

Abstract
Y-box binding protein-1 (YB-1) expression in the mammary gland promotes breast carcinoma that demonstrates a high degree of genomic instability. In the present study, we developed a model of premalignancy to characterize the role of this gene during breast cancer initiation and early progression. Antibody microarray technology was used to ascertain global changes in signal transduction following the conditional expression of YB-1 in human mammary epithelial cells (HMEC). Cell cycle associated proteins were frequently altered with the most dramatic being LIM Kinase 1/2 (LIMK1/2). Consequently, the misexpression of LIMK1/2 was associated with cytokinesis failure that acted as a precursor to centrosome amplification. Detailed investigation revealed that YB-1 localized to the centrosome in a phosphorylation-dependent manner where it complexed with pericentrin and γ-tubulin. This was found to be essential in maintaining the structural integrity and microtubule nucleation capacity of the organelle. Prolonged exposure to YB-1 led to rampant acceleration toward tumourigenesis with the majority of cells acquiring numerical and structural chromosomal abnormalities. Slippage through the G1/S checkpoint due to overexpression of cyclin E promoted continued proliferation of these genomically compromised cells. As malignancy further progressed, we identified a subset of cells harbouring HER2 amplification. Our results recognize YB-1 as a cancer susceptibility gene with the capacity to prime cells for tumourigenesis.
Keywords: YB-1, premalignancy, centrosome, cell cycle, HER2 amplification, breast cancer
Introduction
Cancer arises from the progressive evolution of a cell from normalacy through intermediate premalignant states to finally become invasive and metastasize (Hanahan and Weinberg, 2000). Cells position themselves for this progression to malignancy by accumulating genetic alterations that result in the activation of oncogenes and inactivation of tumour suppressors. Studies of human mammary epithelial cells (HMEC) are beginning to provide key insight into these early genetic events to ascertain their role in fueling breast carcinogenesis (Romanov et al., 2001; Tlsty et al., 2004).
Y-box binding protein-1 (YB-1) is a transcription/translation factor that is overexpressed in a plethora of cancers, including human breast carcinoma (40%) (Habibi et al., 2008; Wu et al., 2006). A protumourigenic role for YB-1 is supported by its ability to directly bind Y-box promoter elements of a variety of genes, notably epidermal growth factor receptor (EGFR), ErbB2 (HER2), cyclin A, and cyclin B1 (Jurchott et al., 2003; Stratford et al., 2007; Wu et al., 2006). Moreover, a function for YB-1 in regulating cell cycle progression is beginning to emerge through its ability to alter the expression of genes involved at the G1/S boundary (Basaki et al., 2010; Yu et al., 2010). The transcriptional activity of YB-1 is dependent upon phosphorylation at its Ser-102 residue mediated by Akt/PKB and even more potently by ribosomal S6 kinase (RSK) (Stratford et al., 2008; Sutherland et al., 2005). To establish the importance of YB-1 in malignant transformation, transgenic mice were developed where expression was targeted to the lactating mammary gland (Bergmann et al., 2005). The resulting mouse mammary tumours formed with 100% penetrance and close examination revealed substantial centrosome amplification and chromosomal instability (Bergmann et al., 2005). Given these findings concurrent with the high prevalence of YB-1 in breast cancer, we hypothesized that it plays an essential role in breast tumourigenesis.
Genomic instability, in the form of alterations to chromosome number and structure, is a characteristic feature of almost all types of cancer (Fukasawa, 2007; Holland and Cleveland, 2009; Nigg, 2002; Nigg and Raff, 2009). However, whether this represents a cause or consequence of tumourigenesis remains mysterious. To address this contentious issue and begin to establish a paradigm for malignant transformation, it has become imperative to study cancer during the earliest premalignant stages, which, to date, have remained wholly uncharacterized. A large body of evidence has recently been compiled indicating that amplification of centrosomes has the potential to cause mitotic defects that lead to chromosomal instability (Basto et al., 2008; Ganem et al., 2009; Nigg, 2006). According to this model, centrosome abnormalities would need to emerge early during neoplastic progression. Ultimately, through Darwinian selection, a karyotype adept at enhancing tumour progression would materialize and expand (Fujiwara et al., 2005; Shi and King, 2005). Of all sporadic breast cancer, 20–30% exhibit amplification of HER2 prompting us to address if this is a common feature of premalignancy that arises through targeted genomic instability preceding clonal outgrowth (Slamon et al., 1987).
Here we examined the role of YB-1 during premalignancy to uncover the molecular events that define the earliest transitions in breast cancer initiation and progression. A comprehensive understanding of these processes will usher the development of novel therapeutics that target the process, rather than the consequences, of tumourigenesis.
Results
YB-1 alters the expression and activity of cell cycle associated proteins
To address the potential contribution of YB-1 in the initiation of tumourigenesis we engineered non-malignant H16N2 HMECs that conditionally expressed the gene under control of a tetracycline-inducible promoter (designated HTRY cells). HMECs containing an inducible LacZ construct served as a matched control (designated HTRZ cells). The ectopic expression level of YB-1 achieved in this model closely recapitulated that observed in established cancer cell lines (Figure S1A). Further characterization revealed that both cell lines were karyotypically normal (data shown below) and possessed similar telomerase activity deeming them genetically stable and thus amenable for investigating early transformation (Figure S1B).
To gain a global understanding into proteome remodeling following YB-1 induction we utilized the Kinexus Kinex™ antibody microarray platform, which allowed us to probe the expression and activation status (levels of phosphorylation) of over 600 proteins concurrently. From this unbiased protein array, we identified 56 proteins with altered expression (Table S1) many of which are fundamental in regulating centrosome dynamics and the cell cycle (Table 1). Identification of known YB-1 transcriptional targets, including HER2, strongly supported the fidelity of the screen. We prioritized subsequent analysis on the active LIM kinases (pLIMK1/2T508/T505)as they exhibited the most profound increase in level (365%) following YB-1 induction. This correlation was validated both by immunoblotting (Figure 1A) and immunofluorescence staining (Figure 1B). In a reciprocal experiment, silencing YB-1 with siRNA in MDA-MB-231 and SUM149 breast cancer cell lines repressed the phosphorylation of LIMK1/2at Thr-508/Thr-505 with minimal effect on total LIMK1 expression (Figure 1C). To confirm these results, we utilized a complimentary pharmacological approach for suppression of YB-1 activity using a signal transduction inhibitor to RSK. Our prior work indicated that inhibition of RSK directly impaired YB-1 phosphorylation and activity (Stratford et al., 2008). Treating MDA-MB-231 cells with the RSK inhibitor BI-D1870 yielded complete suppression of pLIMK1/2T508/T505 highlighting that pYB-1S102 was necessary to promote LIMK1/2 activation (Figure 1D). These data were mirrored using siRNA against RSK1 and RSK2 (Figure S2). Previous reports implicated LIMK1/2 as centrosomal proteins (Chakrabarti et al., 2007; Sumi et al., 2006) and, accordingly, we wanted to examine the localization in our HTRY cell model. At 96 hours post-YB-1 induction we observed punctate pLIMK1/2T508/T505 staining that corresponded to the centrosome as demonstrated by co-localization with the centrosomal marker γ-tubulin (Figure 1E).
Table 1.
Cell cycle associated proteins putatively regulated by YB-1
| Protein | Antibody | Localization | % CFC* |
|---|---|---|---|
| LIMK1/2 | Y507+T508/Y504+T505 | Centrosome | 365 |
| ZAP70 | Y315+Y319 | Centrosome | 230 |
| RSK1/2 | S380/S386 | Kinetochore | 269 |
| CDK1/2 | Y15 | Centrosome | 161 |
| Cdc34 | Pan-specific | Mitotic spindle | 148 |
| Cofilin | Pan-specific | Centrosome | 135 |
| p53 | Pan-specific | Cytoplasm/nucleus | 118 |
| CDK9 | Pan-specific | Nucleus | 116 |
| PP2A | Pan-specific | Centrosome | 102 |
| ZAP70 | Y292 | Centrosome | 102 |
| CDK6 | Pan-specific | Centrosome | 98 |
| PKA | Pan-specific | Centrosome | −67 |
%CFC refers to the percent change from control (uninduced HTRY cells)
Figure 1. YB-1 altered the activity of the centrosomal protein LIMK1/2.
