

Abstract
Kabuki syndrome is a rare, multiple malformation disorder characterized by a distinctive facial appearance, cardiac anomalies, skeletal abnormalities, and mild to moderate intellectual disability. Simplex cases make up the vast majority of the reported cases with Kabuki syndrome, but parent-to-child transmission in more than a half-dozen instances indicates that it is an autosomal dominant disorder. We recently reported that Kabuki syndrome is caused by mutations in MLL2, a gene that encodes a Trithorax-group histone methyltransferase, a protein important in the epigenetic control of active chromatin states. Here, we report on the screening of 110 families with Kabuki syndrome. MLL2 mutations were found in 81/110 (74%) of families. In simplex cases for which DNA was available from both parents, 25 mutations were confirmed to be de novo, while a transmitted MLL2 mutation was found in two of three familial cases. The majority of variants found to cause Kabuki syndrome were novel nonsense or frameshift mutations that are predicted to result in haploinsufficiency. The clinical characteristics of MLL2 mutation-positive cases did not differ significantly from MLL2 mutation-negative cases with the exception that renal anomalies were more common in MLL2 mutation-positive cases. These results are important for understanding the phenotypic consequences of MLL2 mutations for individuals and their families as well as for providing a basis for the identification of additional genes for Kabuki syndrome.
Keywords: Kabuki syndrome, MLL2, ALR, Trithorax group histone methyltransferase
INTRODUCTION
Kabuki syndrome (OMIM#147920) is a rare, multiple malformation disorder characterized by a distinctive facial appearance, cardiac anomalies, skeletal abnormalities, and mild to moderate intellectual disability. It was originally described by Niikawa et al. [1981] and Kuroki et al. [1981] in 1981, and to date, about 400 cases have been reported worldwide [Adam and Hudgins, 2005; Niikawa et al., 1988; White et al., 2004]. The spectrum of abnormalities found in individuals with Kabuki syndrome is diverse, yet virtually all affected persons are reported to have similar facial features consisting of elongated palpebral fissures, eversion of the lateral third of the lower eyelids, and broad, arched eyebrows with lateral sparseness. Additionally, affected individuals commonly have severe feeding problems, failure to thrive in infancy and height around or below the 3rd centile for age in about half of cases.
We recently reported that a majority of cases of Kabuki syndrome are caused by mutations in mixed lineage leukemia 2 (MLL2; OMIM#602113), also known as either MLL4 or ALR [Ng et al., 2010]. MLL2 encodes a SET-domain-containing histone methyltransferase important in the epigenetic control of active chromatin states [FitzGerald and Diaz, 1999]. Exome sequencing revealed that nine of ten individuals had novel variants in MLL2 that were predicted to be deleterious. A single individual had no mutation in the protein-coding exons of MLL2, though in retrospect, his phenotypic features are somewhat atypical of Kabuki syndrome. In a larger validation cohort screened by Sanger sequencing, we found MLL2 mutations in approximately two-thirds of 43 Kabuki cases, suggesting that Kabuki syndrome is genetically heterogeneous.
Herein we report on the results of screening MLL2 for mutations in 110 families with one or more individuals affected with Kabuki syndrome in order to: (1) characterize the spectrum of MLL2 mutations that cause Kabuki syndrome; (2) determine whether MLL2 genotype is predictive of phenotype; (3) assess whether the clinical characteristics of MLL2 mutation-positive cases differ from MLL2 mutation-negative cases; and (4) delineate the subset of Kabuki cases that are MLL2 mutation-negative for further gene discovery studies.
MATERIALS AND METHODS
Subjects
Referral for inclusion into the study required a diagnosis of Kabuki syndrome made by a clinical geneticist. From these cases, phenotypic data were collected by review of medical records, phone interviews, and photographs. These data were summarized by each collaborating clinician and forwarded for review to two of the authors (MB and MH). These data were collected from five different clinical genetics centers in three different countries and over a protracted period of time and forwarded for review to two of the authors (MB and MH). Data on certain phenotypic characteristics including stature, feeding difficulties, and failure to thrive was not uniformly collected or standardized. Therefore, we decided to be conservative in our analysis and use only phenotypic traits that could be represented by discrete variables (i.e., presence or absence) and for which data were available from at least 70% of cases. In addition, these clinical summaries were de-identified and therefore facial photographs were unavailable from most cases studied. Written consent was obtained for all participants who provided identifiable samples. The Institutional Review Boards of Seattle Children’s Hospital and the University of Washington approved all studies. A summary of the clinical characteristics of 53 of these individuals diagnosed with Kabuki syndrome has been reported previously [Ng et al., 2010].
