molecular dynamics simulations (10–11). Moreover, we found the membrane partitioning of two arginine residues to be thermodynamically cooperative. Therefore, our measurements should inform the study of voltage-sensing ion channels, which concertedly move multiple arginines into the hydrophobic interior of membranes during their gating (12, 13).
Results and Discussion
We used the outer membrane phospholipase A (OmpLA) as a transmembrane scaffold on which to introduce amino acid side chains of our choice at various membrane depths. We selected OmpLA because it: (a) spontaneously folds and inserts into lipid membranes from a solubilized unfolded state (14), (b) has a known three-dimensional structure (Fig. 1A) (15), and (c) has enzymatic activity that can be monitored to confirm native-like folding (16, 17). To measure side-chain hydrophobicity in a membrane, we selected the alanine at position 210 in OmpLA’s sequence as a host site for substitution to each of the other 19 amino acids as guests. Position 210 is a lipid-facing exterior residue whose α-carbon is located only 0.2 Å from the midplane between OmpLA’s membrane/water interfacial regions (Fig. 1A). Our membranes were large unilamellar vesicles (LUVs) of 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC).
Fig. 1.

Outer membrane phospholipase A (OmpLA) as a host-guest system for measuring the membrane insertion energies of lipid-exposed amino acid residues. (A) Backbone of OmpLA rendered using PyMol (DeLano Scientific) and Protein Data Bank (PDB) ID code 1qd5 with the α-carbon of the alanine at sequence position 210 shown as a black sphere. Solid horizontal lines represent the boundaries of OmpLA’s transmembrane region. The dashed horizontal line represents the midplane of that region. (B–E) Example denaturation data for A210X sequence variants, each with a fit line from a three-state linear-extrapolation model. Data points represent tryptophan fluorescence emission measurements, normalized such that the emission from folded protein is 1 and the emission from unfolded protein is 0. Data for the wild-type OmpLA is shown in black in each panel. (B) Data for X = F, I, L, M, P, V, W, and Y are shown in orange. (C) Data for X = C and G are shown in gray. (D) Data for X = N, Q, S, and T are shown in teal. (E) Data for X = D, E, H, K, and R are shown in blue.
To determine the energetics of membrane partitioning of the guest amino acids, we adapted soluble protein chemical denaturation methods to measure the stability of folded proteins using tryptophan fluorescence spectroscopy (18, 19). This approach has only previously been attempted with two other transmembrane proteins folded into lipid vesicles (20–23) and it is only valid if folded and unfolded protein populations come to reversible equilibrium at all concentrations of denaturant (18). We verified reversible folding of OmpLA at pH 3.8 (Fig. S1A). We do not yet understand why acidic pH promotes reversible folding of OmpLA or whether this is a common feature for other membrane proteins.
We confirmed the observed folded state of OmpLA was in a native-like transmembrane conformation by verifying that it had enzymatic activity, which requires an acyl-chain of a substrate in the bilayer to bind to a hydrophobic pocket on the transmembrane surface of OmpLA (16, 17). Further, we verified the folded state had its tryptophans embedded in the apolar lipid environment and was protected from protease digestion by being inserted across the membrane (Fig. S2). We determined the unfolded state of OmpLA was not embedded in the membrane and was instead solvated by water by verifying that it was enzymatically inactive, that its tryptophans were in a polar environment whether or not LUVs were present, and it was completely digestible by a protease (Fig. S2).
At equilibrium, OmpLA also adopted a thermodynamic intermediate state at a moderate range of denaturant concentrations, as observed from the fluorescence emission of its tryptophans (Fig. 1 B–E and Fig. S1 B and C). Therefore, we used a three-state linear-extrapolation model to extract a total standard-state Gibbs free energy of unfolding (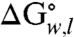 ) for each of the OmpLA sequence variants (Fig. 1 B–E) in the presence of just water (w) and lipid bilayers (l); i.e., with no denaturant present. Because Gibbs free energy is a state-function, the appearance of an intermediate in our denaturant titrations does not affect our total
) for each of the OmpLA sequence variants (Fig. 1 B–E) in the presence of just water (w) and lipid bilayers (l); i.e., with no denaturant present. Because Gibbs free energy is a state-function, the appearance of an intermediate in our denaturant titrations does not affect our total 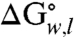 values.
values.