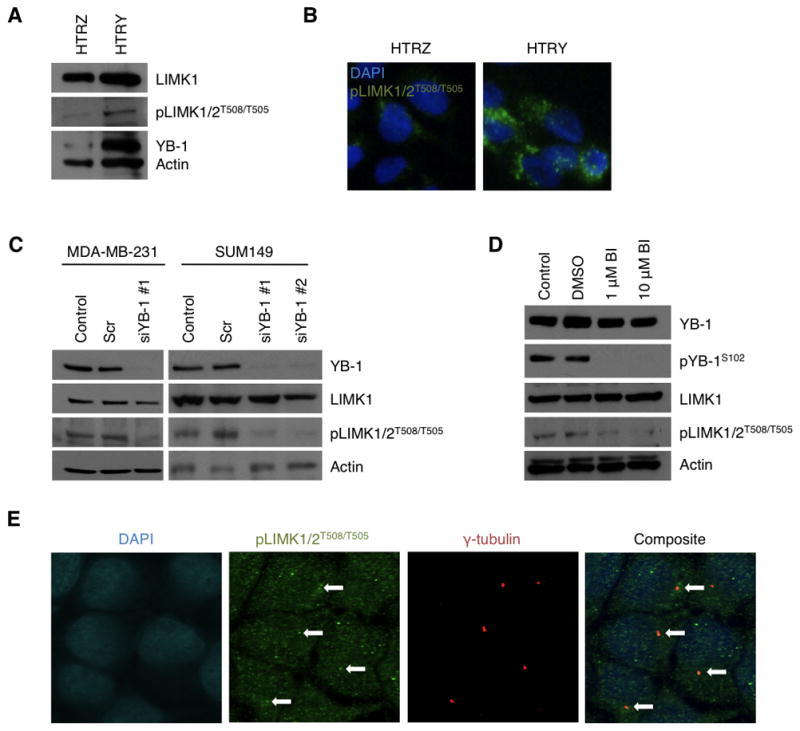
(A) Immunoblotting validated the efficacy of the HTRY cell line to express YB-1 following a 96-hour induction with doxycycline. Concurrent with YB-1 expression was an increase in active LIMK1/2, pLIMK1/2T508/T505. The HTRZ cells did not express YB-1 and exhibited reduced pLIMK1/2T508/T505 expression. (B) These observations were recapitulated in immunofluorescence staining of HTRZ and HTRY cells with pLIMK1/2T508/T505 antibody (green). (C) siYB-1 was used to silence protein expression in MDA-MB-231 and SUM149 cells and at 96 hours post-transfection both total YB-1 and the pool of active pLIMK1/2T508/T505 were depleted, as demonstrated by immunoblotting. (D) Similarly, treatment of MDA-MB-231 cells for 24 hours with BI-D1870 (1 μM or 10 μM) completely inhibited phosphorylation of Ser-102 of YB-1 and Thr-508/Thr-505 of LIMK1/2 as shown by immunoblotting. (E) It was demonstrated that pLIMK1/2T508/T505 (green) co-localized with the centrosomal marker γ-tubulin (red) in HTRY cells as visualized by immunofluorescence.
Cytokinesis failure primes cells for premalignant transformation
With YB-1 regulating a myriad of cell cycle associated genes, we wondered how cells from a non-malignant background would respond to expression of the gene. One of the earliest and most remarkable changes in the HTRY cells following YB-1 induction was the strikingly high incidence of multinucleated cells (Figure 2A). At 48 hours following YB-1 induction, which corresponded roughly to the doubling time of these cells (data not shown), 28% of HTRY cells were binucleate thus indicating a failure of cytokinesis. This was compared to only 5% of HTRZ cells (Figure 2B).
Figure 2. Premalignancy was initiated by pLIMK1/2T508/T505 mislocalization leading to cytokinesis failure.
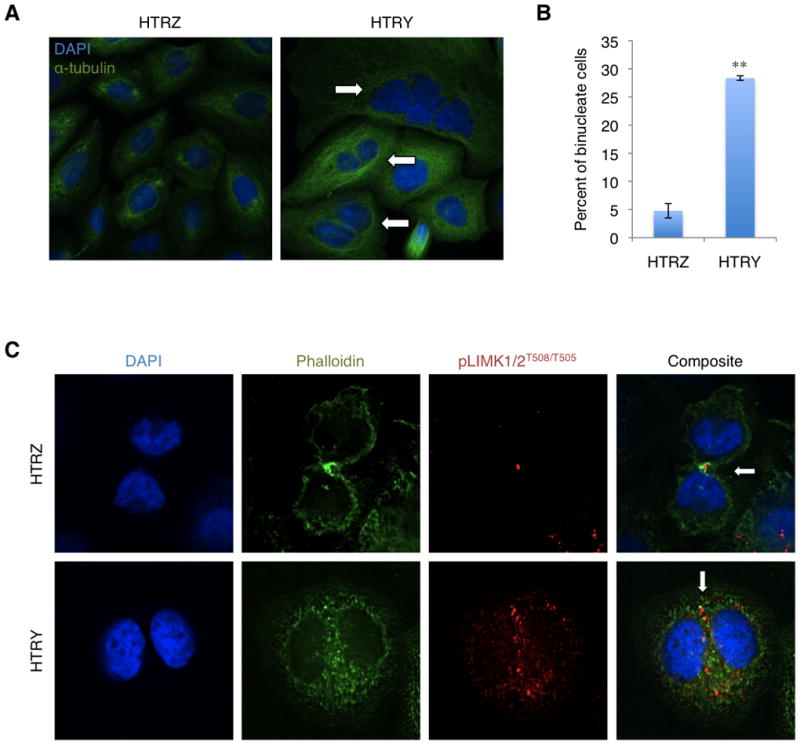
(A) HTRZ and HTRY cells were induced with doxycycline for 48 hours and immunostained with α-tubulin antibody (green) to define cell boundaries. Visually, many HTRY cells were binucleate (arrows) and, thus, (B) we quantified the proportion of HTRZ and HTRY cells displaying this phenotype. 200 cells were assessed in three independent experiments. (C) Immunofluorescence staining was employed to evaluate the spatial localization of pLIMK1/2T508/T505 (red) and phalloidin (green) in cytokinetic HTRZ and HTRY cells following a 48-hour induction with doxycycline. pLIMK1/2T508/T505 was concentrated in the cleavage furrow of HTRZ cells promoting stabilization of the actomyosin contractile ring (arrow). In HTRY cells, pLIMK1/2T508/T505 remained diffuse throughout the cytoplasm and the contractile ring failed to form (arrow).
Previous work has demonstrated that LIMK1 localizes to the cleavage furrow during cytokinesis (Sumi et al., 2006) and its overexpression is associated with polyploidy (Amano et al., 2002). Based on its role in modulating actin dynamics (Bernard, 2007), a likely explanation for the defect in cytokinesis might therefore be that deregulation of LIMK1/2 perturbs the stability of the contractile ring. In cytokinetic HTRZ cells, pLIMK1/2T508/T505 was concentrated in the junction between the two daughter cells as observed by immunofluorescence. Accordingly, F-actin was visualized, using phalloidin, along the cleavage furrow and at the nuclear periphery (Figure 2C). On the other hand, in cytokinetic HTRY cells, pLIMK1/2T508/T505 was strongly expressed but remained diffuse throughout the cytoplasm (Figure 2C). Consequently, the actin cytoskeleton failed to reposition itself for cytokinesis. Another well established protein at the cleavage furrow, polo-like kinase 1 (PLK1), correctly localized in HTRY cells during telophase, however, the actomyosin contractile ring consistently failed to form in these cells (Figure S3). This was in contrast to HTRZ cells, which formed a tight contractile ring with PLK1 characteristically localized to the midzone between the dividing cells. This supports our hypothesis that YB-1 promotes cytokinesis failure through deregulation and subsequent mislocalization of LIMK1/2.