Mutation analysis
Genomic DNA was extracted using standard protocols. Each of the 54 exons of MLL2 was amplified using Taq DNA polymerase (Invitrogen, Carlsbad, CA) following manufacturer’s recommendations and using primers previously reported [Ng et al. 2010]. PCR products were purified by treatment with exonuclease I (New England Biolabs, Inc., Beverly, MA) and shrimp alkaline phosphatase (USB Corp., Cleveland, OH), and products were sequenced using the dideoxy terminator method on an automatic sequencer (ABI 3130xl). The electropherograms of both forward and reverse strands were manually reviewed using CodonCode Aligner (Dedham, MA). Primer sequences and conditions are listed in Supplementary Table I.
For MLL2 mutation-negative samples, DNA was hybridized to commercially available whole-genome tiling arrays consisting of one million oligonucleotide probes with an average spacing of 2.6 kb throughout the genome (SurePrint G3 Human CGH Microarray 1×1M, Agilent Technologies, Santa Clara, CA). Twenty-one probes on this array covered MLL2 specifically. Data were analyzed using Genomics Workbench software according to manufacturer’s instructions.
RESULTS
All 54 protein-coding exons and intron-exon boundaries of MLL2 were screened by Sanger sequencing in a cohort of 110 kindreds with Kabuki syndrome. This cohort included 107 simplex cases (including a pair of monozygotic twins) and three familial (i.e., parent-offspring) cases putatively diagnosed with Kabuki syndrome. Seventy novel MLL2 variants that were inferred to be disease-causing were identified in 81/110 (74%) kindreds (Fig 1 and Supplementary Table II online). These eighty-one mutations included 37 nonsense mutations (32 different sites and five sites with recurrent mutations), three in-frame deletions or duplications (2 different sites and 1 site with a recurrent mutation), 22 frameshifts (22 different sites), 16 missense mutations (11 different sites and four sites with recurrent mutations) and 3 splice consensus site (or intron-exon boundary) mutations. None of these variants were found in dbSNP (build 132), the 1000 Genomes Project pilot data, 190 chromosomes from individuals matched for geographical ancestry. In total, pathogenic variants were found at seventy sites. Additionally, there were ten sites at which recurrent mutations were observed.
Figure 1. Genomic structure and allelic spectrum of MLL2 mutations that cause Kabuki syndrome.

MLL2 is composed of 54 exons that include untranslated regions (orange) and protein coding sequence (blue) including 7 PHD fingers (yellow), FYRN (green), FYRC (green), and a SET domain (red). Arrows indicate the locations of 81 mutations affecting 70 sites found in 110 families with Kabuki syndrome including: 37 nonsense, 22 frameshifts, 16 missense, 3 in-frame deletions/duplications, and 3 splice-site mutations. Asterisks indicate mutations that were confirmed to be de novo and crosses indicate cases for which parental DNA was unavailable.
For 25 simplex cases in which we identified MLL2 mutations, DNA was available from both unaffected parents, and in each case the mutation was confirmed to have arisen de novo (Supplementary Table II online). These included 14 nonsense, five frameshift, three missense, two splice site mutations and one deletion. De novo events were confirmed at six of the 10 sites where recurrent mutations were noted. In addition to the 81 kindreds in which we identified causal MLL2 mutations, we found two MLL2 variants in each of three simplex cases. In each case, neither MLL2 mutation could unambiguously be defined as disease-causing (Supplementary Table II online). In one case, we found both a 21 bp in-frame insertion in exon 39 and a 1 bp insertion in exon 46 predicted to cause a frameshift. However, the unaffected mother also carried the 21 bp insertion suggesting that this is a rare polymorphism, and that the 1 bp deletion is the pathogenic mutation responsible for Kabuki syndrome.
Apparent disease-causing variants were discovered in nearly half (i.e., 22/54) of all protein-coding exons of MLL2 and in virtually every region known to encode a functional domain (Fig 1). However, the distribution of variants appeared non-random as 13 and 12 novel variants were identified in exons 48 and 39, respectively. These sites accounted for 25, or more than one-third, of all the novel MLL2 variants and 31/81 mutations that cause Kabuki syndrome in our cohort. Eleven of the 12 pathogenic variants in exon 39 were nonsense mutations and occurred in regions that encode long polyglutamine tracts.