The differences in the 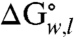 for the side-chain variants at position 210 and the
for the side-chain variants at position 210 and the 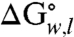 of the wild-type OmpLA (
of the wild-type OmpLA (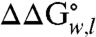 ) comprise a unique whole-protein hydrophobicity scale (Fig. 2 and Table S1). Because the contributions of other residues of OmpLA are subtracted, the
) comprise a unique whole-protein hydrophobicity scale (Fig. 2 and Table S1). Because the contributions of other residues of OmpLA are subtracted, the 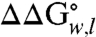 values in the whole-protein scale reflect the energies required to partition just the guest amino acid side chains into the membrane relative to the alanine side chain at position 210 in the wild-type sequence. Therefore, these values should be applicable to membrane proteins of all architectures and will have utility in calibrating force fields or protocols for membrane protein calculations.
values in the whole-protein scale reflect the energies required to partition just the guest amino acid side chains into the membrane relative to the alanine side chain at position 210 in the wild-type sequence. Therefore, these values should be applicable to membrane proteins of all architectures and will have utility in calibrating force fields or protocols for membrane protein calculations.
Fig. 2.

Whole-protein hydrophobicity scale determined from the OmpLA system. The difference in the Gibbs free energy of unfolding (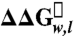 ) of each amino acid variant at position 210 is compared to the wild-type OmpLA. Error bars represent standard errors of the mean from individual titration experiments. The color scheme is the same as in Fig. 1 B–E.
) of each amino acid variant at position 210 is compared to the wild-type OmpLA. Error bars represent standard errors of the mean from individual titration experiments. The color scheme is the same as in Fig. 1 B–E.
Exclusion from water appeared to be the main contribution to the membrane partition energies for the hydrophobic side chains of Ala, Leu, Ile, Met, Phe, Tyr, and Val. We found a strong linear correlation (R = 0.96) between the nonpolar solvent accessible surface areas (ASAs) of those side chains and their values in our whole-protein scale (Fig. S3). Importantly, the ASAs were independent of the particular structural context of OmpLA because they were derived from exposure of the guest side chains in an extended model tripeptide. This independence supports the conclusion that our whole-protein scale is applicable to transmembrane proteins of diverse architecture. Indeed, a traditional hydropathy analysis (3) using our whole-protein scale with the Membrane Protein Explorer (MPEx) (5) application correctly predicted the location of all seven transmembrane α-helices of bovine rhodopsin (Fig. S4).
The correlation of the whole-protein scale and ASAs revealed that each Å2 of surface area of a hydrophobic side chain removed from water and buried into a membrane added 23 cal mol-1 to the stability of OmpLA. This value is a quantitative description of the hydrophobic effect and constitutes an atomic solvation parameter (ASP) for transferring nonpolar side chains from water into a lipid bilayer. Our ASP matches the ASP previously found for transferring nonpolar side chains from water into the nonpolar solvent octanol (24) as well as that for burying nonpolar side chains into the hydrophobic interior of a water-soluble protein (9). Therefore, the hydrophobic effect stabilizes both membrane and soluble proteins to an equivalent degree. We also used our ASP to calculate an insertion energy for the alanine side chain by multiplying it by alanine’s nonpolar ASA (69 Å2). By shifting the whole-protein scale by alanine’s insertion energy, we can express a scale for all twenty side chains (Table S1) that does not use any particular one of them as a reference. It should be noted that this side-chain scale does not include a contribution of the peptide bond.
Using our thermodynamically rigorous host-guest system, we were well positioned to investigate the energetic cost of water-to-membrane partitioning of arginine. This cost has been a matter of controversy because it concerns the ability of the arginine-rich S4 helix of KvAP and related ion channels to sense membrane depolarization and mediate channel gating (10, 11, 13, 25, 26). Earlier molecular dynamics simulations (10, 11) calculated the total cost of moving a single arginine to the middle of a membrane to be 14–17 kcal mol-1. If the side chain snorkeled toward the water/bilayer interface, the calculated cost would still be 6–7 kcal mol-1 (8). The energetics of water-to-membrane partitioning of arginine would also depend on possible rearrangement of other parts of the protein and/or a deformation of the membrane to reduce its hydrophobic thickness (10, 11, 27, 28). However, no such energetics have been measured with a whole protein until the present study. The previous translocon scale had a modest value for arginine (7), but it did not represent the water-to-bilayer transition (8). Our measurement for arginine here does represent a water-to-bilayer transition and it also is quite modest: 2.1 ± 0.1 kcal mol-1 (Table S1). We speculate that at least part of the discrepancy between our experimental value and the previous theoretical values may be due to differences in the hydrophobic thicknesses of the lipid bilayers used in the different systems and may be resolved by further calculations that are matched to our experimental conditions.