Cell cycle checkpoint slippage potentiates centrosome amplification leading to aneuploidy
Cytokinesis failure can lead to both centrosome amplification and production of tetraploid cells, which could set the stage for the development of tumour cells (Fujiwara et al., 2005). We examined whether YB-1 expression allowed for cells arrested in cytokinesis to slip through cell cycle checkpoints and re-enter mitosis with multiple centrosomes and a compromised genome. At 96 hours post-YB-1 induction, HTRY cells demonstrated centrosome amplification leading to multipolar spindle formation in mitosis (Figure 3A). Consequently, kinetochore bi-orientation was not established resulting in failed segregation, which manifested as lagging chromosomes at the metaphase plate and micronuclei (Figure 3A). Quantifying the proportion of tetraploid cells (>4N DNA content) and those with supernumerary centrosomes (>2 centrosomes) revealed a significant increase of 4.5-fold and 12.2-fold, respectively, between the HTRY and HTRZ cells (Figure 3B). Deeper interrogation uncovered that the amplified centrosomes contained an excess of mother, but not daughter, centrioles (Figure S4A). Specifically, the 1:1 mother:daughter centriole ratio observed in the HTRZ cells approached 3:1 in the HTRY cells (Figure S4B). Having established the importance of pYB-1S102 at the centrosome it is not surprising that the described phenotype was contingent upon YB-1 Ser-102 phosphorylation as transient expression of YB-1S102D in HTRZ cells could recapitulate the phenotype whereas YB-1S102A could not (Figure S5A). Moreover, the incidence of aneuploidy and centrosome amplification was most profound in YB-1S102D overexpressing MDA-MB-231 cells by a significant margin (Figure S5B).
Figure 3. Centrosome amplification and aneuploidy emerged as a consequence of cytokinesis failure and cell cycle checkpoint slippage.

(A) At 96 hours post-YB-1 induction, HTRY cells demonstrated an amplification of centrosomes promoting multipolar spindle formation during mitosis and, as a result, lagging chromosomes and micronuclei (arrows). DAPI (blue) was used to visualize the nuclei, α-tubulin (green) labeled microtubules, and pericentrin (red) or γ-tubulin (red) served as markers for centrosomes. (B) At the same time point, we quantified the extent of polyploidy and centrosome amplification in 500 HTRZ and HTRY cells. DNA content was measured using Cellomics while the number of centrosomes per cell was counted manually. The data represent a compilation of three independent experiments. (C) Transient transfection of YB-1 into mammary epithelial 184-hTERT cells for 96 hours promoted polyploidy and centrosome amplification as measured by nuclear size/DAPI intensity and pericentrin immunofluorescence, respectively. Data was acquired from six random microscope fields and is presented as the mean and standard deviation from three independent experiments. Representative images portrayed genomic instability, while immunoblotting confirmed FLAG:YB-1 transgene expression in the 184-hTERT cells. (D) The proportion of HTRZ and HTRY cells in each phase of the cell cycle was ascertained by assessing DNA content using Cellomics on asynchronous populations of cells induced for 96 hours. Variation between three separate experiments is indicated. (E) To determine if cells with G2/M DNA content (4N) were truly in M-phase, we quantified the proportion of HTRZ and HTRY cells positive for pHistone H3S10 immunofluorescence (green). Data was collected from five unique microscope fields and is presented as the mean and standard deviation from three independent experiments. Representative images are shown. (F) In defining a mechanism for cell cycle checkpoint slippage, immunoblot analysis illustrated how YB-1 expression led to an enhancement in signal transduction through the RSK/p27Kip1 pathway thereby promoting cyclin E/CDK2 overexpression.
Due to HPV-16 E6/E7 immortalization, the p53 and retinoblastoma (Rb) tumour suppressor genes are inactivated in the HTRZ and HTRY cells. To ascertain if this background was necessary to generate the abnormal phenotypes observed in HTRY cells, we transfected YB-1 into the 184-hTERT cell line. The parental cells, which express no YB-1, have been extensively characterized to be chromosomally stable with a nearly normal karyotype (Raouf et al., 2005). Transient expression of YB-1 for 96-hours was sufficient to promote polyploidy and centrosome amplification (Figure 3C). We also observed early indicators of genomic instability, including lagging chromosomes and micronuclei (Figure 3C). The faithful recapitulation between phenotypes observed between the HTRY and 184-hTERT-YB-1 cells indicates that YB-1 expression alone is sufficient to drive genomic instability without the requirement of secondary gene deregulation.
One would expect that given the centrosome amplification coupled with aneuploid DNA content that the YB-1 induced cells would be subject to cell cycle arrest. To our surprise, only 18% of HTRY cells were classified as being in G1-phase of the cell cycle based on DNA content. This was in stark contrast to 66% of HTRZ cells (Figure 3D), strongly supporting the notion that HTRY cells resist anti-proliferative signals and slip through the G1/S checkpoint. Consistent with our findings of a cytokinesis defect, 62% of HTRY cells were in G2/M-phase compared to 14% of HTRZ cells (Figure 3D). To differentiate between cells arrested at the G2 checkpoint and those that have progressed into M-phase we quantified pHistone H3S10 by immunofluorescence. In agreement with increased DNA content, 23% of HTRY cells were positive for pHistone H3S10, thus truly in mitosis, compared to 3% of HTRZ cells (Figure 3E). This implies that overcoming the cytokinesis defect maybe a rate-limiting step in tumourigenesis.
We next assessed changes in signal transduction to define a mechanism to explain the observed slippage through the G1/S checkpoint. Following 96 hours of YB-1 induction, strong expression of HER2 was detected correlative with RSK activation. Accordingly, p27Kip1, an inhibitory target of RSK and negative regulator of cyclin E/CDK2, was suppressed (Figure 3F). These data demonstrate that YB-1 deregulates the cell cycle by altering signal transduction to favour a proliferative program.
Identification of YB-1 as a centrosomal protein
The data described above establishes the importance of YB-1 in regulating centrosomal proteins with a role in promoting amplification of the organelle during premalignancy. On this basis, we next explored whether YB-1 was itself directly associated with the centrosome. Co-localization between pYB-1S102 and pericentrin, a centrosomal marker, was observed in both interphase and mitotic HTRY cells using immunofluorescence (Figure 4A). Notably, during metaphase, pYB-1S102 was expressed along the entire length of the mitotic spindle. In MDA-MB-231 cells, pYB-1S102 was found to co-localize with pericentrin confirming its association with centrosomes was not unique to our inducible system but rather extended to established cancer cell lines (Figure S6A). To validate YB-1 as a bona fide centrosomal protein we mapped both FLAG:YB-1 (Figure S6B) and GFP:YB-1 (Figure S6C) to the centrosome using antibody directed against the FLAG epitope and direct immunofluorescence, respectively.
Figure 4. pYB-1S102 localized to the centrosomes throughout the cell cycle.

(A) Immunofluorescence staining with antibodies against pYB-1S102 (green) and pericentrin (red) demonstrated that pYB-1S102 was localized to the centrosomes in both interphase and mitotic HTRY cells (arrows). pYB-1S102 was predominately nuclear, however, it dissociated from DNA at metaphase and extended along the length of the mitotic spindle. (B) To evaluate the importance of YB-1 Ser-102 phosphorylation for its centrosomal localization, we generated MDA-MB-231 cells stably expressing FLAG:YB-1WT, FLAG:YB-1S102D, and FLAG:YB-1S102A protein. Immunoblotting confirmed ectopic expression of the tagged-proteins. (C, D) Quantification of FLAG (green) and pericentrin (red) co-localization in 250 interphase cells revealed that FLAG:YB-1WT and FLAG:YB-1S102D proteins, but not FLAG:YB-1S102A protein, localized to the centrosomes (arrows). Data represents a compilation from three independent experiments.
To ascertain whether phosphorylation was a prerequisite for YB-1 centrosomal localization, we generated MDA-MB-231 cells stably expressing FLAG:YB-1WT, FLAG:YB-1S102D, and FLAG:YB-1S102A protein (Figure 4B). Double immunofluorescence using anti-FLAG and anti-pericentrin antibodies revealed that the centrosomal localization of YB-1 was contingent upon phosphorylation at the Ser-102 residue. FLAG:YB-1S102D protein, which mimicked constitutively phosphorylated YB-1, was detected in 93.5% of centrosomes. In contrast, the non-phosphorylatable FLAG:YB-1S102A protein failed to localize to the centrosomes (Figures 4C and 4D). Collectively, these data provide insight into the dependence on phosphorylation for the trafficking, retention, and/or function of YB-1 at the centrosome.