Four of the families studied herein had two individuals affected with Kabuki syndrome. A pair of monozygous twins with a c.15195G>A mutation were concordant for mild developmental delay, congenital heart disease, preauricular pits and palatal abnormalities, but discordant for hearing loss, and a central nervous system malformation. Concordance for mild developmental delay between an affected parent and child was observed in two families with MLL2 mutations, one with a nonsense mutation, c.13579A>T, p.K4527X, and the other with a missense mutation, c.16391C>T, p.T5464M that was also found in a simplex case. No MLL2 mutation was found in the remaining affected parent and child pair.
To examine the relationship between genotype and phenotype, we first compared the frequency of developmental delay, congenital heart disease, cleft lip and/or palate, and structural renal defects between MLL2 mutation-positive vs. MLL2 mutation-negative cases. No significant difference was observed between groups for three of these four phenotypes (Table I, a). However, renal anomalies were observed in 47% (31/66 cases) of MLL2 mutation-positive cases compared to 14% (2/14 cases) of MLL2 mutation-negative cases and this difference was statistically significant (χ=5.1, df=1, p=0.024). In 35 cases in two clinical cohorts for whom more complete phenotypic data were available, short stature was observed in 54% (14/26) of MLL2 mutation-positive cases compared to 33% (3/19 cases) of MLL2 mutation-negative cases. We also divided the MLL2 mutation-positive cases into those with nonsense and frameshift mutations and those with missense mutations and compared the frequency of developmental delay, congenital heart disease, cleft lip and/or palate, and structural renal defects between groups. No significant differences were observed between groups (Table I, b).
Table I.
Phenotypic traits grouped by MLL2 mutation status (a) and type (b)
| Trait | MLL2 + | MLL2 − |
|---|---|---|
| Intellectual Disability | 74/74 (100%) | 19/20 (95%) |
| Mild | 51/74 (69%) | 10/20 (50%) |
| Moderate | 18/74 (24%) | 4/20 (20%) |
| Severe | 4/74 (5%) | 3/20 (15%) |
| Cleft palate, CL/CP | 29/72 (40%) | 8/18 (44%) |
| Congenital heart defect | 36/71 (51%) | 8/19 (42%) |
| Renal abnormality | 31/66 (47%) | 2/14 (14%) |
| Trait | Truncating (N=59) | Missense (N=16) |
| Intellectual disability | 54/54 (100%) | 15/15 (100%) |
| Mild | 36/54 (67%) | 11/15 (73 %) |
| Moderate | 13/54 (24%) | 4/15 (27%) |
| Severe | 5/54 (9%) | 0/15 |
| Cleft palate, CL/CP | 23/54 (43%) | 3/14 (21%) |
| Congenital heart defect | 30/54 (55%) | 4/13 (30%) |
| Renal anomaly | 9/44 (20%) | 2/12 (17%) |
In 26 independent cases of Kabuki syndrome, including one parent-offspring pair, no MLL2 mutation was identified. Both persons in the mother-child pair had facial characteristics consistent with Kabuki syndrome (Fig 2), mild developmental delay, and no major malformations. The mother is of Cambodian ancestry and her daughter is of Cambodian and European American ancestry. In general, most of the MLL2 mutation-negative Kabuki cases had facial characteristics (Fig 3) similar to those of the MLL2 mutation-positive Kabuki cases, and a similar pattern of major malformations (Table I) with the exception of fewer renal abnormalities.
Figure 2.

Facial photographs of mother and daughter with Kabuki syndrome in whom no causative mutation in MLL2 was identified. Both have mild developmental delay and no known major malformations.
Figure 3.