To address the effects of bilayer depth on side-chain partitioning, we studied side chains at different positions on the OmpLA scaffold. In addition to position 210, we engineered both arginine and leucine at five additional locations in the OmpLA sequence (positions 120, 164, 212, 214, and 223) and compared the resulting variants to alanine variants at the same positions. Arginine partition energies were most unfavorable at the middle of the membrane (Fig. 3 and Table S2) and displayed a shape with a depth-dependence that recapitulated the trends observed in earlier molecular dynamics simulations (8, 10, 11, 27, 28) and in an earlier depth analysis of the translocon scale (6, 7). Leucine partition energies had an opposite response—they were more favorable closer to the middle of the membrane.
Fig. 3.

Energetics of side-chain partitioning varies by depth in the membrane. The OmpLA host-guest system is shown similarly as in Fig. 1 with the α-carbons of sequence positions 120, 164, 210, 212, 214, and 223 shown as black spheres. The membrane depth of those five α-carbons versus the 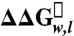 of leucine and arginine variants (compared to alanine variants) is shown aligned with the OmpLA image. Normal distributions fit to the leucine and arginine data are also shown. Error bars represent standard errors of the mean from individual titration experiments.
of leucine and arginine variants (compared to alanine variants) is shown aligned with the OmpLA image. Normal distributions fit to the leucine and arginine data are also shown. Error bars represent standard errors of the mean from individual titration experiments.
We also examined a double-arginine variant (A210R, G212R) to address the issue of tandem membrane insertion of multiple arginine side chains that could occur with KvAP channel gating. Fig. 4 and Table S2 show that the 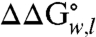 of the double-arginine variant is 1.6 kcal mol-1 less than the sum of the
of the double-arginine variant is 1.6 kcal mol-1 less than the sum of the 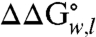 for each single-arginine variant (A210R and G212R). Therefore, there is cooperativity between the two arginines that reduces their overall energetic cost to be in the membrane. The source of this cooperativity could be the ability of the two arginines to share access to a deformation or water penetration in the adjacent lipid bilayer. Therefore, our results suggest that the multiple arginines of the S4 helix could function together to suppress their energetic burden to pass across the apolar interior of the membrane during gating of the KvAP channel.
for each single-arginine variant (A210R and G212R). Therefore, there is cooperativity between the two arginines that reduces their overall energetic cost to be in the membrane. The source of this cooperativity could be the ability of the two arginines to share access to a deformation or water penetration in the adjacent lipid bilayer. Therefore, our results suggest that the multiple arginines of the S4 helix could function together to suppress their energetic burden to pass across the apolar interior of the membrane during gating of the KvAP channel.
Fig. 4.

A double-mutant cycle reveals that two arginines cooperate energetically to partition into a membrane. The OmpLA system is shown similarly as in Fig. 1 with the atoms of the side chains varied in the cycle shown as spheres. The arginine side-chain orientations shown are for illustration of size and are not intended to depict any known side-chain conformations relative to the lipid bilayer in our experiments.
The power of our whole-protein hydrophobicity scale is that it unambiguously reflects the thermodynamics of water-to-bilayer partitioning of side chains in the context of a native transmembrane protein spanning a phospholipid bilayer. The earlier translocon scale (7) did not reflect a system verifiably at equilibrium, and its values are relevant to a different event: translocon-to-bilayer partitioning (8). The Wimley White water-to-octanol scale (24) did derive from equilibrium thermodynamic measurements, however, it represented the protein by short peptides and it approximated the lipid membrane environment with octanol. Our observations reveal that both of those previous scales would undervalue the energetics of most amino acids if they were used to represent water-to-bilayer side-chain partitioning (Fig. S5).