To functionally examine the role of YB-1 at the centrosome we began by performing co-immunoprecipitation experiments, which revealed physical association between FLAG:YB-1 and the centrosomal proteins pericentrin and γ-tubulin (Figure 5A). We further queried whether LIMK1 was part of a YB-1 centrosomal complex; however, the two proteins failed to co-precipitate (data not shown). Next, we silenced YB-1 in MDA-MB-231 cells using two independent siRNA sequences (siYB-1#1 and siYB-1#2), which in turn, was found to yield substantial enlargement and morphological changes of the centrosomes as visualized by immunofluorescence targeting pericentrin (Figure 5B). Further investigation uncovered that these centrosomes harboured large clusters of γ-tubulin ring complexes (Figure 5B). We quantified a 3.3 to 4.1-fold increase in centrosome area at 96 hours post-siYB-1 transfection relative to mock-transfected cells (Figure 5C). Importantly, because Ser-102 phosphorylation was a necessity for YB-1 to localize at the centrosome, we wanted to assess if the mere inhibition of protein activity would be sufficient to alter centrosome structure. In agreement with the siRNA experiments, treating cells with 1 μM or 10 μM BI-D1870 for 24 hours prompted the emergence of cells containing enlarged centrosomes with numerous γ-tubulin ring complexes (Figure S7A). Specifically, the centrosomes increased in area by up to 2.9-fold relative to the DMSO-treated controls (Figure S7B). In further support, we observed increased centrosomal area in MDA-MB-231 cells stably expressing YB-1S102A mutant protein (Figure S7C). Finally, to analyze if the changes to centrosome structure correlated with altered function we performed microtubule-regrowth assays to detect defects in centrosome-mediated microtubule nucleation and anchoring. The assay was used to assess a fundamental parameter of centrosome function, that is, the ability to regrow microtubules following depolymerization. Displacement of YB-1 from the centrosome following siRNA silencing clearly delayed microtubule-regrowth as asters of short microtubules only began to emerge after a five minute regrowth as opposed to one minute in mock transfected cells (Figure 5D). This clearly demonstrates that loss of YB-1 perturbs normal centrosome function. Taken together, these results provide strong evidence for a previously uncharacterized, yet essential, role for YB-1at the centrosome.
Figure 5. YB-1 altered the architecture and microtubule nucleataion capacity of centrosomes by directly binding pericentrin and γ-tubulin.

(A) Co-immunoprecipitation of FLAG:YB-1 with γ-tubulin and pericentrin in MDA-MB-231 cells revealed direct association between the proteins. (B) To elucidate a role for YB-1 at the centrosomes, two unique siRNA oligos targeting YB-1 (siYB-1#1 and siYB-1#2) were transfected into MDA-MB-231 cells. At 96 hours post-transfection cells were analyzed by immunofluorescence staining with pericentrin (red; upper panel) and γ-tubulin (red; lower panel) to assess changes in centrosome organization. Immunoblotting confirmed YB-1 knockdown. (C) Centrosome area was measured using Image Pro Analyzer software and is represented relative to the non-transfected control. 100 centrosomes from G1-phase cells were measured across three independent experiments. (D) The function of YB-1 in microtubule nucleation at the centrosome was ascertained using the microtubule regrowth assay. Scrambled and siYB-1 transfected MDA-MB-231 cells were stained with α-tubulin (green) and γ-tubulin (red) antibodies at 1, 5, or 10 minutes after regrowth.
Genomic instability arises during premalignancy to generate clones with strong tumourigenic potential
To better understand the aneuploidy and chromosomal instability that emerge as a consequence of centrosome amplification during premalignancy, we assessed metaphase chromosomes. The majority of uninduced HTRZ and HTRY cells, as well as induced HTRZ cells, had a normal diploid karyotype. A small subset were classified as “near diploid” (40 to 52 chromosomes; Figure 6A). In stark contrast, 94% of induced HTRY cells were aneuploid (Figure 6A). Further to these numerical abnormalities, structural chromosome aberrations were readily detected in the induced HTRY cells. Dramatic increases of ≥5.5-fold in the appearance of dicentric chromosomes and double minutes were observed in the HTRY spreads compared to those from HTRZ cells. Most strikingly, there was a 17.6-fold increase in acentric pairs and 13.2-fold increase in acentric fragments between HTRY and HTRZ spreads indicating that YB-1 promoted extensive chromosome breakage (Figure 6B). In addition, defective sister chromatid cohesion was detected in the HTRY cells. Almost half of these cells exhibited a lack of primary constriction, identified by primary constriction gaps (PCGs) between sister chromatids at metaphase, compared to 11% of HTRZ cells (Figure S8).
Figure 6. Numerical and structural chromosomal aberrations materialized as a consequence of YB-1 expression.

(A, B) Metaphase spreads were performed on HTRZ and HTRY cells following a 96-hour induction with doxycycline. Uninduced cells served as controls. (A) The number of chromosomes and (B) structural chromosomal abnormalities were evaluated in 200 metaphase spreads from both HTRZ and HTRY cells. Three independent experiments were performed. Representative images are shown. (C) To determine if a recurrent pattern of amplification at the HER2 locus could be detected, we performed FISH analysis on interphase cells. Following a 96-hour induction, HTRZ and HTRY cells were hybridized with HER2 (red) and CEP17 (green) probes. 100 cells were assessed for low-copy and high-copy gene amplification. Representative images are shown.
To address whether genomic instability initiated by YB-1 could promote an optimal karyotypic composition for tumourigenesis, the frequency of HER2 amplification was measured using fluorescence in situ hybridization (FISH). We uncovered that 11% of HTRY cells were positive for HER2 amplification (HER2:CEP17 >2.2). No HTRZ cells exhibited HER2 amplification (Figure 6C). Upon a more rigorous assessment we noted that 20% of the HTRY cell population contained low-level HER2 amplification (HER2:CEP17 between 1.5 and 2.2; Figure 6C). The HER2 amplification in HTRY cells was largely undetected until they reached tetraploid DNA content. At this time there was an apparent relaxation in the mechanisms safeguarding genomic stability and the number of HER2 signals began to exceed the number of centromeres, a hallmark of gene amplification (Figure S9A and S9B). Collectively, these data indicate that a subset of premalignant cells have an amplification at the HER2 locus that could enhance their tumourigenic potential.
We conclude from our data that YB-1 is instrumental in activating a tumourigenic program that manifests as a cytokinesis defect and progresses toward the emergence of HER2-positive cancer (Figure 7). From this, we have proposed a model of premalignant progression.
Figure 7. Proposed model for how YB-1 instigates premalignancy.
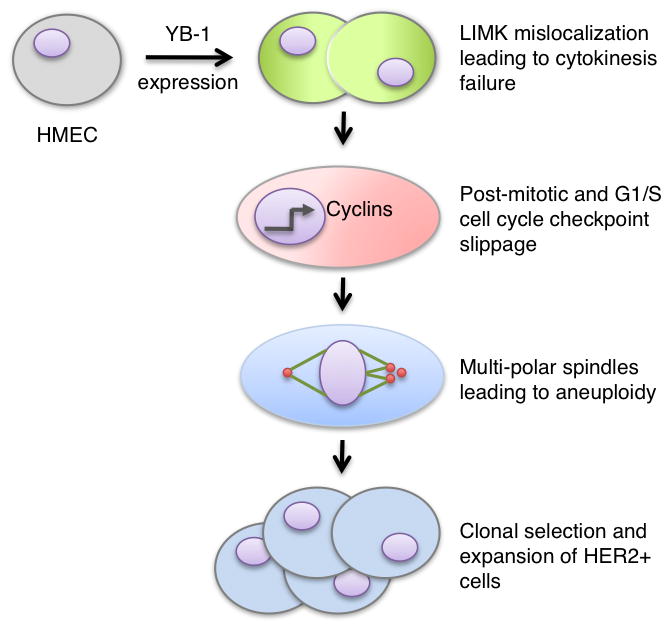
Targeted YB-1 expression in non-tumourigenic HMECs prompted a strong enhancement in LIMK1/2 activity that resulted in a cytokinesis defect. Concurrently, YB-1 altered signal transduction allowing cells to slip through the G1/S checkpoint. The resulting centrosome amplification led to multipolar spindles during mitosis, which promoted aneuploidy. With sustained YB-1 expression, a population of cells containing HER2 amplification emerged.
Discussion
In the present study we propose a distinctive model of breast cancer premalignancy whereby YB-1 enables the evolution of human mammary epithelial cells toward a tumourigenic fate. A cytokinesis defect acted as the initial trigger for transformation promoting centrosome amplification and aneuploidy, which were potentiated by cell cycle checkpoint slippage. In turn, we identified a small population of cells harbouring amplification at the HER2 locus. These studies provide significant insight into the process of tumour initiation and demonstrate how YB-1 alone can initiate a program that primes cells for tumourigenesis.
Although YB-1 upregulation is well characterized in breast cancer cell lines and advanced stage primary tumours (Habibi et al., 2008; Janz et al., 2002; Kohno et al., 2003; To et al., 2010), a role for the gene in tumour initiation and premalignant progression is unknown. Chromosomal aberrations observed in YB-1 transgenic mice prompted us to address if ectopic YB-1 expression in genetically stable HMECs acted to directly destabilize the genome as a prelude to malignancy (Bergmann et al., 2005). Here we have demonstrated that expression of the gene promoted gross alterations to the centrosomal milieu and, ultimately, led to centrosome amplification. Most notably, a strong activation of LIMK1/2 was detected at the centrosomes. Likewise, in prostate cancer LIMK is expressed at the centrosome and has been linked to chromosomal instability and metastasis (Chakrabarti et al., 2007; Davila et al., 2007; Yoshioka et al., 2003). An important finding of this study was that cytokinesis failure is the predominant mechanism for the amplification of centrosomes during premalignancy. We identified that sustained upregulation and mislocalization of active LIMK1/2 by YB-1 was sufficient to induce a cytokinesis defect. This could be attributed to enhanced actin polymerization at the cleavage furrow (Yang et al., 2004). Given these results we propose that YB-1 causes early changes in cytokinesis and centrosomal architecture that lead to eventual chromosomal instability.
In this work we have established YB-1 as a centrosomal protein. This was found to be contingent upon phosphorylation of the Ser-102 residue in the cold shock domain (CSD) implying that this domain is minimally required for centrosomal trafficking. Especially interesting is the fact that the CSD is necessary for binding oligonucleotides, including RNA due to the presence of two RNP motifs (Bouvet et al., 1995). As the centrosome contains an intrinsic compliment of RNA (Alliegro et al., 2006) it is possible that YB-1 is involved in regulating their translation. YB-1 has already been shown to induce cap-dependent translation of RNA giving credence to this hypothesis (Evdokimova et al., 2009). A second function for YB-1 at the centrosome may be to mediate protein stability via physical association. It has been demonstrated that YB-1 interacts with a myriad of proteins, including PCNA, MSH2, and DNA polymerase δ, via B/A repeats residing in the C-terminal domain (Gaudreault et al., 2004; Ise et al., 1999). It is tempting to speculate that centrosomal proteins may represent an underappreciated pool with strong capacity to promote tumourigenesis by their inherent ability to directly interface with the genome. In support of this, deregulation of centrosomal proteins including Aurora A, BRCA1, and PLK1 all promote genomic instability with eventual cellular transformation (Scully, 2000; Takai et al., 2005; Wang et al., 2006).
Studies of human mammary epithelial cells (HMEC) have proven effective in providing key insights into the early genetic events that fuel breast carcinogenesis (Dimri et al., 2005; Dumont et al., 2009; Elenbaas et al., 2001; Romanov et al., 2001; Tlsty et al., 2004). Here we report that YB-1 expression in this model leads to catastrophic genetic changes that if left unchecked could allow for the replication of cells containing vast chromosomal amplifications and rearrangements. Because YB-1 expressing HTRY cells fail to arrest following genomic destabilization it suggests that mutant cells are able to escape the necessary checkpoints needed to eliminate such renegade cells. Permissiveness through the cell cycle could relate to direct YB-1 transcriptional targets, such as CCNB1, CDC6, PCNA, and TOPO2 (Basaki et al., 2010; Jurchott et al., 2003; Shibao et al., 1999; Yu et al., 2010). We chose to address the possibility that YB-1 permits the expansion of cells harbouring specific amplifications common to breast cancer. Notably, we describe HER2 as being amplified in a small subset of HTRY cells. We speculate that over time this population of cells would clonally expand due to the distinct survival advantage brought about by HER2 overexpression. Moreover, this study furthers our understanding of the relationship between YB-1 and HER2 as previously described by our laboratory (Dhillon et al., 2010; Lee et al., 2008; Wu et al., 2006). Our previous studies show that YB-1 directly binds to the HER2 promoter in cells where the gene is known to be amplified (Wu et al., 2006). One could envisage that YB-1 is permissive for allowing cells with HER2 amplification to slip through the cell cycle checkpoints. Following this YB-1 is poised to increase the expression of HER2 by binding directly to its promoter. This too may explain why YB-1 and HER2 are highly expressed in ~65% of primary breast tumours (Habibi et al., 2008). Future work will focus on identifying additional genomic rearrangements that frequently materialize during premalignancy.
We report that increased expression of YB-1 is as a single event sufficient to uncouple genomic integrity and cell cycle progression during breast cancer premalignancy. In summary, our findings argue that YB-1 plays a principal role in the early evolution of cancer and thus represents a promising biomarker and therapeutic target.
Materials and methods
Cell culture and drug treatments
H16N2 human mammary epithelial cells with tetracycline-inducible YB-1 (HTRY) or LacZ (HTRZ) were generated using the T-Rex system as previously described (Band et al., 1990; Berquin et al., 2005). The cells were cultured in Ham’s F12 media and induction was achieved through the addition of 1 μg/mL doxycycline (Calbiochem, Gibbstown, NJ, USA). The human mammary epithelial 184-hTERT cell line (a gift from Dr. J. Carl Barrett, National Institutes of Health, Bethesda, MD) was cultured in supplemented HuMEC media (Invitrogen, Burlington, ON, Canada). LCC6, MDA-MB-231 (American Tissue culture collection, Manassas, VA, USA), and SUM149 (Asterand, Detroit, MI, USA) breast cancer cell lines were cultured as recommended.
For treatment with BI-D1870, MDA-MB-231 cells were seeded at a density of 3 × 105 cells in a six-well plate. Subsequently, cells were treated with a DMSO vehicle or BI-D1870 (1 μM or 10 μM) for 24 hours.
Kinexus Kinex™ antibody mircoarray
HTRY cells were induced for 96 hours. Comparisons were made to cells not treated with doxycycline. Protein was sent to Kinexus Bioinformatics Corporation (Vancouver, BC, Canada) for hybridization and analysis using the Kinex™ KAM-1.1 antibody microarray.
Immunoblotting, immunoprecipitation, and immunofluorescence
Immunoblotting, immunoprecipitation, and immunofluorescence were performed as described previously (Finkbeiner et al., 2009; Stratford et al., 2008; Wu et al., 2006). The origin and dilutions of all antibodies used in this study are detailed in Table S2. For immunoprecipitation, 500 μg of cell lysate was pre-cleared with 35 μl of protein G agarose (Sigma, St. Louis, MO, USA) prior to overnight antibody incubation. The proteins were retrieved through the addition of protein G agarose for 2 hours and eluted in 5× SDS-sample loading buffer heated to 100°C for 5 minutes. For immunofluorescence staining, antibodies were diluted in ICC buffer (10% BSA, 2% goat serum, 1% saponin in PBS), and all incubations were carried out at room temperature for one hour with three washes in PBS following each of the incubations. Cells were mounted using ProLong Gold antifade reagent containing DAPI (Invitrogen). Images were acquired using an Olympus FV1000 laser scanning confocal microscope, a DeltaVision personalDV live cell imaging microscope, or an Olympus BX61 epifluorescence microscope and analyzed with ImageJ 1.43 (National Institutes of Health, USA).
siRNA and plasmid transfections
Cells were transfected with 20 nM of siRNA to RSK1, RSK2, YB-1, or scrambled control using Lipofectamine RNAiMAX (Invitrogen). The siRNA target sequences are provided in Table S3. The empty vector, YB-1WT, YB-1S102A, and YB-1S102D constructs have previously been described (Finkbeiner et al., 2009; Sutherland et al., 2005; Wu et al., 2006). Plasmid transfections were performed using 4 μg of DNA and carried out with Lipofectamine 2000 (Invitrogen). Stable transfectants were generated and selected in G418 (400 μg/mL). The GFP:YB-1 construct (Guay et al., 2006), was transfected into MDA-MB-231 cells by electroporation with Amaxa Nucleofactor Kit V using the manufacturer’s recommendations (Lonza, Walkersville, MD, USA).