Two exceptions are aspartic acid and glutamic acid, whose energetics are underrepresented in our whole-protein scale because the pH of our experiments was 3.8, which is close to the pKa values for model compounds of Asp and Glu side chains (29). It is reasonable to expect that a significant population of the guest Asp and Glu side chains in our system were protonated and thereby more easily partitioned into the membrane than they would have at normal physiological pH. Therefore, our results for Asp and Glu may help explain why certain membrane active peptides, such as bacterial toxins (30), can partition across membranes only at low pH. This phenomenon of acidic membrane partitioning should be considered in the study of how protein–lipid interactions affect the many types of tumors that feature acidic extracellular pH (31).
Our measurements here provide water-to-bilayer transfer free energies of amino acid side chains determined from the spontaneous insertion of a whole transmembrane protein into membranes. Therefore, our whole-protein free energy measurements are highly applicable to models of spontaneous changes in protein–lipid interactions, such as models of channel gating. To completely describe such events, however, more than just equilibrium energies should be known. Channel gating is a dynamic and kinetic event, so the activation energy barriers to amino acid side-chain insertion into membranes should be determined. Our system of using a scaffold transmembrane protein for guest amino acids should also allow us to measure those kinetic barriers.
Methods
Protein Folding denaturant Titrations.
The wild-type OmpLA and its sequence variants were engineered, expressed, and purified as previously described (17). Prior to each experiment, we made fresh unfolded protein stocks dissolved in 8 M guanidine HCl (UltraPure powder, Invitrogen). The background buffer for all fluorescence experiments was 100 mM citrate (Sigma) and 2 mM EDTA (Sigma), pH 3.8. For the variant A210C, we included 1 mM dithiothreitol (DTT, from Sigma) in the buffers. We handled lipids (Avanti Polar Lipids) and prepared LUVs as previously described (14), but with 21 passes through two stacked polycarbonate filters having a 0.1 μM pore size.
We set up protein folding and unfolding reactions in three steps. The first step was a dilution of the unfolded protein stocks to a final guanidine concentration of 2.5 M and a final protein concentration of 6.0 μM with 1.4 mM 3-N,N-Dimethylmyristyl-ammonio [propanesulfonate (SB3-14, from Sigma)]. The SB3-14 prevented protein aggregation before the LUVs were added. The second step was a threefold dilution of the protein/detergent mixture into LUVs of DLPC to attain a 2,000∶1 lipid-protein ratio and to take the SB3-14 below its critical micelle concentration. For titrations for free energy measurements (Fig. 1), this second step kept the protein unfolded at 5.0 M guanidine HCl. For reversible folding verification (Fig. S1A), we prepared two reactions at the second step: one allowing the protein to fold at 2.5 M guanidine HCl and the other keeping the protein unfolded at 5.0 M guanidine HCl. We incubated all samples from the second step at 37 °C with gentle rotation for 5 h. The third step was a fivefold dilution into buffer/guanidine HCl mixtures to attain different final concentrations of guanidine HCl. The final protein concentration after the third step was 400 nM and the final lipid concentration was 800 μM. We incubated the titration samples from the third step at 37 °C with gentle rotation for 40–50 h before fluorescence experiments. For free energy measurements, we carried out three titrations of the wild-type OmpLA and two titrations for each of the sequence variants. Each titration was an independent experiment with its own protein, lipid, denaturant, and buffer stocks.
Tryptophan Fluorescence Emission Spectroscopy.
Tryptophan fluorescence emission was monitored on an ISS PC1 photon counting spectrofluorometer. The excitation wavelength was 295 nm and the path length was 1 cm. We used an excitation polarizer at 90° with 2.4 mm slits and an emission polarizer at 0° with 2.0 mm slits (32, 33). For samples used for free energy measurements, we collected 350 readings of emission intensity at 330 nm with 0.3 s of signal averaging for each reading. For samples used to show full tryptophan spectral properties (Figs. S1A and S2B), we averaged four emission scans from 280 to 400 nm at 1 nm resolution. We found the wavelength position of maximum intensity (λmax) by fitting spectra to a log-normal function (32, 33).
Free Energy Measurements.