Microtubule regrowth assay
siRNA transfected MDA-MB-231 cells were treated with 5 μM nocodazole for 1 hour to depolymerize all microtubules. Nocodazole was then removed by washing twice with DMEM. At 1, 5, and 10 minutes after regrowth, the cells were fixed with 100% methanol and stained with α-tubulin and γ-tubulin antibodies.
Cell cycle analysis
HTRZ and HTRY cells were seeded in 96 well plates and induced for 96 hours. The cells were subsequently fixed using 2% paraformaldehyde and stained with Hoechst 33342 (1ug/ml; Sigma) for 30 minutes. Based on total nuclear Hoechst intensity, the proportion of cells in each stage of the cell cycle was analyzed by Cell Cycle Bioapplication software on a high content screening instrument (ArrayScan®, Thermo Fisher Scientific).
Chromosome spreads
Following a 96 hour induction, HTRZ and HTRY cells were treated with 0.1 μg/mL colcemid (Invitrogen) for 2 hours. Mitotic chromosomes were resuspended in hypotonic solution (75 mM KCl) for 20 minutes and fixed using methanol:glacial acetic acid (3:1) as previously described (Barber et al., 2008). Metaphase chromosomes were imaged using a Zeiss Axioplan digital imaging microscope and analyzed with Metamorph imaging software (Universal Imaging Corp, Downingtown, PA, USA). For analysis, we assessed chromosomal abnormalities based on their incidence with “mild” referring to less than five occurrences in a spread, “moderate” between five and twenty, and “severe” greater than twenty. Primary constriction gaps (PCGs) were defined as a clear separation between DAPI stained sister chromatids. The severity ranged from only one or two chromosomes in a spread exhibiting a gap (PCGI), to between three and ten chromosomes (PCGII), to no semblance of cohesion (PCGIII).
HER2 FISH
Asynchronous HTRZ and HTRY cells were prepared for chromosome analysis as described above. Interphase cells were hybridized with LSI HER2 and CEP17 probe using the PathVysion HER2 DNA Probe Kit at the Centre for Translational and Applied Genomics (Vancouver, BC, Canada). Analysis of FISH signals was performed in 100 randomly selected cells. HER2 amplification was defined as a HER2:CEP17 ratio of greater than 2.2. A HER2:CEP17 ratio <1.5 was considered negative for HER2 amplification while a ratio near the cut-off (1.5 – 2.2) was interpreted as intermediate amplification.
Telomerase assay
The telomerase activity in 1 μg of cell lysate from HTRZ and HTRY cells was measured using the Quantitative Telomerase Detection Kit (Allied Biotech, Vallejo, CA, USA) following the manufacturers instructions. Each sample was analyzed in triplicate. A no template control and cell lysate from telomerase positive cells (MDA-MB-231) were included in each experiment.
Statistical analysis
Data from at least three independent experiments are reported as mean ± standard deviation. Significance was examined using a paired Student’s t-test, where *P <0.05, **P <0.01, and ***P <0.001.
Supplementary Material
(A) Immunoblotting was performed to ensure that the level of YB-1 protein in HTRY cells, following a 96-hour induction, was similar to that measured across a panel of established breast cancer cell lines (SUM149, MDA-MB-231, and LCC6). HTRZ cells did not express YB-1. (B) Telomerase activity assay depicting a similar amount of telomerase activity in lysates from induced HTRZ and HTRY cells.
To validate that the decrease in LIMK1/2 phosphorylation following treatment with BI-D1870 was due to inhibition of YB-1 activity and not an off-target effect of the drug, we treated MDA-MB-231 cells with siRNA against RSK1 and RSK2. The decrease in both pYB-1S102 and pLIMK1/2T508/T505 following knockdown with RSK1+2 recapitulate the data obtained with BI-D1870.
We assessed changes in the localization of PLK1, a protein that is known to localize to the cleavage furrow, following YB-1 induction in HTRZ and HTRY cells by immunofluorescence. In contrast to pLIMK1/2T508/T505, PLK1 (red) localization at cytokinesis was as expected and not altered by YB-1 expression in HTRY cells. The actomyosin contractile ring can clearly be visualized using phalloidin (green) in HTRZ, but not HTRY, cells.
(A) HTRZ and HTRY cells were induced for 96 hours and subsequently analyzed by immunofluorescence using antibodies specific for mother (cenexin; green) and daughter (CNTROB; red) centrioles. (B) The number of mother and daughter centrioles was quantified in 50 cells. Data is presented as the mean and standard deviation of three independent experiments.
(A) To confirm the importance of YB-1 Ser-102 phosphorylation in the emergence of centrosome amplification and aneuploidy, HTRZ cells were transiently transfected with YB-1 wild-type (WT-YB-1) or mutant (S102D-YB-1 and S102A-YB-1) DNA. At 96 hours post-transfection, the number of multinucleated cells and those with amplified centrosomes were quantified. 500 cells were assessed across three independent experiments. (B) MDA-MB-231 cells were transfected with YB-1 wild-type or mutant DNA and stable clones were generated. The extent of polyploidy and centrosome amplification was assessed in 500 cells from three unique experiments.
(A) Immunofluorescence staining in MDA-MB-231 cells with antibodies targeting pYB-1S102 (green) and pericentrin (red) demonstrated that pYB-1S102 was localized to the centrosomes in an established cancer cell line. (B) To confirm YB-1 as a centrosomal protein, we generated MDA-MB-231 cells stably expressing FLAG:YB-1. Immunofluorescence staining with antibodies directed against FLAG (green) and pericentrin (red) confirmed YB-1 centrosomal localization. (C) Furthermore, MDA-MB-231 cells were transfected with full length GFP:YB-1 (green) via electroporation. By direct immunofluorescence, co-localization between YB-1 and pericentrin (red) was evident at the centrosomes.
(A) To specifically assess the role of phosphorylated YB-1 in promoting abnormal centrosome architecture, MDA-MB-231 cells were treated with either a DMSO vehicle or BI-D1870 (1 μM or 10 μM) for 24 hours. Immunofluorescence staining targeting pericentrin (red; upper panel) and γ-tubulin (red; lower panel) was used to visualize changes in centrosome organization. Immunoblotting confirmed suppression of pYB-1S102, but not total YB-1. (B) Centrosome area was measured using Image Pro Analyzer software and is represented relative to the untreated control. 100 centrosomes from G1-phase cells were measured across three independent experiments. (C) Immunofluorescence targeting pericentrin (red) to assess centrosomes in MDA-MB-231 cells stably expressing FLAG:YB-1WT, FLAG:YB-1S102D, or FLAG:YB-1S102A protein. Expression of YB-1S102A mutant protein, which does not localize to the centrosomes, yielded enlargement and disorganization similar to that observed following siYB-1 or BI-D1870 treatment.
Defects in sister chromatid cohesion were assessed in 200 metaphase spreads from HTRZ and HTRY cells following a 96-hour induction with doxycycline. Representative images demonstrate normal primary constriction cohesion and the various classes of defective cohesion: PCGI (mild), PCGII (moderate), and PCGIII (severe).
(A, B) Following a 96-hour induction, HTRZ and HTRY cells were hybridized with HER2/CEP17 probe and the number of signals were counted in 100 interphase cells. (A) HTRZ cells predominately exhibited a HER2:CEP17 ratio of 2:2, which is consistent with no amplification. (B) Amplification of HER2 relative to CEP17 (HER2:CEP17 >1.0) was detected in HTRY cells only after they reached tetraploid DNA content ( 4 copies of CEP17).
Table S1. Proteins regulated by YB-1 based on Kinex antibody microarray analysis
* %CFC refers to the percent change from control (uninduced HTRY cells)
Table S2. List of antibodies, dilutions, and suppliers used for immunoblotting (IB) and immunofluorescence (IF)
Table S3. siRNA target sequences
Acknowledgments
We thank Ching-Shih Chen (The Ohio State University) for providing BI-D1870 and Michel Lebel (l’Universite Laval) for providing plasmid encoding GFP-tagged YB-1. This study was supported by the National Institutes of Health RO1 CA114017 (SED, IMB), the Michael Smith Foundation for Health Research (AHD), and the Canadian Institutes of Health Research (AHD).
Footnotes
Conflict of interest:
The authors declare no conflict of interest.
References
- Alliegro MC, Alliegro MA, Palazzo RE. Centrosome-associated RNA in surf clam oocytes. Proc Natl Acad Sci U S A. 2006;103:9034–9038. doi: 10.1073/pnas.0602859103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano T, Kaji N, Ohashi K, Mizuno K. Mitosis-specific activation of LIM motif-containing protein kinase and roles of cofilin phosphorylation and dephosphorylation in mitosis. J Biol Chem. 2002;277:22093–22102. doi: 10.1074/jbc.M201444200. [DOI] [PubMed] [Google Scholar]
- Band V, Zajchowski D, Kulesa V, Sager R. Human papilloma virus DNAs immortalize normal human mammary epithelial cells and reduce their growth factor requirements. Proc Natl Acad Sci U S A. 1990;87:463–467. doi: 10.1073/pnas.87.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A. 2008;105:3443–3448. doi: 10.1073/pnas.0712384105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basaki Y, Taguchi K, Izumi H, Murakami Y, Kubo T, Hosoi F, et al. Y-box binding protein-1 (YB-1) promotes cell cycle progression through CDC6-dependent pathway in human cancer cells. Eur J Cancer. 2010;46:954–965. doi: 10.1016/j.ejca.2009.12.024. [DOI] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, et al. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann S, Royer-Pokora B, Fietze E, Jurchott K, Hildebrandt B, Trost D, et al. YB-1 provokes breast cancer through the induction of chromosomal instability that emerges from mitotic failure and centrosome amplification. Cancer Res. 2005;65:4078–4087. doi: 10.1158/0008-5472.CAN-04-4056. [DOI] [PubMed] [Google Scholar]
- Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071–1076. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Berquin IM, Pang B, Dziubinski ML, Scott LM, Chen YQ, Nolan GP, et al. Y-box-binding protein 1 confers EGF independence to human mammary epithelial cells. Oncogene. 2005;24:3177–3186. doi: 10.1038/sj.onc.1208504. [DOI] [PubMed] [Google Scholar]
- Bouvet P, Matsumoto K, Wolffe AP. Sequence-specific RNA recognition by the xenopus Y-box proteins. an essential role for the cold shock domain. J Biol Chem. 1995;270:28297–28303. doi: 10.1074/jbc.270.47.28297. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, Jones JL, Oelschlager DK, Tapia T, Tousson A, Grizzle WE. Phosphorylated LIM kinases colocalize with gamma-tubulin in centrosomes during early stages of mitosis. Cell Cycle. 2007;6:2944–2952. doi: 10.4161/cc.6.23.4957. [DOI] [PubMed] [Google Scholar]
- Davila M, Jhala D, Ghosh D, Grizzle WE, Chakrabarti R. Expression of LIM kinase 1 is associated with reversible G1/S phase arrest, chromosomal instability and prostate cancer. Mol Cancer. 2007;6:40. doi: 10.1186/1476-4598-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon J, Astanehe A, Lee C, Fotovati A, Hu K, Dunn SE. The expression of activated Y-box binding protein-1 serine 102 mediates trastuzumab resistance in breast cancer cells by increasing CD44+ cells. Oncogene. 2010;29:6294–6300. doi: 10.1038/onc.2010.365. [DOI] [PubMed] [Google Scholar]
- Dimri G, Band H, Band V. Mammary epithelial cell transformation: Insights from cell culture and mouse models. Breast Cancer Res. 2005;7:171–179. doi: 10.1186/bcr1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont N, Crawford YG, Sigaroudinia M, Nagrani SS, Wilson MB, Buehring GC, et al. Human mammary cancer progression model recapitulates methylation events associated with breast premalignancy. Breast Cancer Res. 2009;11:R87. doi: 10.1186/bcr2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell. 2009;15:402–415. doi: 10.1016/j.ccr.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Finkbeiner MR, Astanehe A, To K, Fotovati A, Davies AH, Zhao Y, et al. Profiling YB-1 target genes uncovers a new mechanism for MET receptor regulation in normal and malignant human mammary cells. Oncogene. 2009;28:1421–1431. doi: 10.1038/onc.2008.485. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer. 2007;7:911–924. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreault I, Guay D, Lebel M. YB-1 promotes strand separation in vitro of duplex DNA containing either mispaired bases or cisplatin modifications, exhibits endonucleolytic activities and binds several DNA repair proteins. Nucleic Acids Res. 2004;32:316–327. doi: 10.1093/nar/gkh170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guay D, Gaudreault I, Massip L, Lebel M. Formation of a nuclear complex containing the p53 tumor suppressor, YB-1, and the werner syndrome gene product in cells treated with UV light. Int J Biochem Cell Biol. 2006;38:1300–1313. doi: 10.1016/j.biocel.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Habibi G, Leung S, Law JH, Gelmon K, Masoudi H, Turbin D, et al. Redefining prognostic factors for breast cancer: YB-1 is a stronger predictor of relapse and disease-specific survival than estrogen receptor or HER-2 across all tumor subtypes. Breast Cancer Res. 2008;10:R86. doi: 10.1186/bcr2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ise T, Nagatani G, Imamura T, Kato K, Takano H, Nomoto M, et al. Transcription factor Y-box binding protein 1 binds preferentially to cisplatin-modified DNA and interacts with proliferating cell nuclear antigen. Cancer Res. 1999;59:342–346. [PubMed] [Google Scholar]
- Janz M, Harbeck N, Dettmar P, Berger U, Schmidt A, Jurchott K, et al. Y-box factor YB-1 predicts drug resistance and patient outcome in breast cancer independent of clinically relevant tumor biologic factors HER2, uPA and PAI-1. Int J Cancer. 2002;97:278–282. doi: 10.1002/ijc.1610. [DOI] [PubMed] [Google Scholar]
- Jurchott K, Bergmann S, Stein U, Walther W, Janz M, Manni I, et al. YB-1 as a cell cycle-regulated transcription factor facilitating cyclin A and cyclin B1 gene expression. J Biol Chem. 2003;278:27988–27996. doi: 10.1074/jbc.M212966200. [DOI] [PubMed] [Google Scholar]
- Kohno K, Izumi H, Uchiumi T, Ashizuka M, Kuwano M. The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays. 2003;25:691–698. doi: 10.1002/bies.10300. [DOI] [PubMed] [Google Scholar]
- Lee C, Dhillon J, Wang MY, Gao Y, Hu K, Park E, et al. Targeting YB-1 in HER-2 overexpressing breast cancer cells induces apoptosis via the mTOR/STAT3 pathway and suppresses tumor growth in mice. Cancer Res. 2008;68:8661–8666. doi: 10.1158/0008-5472.CAN-08-1082. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer. 2006;119:2717–2723. doi: 10.1002/ijc.22245. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosome aberrations: Cause or consequence of cancer progression? Nat Rev Cancer. 2002;2:815–825. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–678. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Raouf A, Brown L, Vrcelj N, To K, Kwok W, Huntsman D, et al. Genomic instability of human mammary epithelial cells overexpressing a truncated form of EMSY. J Natl Cancer Inst. 2005;97:1302–1306. doi: 10.1093/jnci/dji254. [DOI] [PubMed] [Google Scholar]
- Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, Tlsty TD. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature. 2001;409:633–637. doi: 10.1038/35054579. [DOI] [PubMed] [Google Scholar]
- Scully R. Role of BRCA gene dysfunction in breast and ovarian cancer predisposition. Breast Cancer Res. 2000;2:324–330. doi: 10.1186/bcr76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, King RW. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature. 2005;437:1038–1042. doi: 10.1038/nature03958. [DOI] [PubMed] [Google Scholar]