We globally fit all sets of titration data for all proteins to a three-state linear-extrapolation model (34) using Igor Pro v6.12 (WaveMetrics) to find a common measure of the two m-values in the model (see SI Text). To find total free energies of unfolding for the proteins, we summed the two 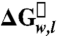 values for each set of titration data included in the fit. Our measured free energy of unfolding for the wild-type OmpLA (32.4 ± 0.1 kcal mol-1) is about two- to threefold higher than previously reported values for OmpA (35) and PagP (23), which are smaller proteins. Thus, the energy measurements for all three proteins may be consistent considering the different sizes of the proteins and also the variation in the lipid systems used to make the measurements.
values for each set of titration data included in the fit. Our measured free energy of unfolding for the wild-type OmpLA (32.4 ± 0.1 kcal mol-1) is about two- to threefold higher than previously reported values for OmpA (35) and PagP (23), which are smaller proteins. Thus, the energy measurements for all three proteins may be consistent considering the different sizes of the proteins and also the variation in the lipid systems used to make the measurements.
Membrane Depth Measurements.
We determined where the membrane midplane should intersect OmpLA by using its atomic coordinates in PDB ID code 1qd5 (Fig. 1A). First, we defined a boundary plane for each side of OmpLA’s hydrophobic surface (extracellular and intracellular) by averaging the set of planes that are formed by each triad of polar atoms of OmpLA’s two aromatic “girdles” of residues (i.e., the OH atoms of tyrosines and the NE1 atoms of tryptophans) (36). The distance between the two boundary planes (24.4 Å) was similar to a previously calculated value for OmpLA’s hydrophobic thickness (23.2 ± 1.5 Å) (37). We placed the membrane midplane halfway between the boundary planes. We assigned the membrane depths of OmpLA’s residues as the distance between their α-carbons to the midplane along a normal to the midplane. The depths for the variant positions in this work were: -4.0 Å for position 120, -10.1 Å for position 164, 0.2 Å for position 210, 5.3 Å for position 212, 8.8 Å for position 214, and 7.1 Å for position 223.
Supplementary Material
Acknowledgments.
This work was supported by grants from the National Science Foundation (MCB0423807, MCB0919868) and the National Institutes of Health (R01 GM079440, T32 GM008403).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
See Commentary on page 10027.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1103979108/-/DCSupplemental.
References
- 1.Tanford C. The hydrophobic effect and the organization of living matter. Science. 1978;200:1012–1018. doi: 10.1126/science.653353. [DOI] [PubMed] [Google Scholar]
- 2.White SH, Wimley WC. Membrane protein folding and stability: Physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 3.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 4.Bernsel A, et al. Prediction of membrane-protein topology from first principles. Proc Natl Acad Sci USA. 2008;105:7177–7181. doi: 10.1073/pnas.0711151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snider C, Jayasinghe S, Hristova K, White SH. MPEx: A tool for exploring membrane proteins. Protein Sci. 2009;18:2624–2628. doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hessa T, et al. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- 7.Hessa T, et al. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 8.Schow EV, et al. Arginine in membranes: The connection between molecular dynamics simulations and translocon-mediated insertion experiments. J Membr Biol. 2011;283:35–48. doi: 10.1007/s00232-010-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chothia C. Hydrophobic bonding and accessible surface area in proteins. Nature. 1974;248:338–339. doi: 10.1038/248338a0. [DOI] [PubMed] [Google Scholar]
- 10.Dorairaj S, Allen TW. On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc Natl Acad Sci USA. 2007;104:4943–4948. doi: 10.1073/pnas.0610470104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacCallum JL, Bennett WF, Tieleman DP. Partitioning of amino acid side chains into lipid bilayers: results from computer simulations and comparison to experiment. J Gen Physiol. 2007;129:371–377. doi: 10.1085/jgp.200709745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Y, et al. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- 13.Ruta V, Chen J, MacKinnon R. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 2005;123:463–475. doi: 10.1016/j.cell.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 14.Burgess NK, Dao TP, Stanley AM, Fleming KG. β-Barrel proteins that reside in the Escherichia coli outer membrane in vivo demonstrate varied folding behavior in vitro. J Biol Chem. 2008;283:26748–26758. doi: 10.1074/jbc.M802754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snijder HJ, et al. Structural evidence for dimerization-regulated activation of an integral membrane phospholipase. Nature. 1999;401:717–721. doi: 10.1038/44890. [DOI] [PubMed] [Google Scholar]