- Shibao K, Takano H, Nakayama Y, Okazaki K, Nagata N, Izumi H, et al. Enhanced coexpression of YB-1 and DNA topoisomerase II alpha genes in human colorectal carcinomas. Int J Cancer. 1999;83:732–737. doi: 10.1002/(sici)1097-0215(19991210)83:6<732::aid-ijc6>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Stratford AL, Fry CJ, Desilets C, Davies AH, Cho YY, Li Y, et al. Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res. 2008;10:R99. doi: 10.1186/bcr2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford AL, Habibi G, Astanehe A, Jiang H, Hu K, Park E, et al. Epidermal growth factor receptor (EGFR) is transcriptionally induced by the Y-box binding protein-1 (YB-1) and can be inhibited with iressa in basal-like breast cancer, providing a potential target for therapy. Breast Cancer Res. 2007;9:R61. doi: 10.1186/bcr1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi T, Hashigasako A, Matsumoto K, Nakamura T. Different activity regulation and subcellular localization of LIMK1 and LIMK2 during cell cycle transition. Exp Cell Res. 2006;312:1021–1030. doi: 10.1016/j.yexcr.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Sutherland BW, Kucab J, Wu J, Lee C, Cheang MC, Yorida E, et al. Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene. 2005;24:4281–4292. doi: 10.1038/sj.onc.1208590. [DOI] [PubMed] [Google Scholar]
- Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (plks) and cancer. Oncogene. 2005;24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- Tlsty TD, Crawford YG, Holst CR, Fordyce CA, Zhang J, McDermott K, et al. Genetic and epigenetic changes in mammary epithelial cells may mimic early events in carcinogenesis. J Mammary Gland Biol Neoplasia. 2004;9:263–274. doi: 10.1023/B:JOMG.0000048773.95897.5f. [DOI] [PubMed] [Google Scholar]
- To K, Fotovati A, Reipas KM, Law JH, Hu K, Wang J, et al. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010;70:2840–2851. doi: 10.1158/0008-5472.CAN-09-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, et al. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 2006;25:7148–7158. doi: 10.1038/sj.onc.1209707. [DOI] [PubMed] [Google Scholar]
- Wu J, Lee C, Yokom D, Jiang H, Cheang MC, Yorida E, et al. Disruption of the Y-box binding protein-1 results in suppression of the epidermal growth factor receptor and HER-2. Cancer Res. 2006;66:4872–4879. doi: 10.1158/0008-5472.CAN-05-3561. [DOI] [PubMed] [Google Scholar]
- Yang X, Yu K, Hao Y, Li DM, Stewart R, Insogna KL, et al. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol. 2004;6:609–617. doi: 10.1038/ncb1140. [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Foletta V, Bernard O, Itoh K. A role for LIM kinase in cancer invasion. Proc Natl Acad Sci U S A. 2003;100:7247–7252. doi: 10.1073/pnas.1232344100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YN, Yip GW, Tan PH, Thike AA, Matsumoto K, Tsujimoto M, et al. Y-box binding protein 1 is up-regulated in proliferative breast cancer and its inhibition deregulates the cell cycle. Int J Oncol. 2010;37:483–492. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Immunoblotting was performed to ensure that the level of YB-1 protein in HTRY cells, following a 96-hour induction, was similar to that measured across a panel of established breast cancer cell lines (SUM149, MDA-MB-231, and LCC6). HTRZ cells did not express YB-1. (B) Telomerase activity assay depicting a similar amount of telomerase activity in lysates from induced HTRZ and HTRY cells.
To validate that the decrease in LIMK1/2 phosphorylation following treatment with BI-D1870 was due to inhibition of YB-1 activity and not an off-target effect of the drug, we treated MDA-MB-231 cells with siRNA against RSK1 and RSK2. The decrease in both pYB-1S102 and pLIMK1/2T508/T505 following knockdown with RSK1+2 recapitulate the data obtained with BI-D1870.
We assessed changes in the localization of PLK1, a protein that is known to localize to the cleavage furrow, following YB-1 induction in HTRZ and HTRY cells by immunofluorescence. In contrast to pLIMK1/2T508/T505, PLK1 (red) localization at cytokinesis was as expected and not altered by YB-1 expression in HTRY cells. The actomyosin contractile ring can clearly be visualized using phalloidin (green) in HTRZ, but not HTRY, cells.
(A) HTRZ and HTRY cells were induced for 96 hours and subsequently analyzed by immunofluorescence using antibodies specific for mother (cenexin; green) and daughter (CNTROB; red) centrioles. (B) The number of mother and daughter centrioles was quantified in 50 cells. Data is presented as the mean and standard deviation of three independent experiments.
(A) To confirm the importance of YB-1 Ser-102 phosphorylation in the emergence of centrosome amplification and aneuploidy, HTRZ cells were transiently transfected with YB-1 wild-type (WT-YB-1) or mutant (S102D-YB-1 and S102A-YB-1) DNA. At 96 hours post-transfection, the number of multinucleated cells and those with amplified centrosomes were quantified. 500 cells were assessed across three independent experiments. (B) MDA-MB-231 cells were transfected with YB-1 wild-type or mutant DNA and stable clones were generated. The extent of polyploidy and centrosome amplification was assessed in 500 cells from three unique experiments.
(A) Immunofluorescence staining in MDA-MB-231 cells with antibodies targeting pYB-1S102 (green) and pericentrin (red) demonstrated that pYB-1S102 was localized to the centrosomes in an established cancer cell line. (B) To confirm YB-1 as a centrosomal protein, we generated MDA-MB-231 cells stably expressing FLAG:YB-1. Immunofluorescence staining with antibodies directed against FLAG (green) and pericentrin (red) confirmed YB-1 centrosomal localization. (C) Furthermore, MDA-MB-231 cells were transfected with full length GFP:YB-1 (green) via electroporation. By direct immunofluorescence, co-localization between YB-1 and pericentrin (red) was evident at the centrosomes.
(A) To specifically assess the role of phosphorylated YB-1 in promoting abnormal centrosome architecture, MDA-MB-231 cells were treated with either a DMSO vehicle or BI-D1870 (1 μM or 10 μM) for 24 hours. Immunofluorescence staining targeting pericentrin (red; upper panel) and γ-tubulin (red; lower panel) was used to visualize changes in centrosome organization. Immunoblotting confirmed suppression of pYB-1S102, but not total YB-1. (B) Centrosome area was measured using Image Pro Analyzer software and is represented relative to the untreated control. 100 centrosomes from G1-phase cells were measured across three independent experiments. (C) Immunofluorescence targeting pericentrin (red) to assess centrosomes in MDA-MB-231 cells stably expressing FLAG:YB-1WT, FLAG:YB-1S102D, or FLAG:YB-1S102A protein. Expression of YB-1S102A mutant protein, which does not localize to the centrosomes, yielded enlargement and disorganization similar to that observed following siYB-1 or BI-D1870 treatment.
Defects in sister chromatid cohesion were assessed in 200 metaphase spreads from HTRZ and HTRY cells following a 96-hour induction with doxycycline. Representative images demonstrate normal primary constriction cohesion and the various classes of defective cohesion: PCGI (mild), PCGII (moderate), and PCGIII (severe).
(A, B) Following a 96-hour induction, HTRZ and HTRY cells were hybridized with HER2/CEP17 probe and the number of signals were counted in 100 interphase cells. (A) HTRZ cells predominately exhibited a HER2:CEP17 ratio of 2:2, which is consistent with no amplification. (B) Amplification of HER2 relative to CEP17 (HER2:CEP17 >1.0) was detected in HTRY cells only after they reached tetraploid DNA content ( 4 copies of CEP17).
Table S1. Proteins regulated by YB-1 based on Kinex antibody microarray analysis
* %CFC refers to the percent change from control (uninduced HTRY cells)
Table S2. List of antibodies, dilutions, and suppliers used for immunoblotting (IB) and immunofluorescence (IF)
Table S3. siRNA target sequences