- 16.Dekker N, Tommassen J, Lustig A, Rosenbusch JP, Verheij HM. Dimerization regulates the enzymatic activity of Escherichia coli outer membrane phospholipase A. J Biol Chem. 1997;272:3179–3184. doi: 10.1074/jbc.272.6.3179. [DOI] [PubMed] [Google Scholar]
- 17.Stanley AM, Fleming KG. The role of a hydrogen bonding network in the transmembrane β-barrel OMPLA. J Mol Biol. 2007;370:912–924. doi: 10.1016/j.jmb.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Santoro MM, Bolen DW. Test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]
- 19.Street TO, Courtemanche N, Barrick D. Protein folding and stability using denaturants. Methods Cell Biol. 2008;84:295–325. doi: 10.1016/S0091-679X(07)84011-8. [DOI] [PubMed] [Google Scholar]
- 20.Hong H, Tamm LK. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc Natl Acad Sci USA. 2004;101:4065–4070. doi: 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pocanschi CL, Patel GJ, Marsh D, Kleinschmidt JH. Curvature elasticity and refolding of OmpA in large unilamellar vesicles. Biophys J. 2006;91:L75–L77. doi: 10.1529/biophysj.106.091439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez KM, Gable JE, Schlamadinger DE, Kim JE. Effects of tryptophan microenvironment, soluble domain, and vesicle size on the thermodynamics of membrane protein folding: lessons from the transmembrane protein OmpA. Biochemistry. 2008;47:12844–12852. doi: 10.1021/bi800860k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huysmans GHM, Baldwin SA, Brockwell DJ, Radford SE. The transition state for folding of an outer membrane protein. Proc Natl Acad Sci USA. 2010;107:4099–4104. doi: 10.1073/pnas.0911904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wimley WC, Creamer TP, White SH. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 1996;35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- 25.Hessa T, White SH, von Heijne G. Membrane insertion of a potassium-channel voltage sensor. Science. 2005;307:1427. doi: 10.1126/science.1109176. [DOI] [PubMed] [Google Scholar]
- 26.Roux B. Lonely arginine seeks friendly environment. J Gen Physiol. 2007;130:233–236. doi: 10.1085/jgp.200709819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freites JA, Tobias DJ, von Heijne G, White SH. Interface connections of a transmembrane voltage sensor. Proc Natl Acad Sci USA. 2005;102:15059–15064. doi: 10.1073/pnas.0507618102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bond PJ, Sansom MSP. Bilayer deformation by the Kv channel voltage sensor domain revealed by self-assembly simulations. Proc Natl Acad Sci USA. 2007;104:2631–2636. doi: 10.1073/pnas.0606822104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nozaki Y, Tanford C. Examination of titration behavior. Methods Enzymol. 1967;11:715–734. [Google Scholar]
- 30.O’Keefe DO, Cabiaux V, Choe S, Eisenberg D, Collier J. pH-dependent insertion of proteins into membranes: B-chain mutation of diphtheria toxin that inhibits membrane translocation, Glu-349 → Lys. Proc Natl Acad Sci USA. 1992;89:6202–6206. doi: 10.1073/pnas.89.13.6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andreev OA, et al. Mechanism and uses of a membrane peptide that targets tumors and other acidic tissues in vivo. Proc Natl Acad Sci USA. 2007;104:7893–7898. doi: 10.1073/pnas.0702439104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moon CP, Fleming KG. Using tryptophan fluorescence to measure the stability of membrane proteins folded in liposomes. Methods Enzymol. 2011;492:189–211. doi: 10.1016/B978-0-12-381268-1.00018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ladokhin AS, Jayasinghe S, White SH. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal Biochem. 2000;285:235–245. doi: 10.1006/abio.2000.4773. [DOI] [PubMed] [Google Scholar]
- 34.Latypov RF, Cheng H, Roder NA, Zhang J, Roder H. Structural characterization of an equilibrium unfolding intermediate in Cytochrome c. J Mol Biol. 2006;357:1009–1025. doi: 10.1016/j.jmb.2006.01.055. [DOI] [PubMed] [Google Scholar]
- 35.Hong H, Park S, Flores-Jiménez RH, Rinehart D, Tamm LK. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J Am Chem Soc. 2007;129:8320–8327. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- 36.Lee AG. Lipid-protein interactions in biological membranes: A structural perspective. Biochim Biophys Acta. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 37.Lomzie AL, Pogozheva ID, Lomzie MA, Mosberg HI. Positioning of proteins in membranes: A computational approach. Protein Sci. 2006;15:1318–1333. doi: 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.