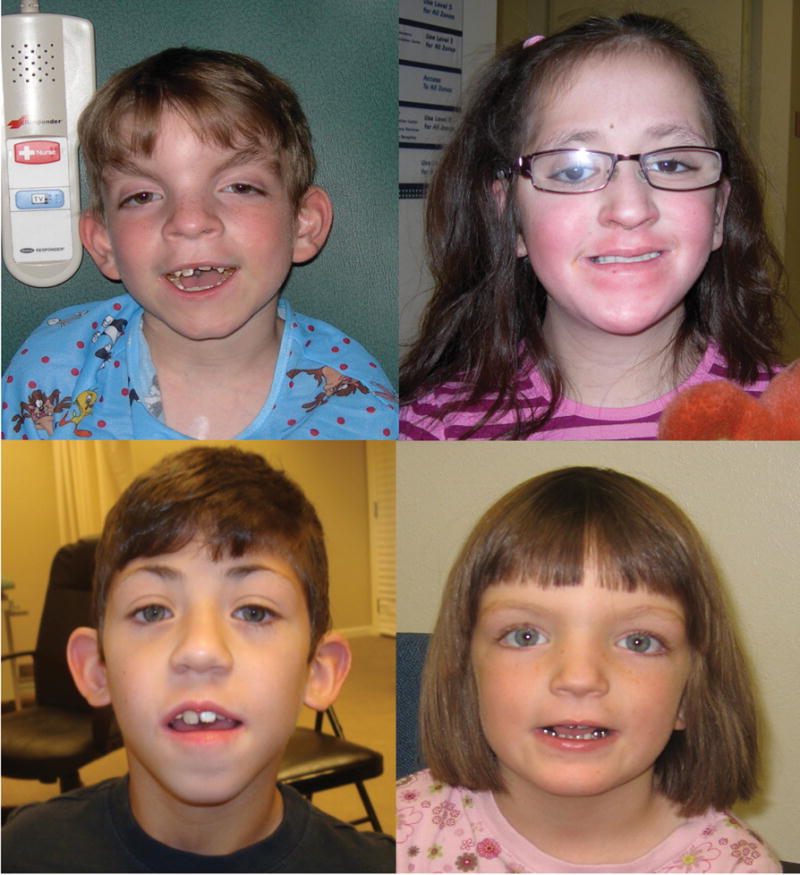
Facial photographs of four children diagnosed with Kabuki syndrome in whom no causative mutation in MLL2 was found.
We screened the MLL2 mutation-negative cases by aCGH for large deletions or duplications that encompassed MLL2. Abnormalities were found in four cases. In one case, a 1.87 kb deletion of chromosome 5 (hg18, chr5:175,493,803-177,361,744) that included NSD1 and had breakpoints in flanking segmental duplications identical to the microdeletion commonly found in Sotos syndrome, was found. This suggests that this individual has Sotos syndrome, not Kabuki syndrome [Kurotaki et al., 2002]. A second case had a novel 977-kb deletion of chromosome 19q13 (hg18, chr19:61,365,420–62,342,064) encompassing 20 genes. The majority of genes within the deleted region are zinc finger genes, some of which are known to be imprinted in both human and mouse. A third case had a complex translocation t(8;18)(q22;q21). Finally, a fourth case was found to have extra material for the entire chromosome 12. Average log2 ratio across chromosome 12 was 0.49, most likely representing mosaic aneuploidy of chromosome 12. No aCGH abnormalities were observed in 21 cases and aCGH failed for one case.
DISCUSSION
We have expanded the spectrum of mutations in MLL2 that cause Kabuki syndrome and explored the relationship between MLL2 genotype and some of the major, objective phenotypic characteristics of Kabuki syndrome. The majority of variants found to cause Kabuki syndrome are either novel nonsense or frameshift mutations, and appear to arise de novo. While mutations that cause Kabuki syndrome are found throughout the MLL2 gene, there appear to be at least two exons (39 and 48) in which mutations are identified with a considerably higher frequency. Mutations in these three exons account for nearly half of all mutations found in MLL2, while the length of these exons represents ~35% of the MLL2 open reading frame (ORF). Furthermore, exon 48, the exon in which mutations are most common, comprises only ~7% of the MLL2 ORF. Exon 39 contains several regions that encode long polyglutamine tracts suggesting the presence of a mutational hotspot, although no such explanation is obvious for exon 48. A stepwise approach in which these regions are the first screened might be a reasonable approach to diagnostic testing. However, capture of all introns, exons, and nearby MLL2 regulatory regions followed by next-generation sequencing would be more comprehensive and likely to be less costly over the long term.
Comparison of four of the objective clinical characteristics of MLL2 mutation-negative versus MLL2 mutation-positive cases allowed us to explore both the relationship between MLL2 genotype and Kabuki phenotype and the phenotype of MLL2 mutation-negative cases. Overall, the clinical characteristics of MLL2 mutation-positive cases did not differ significantly from MLL2 mutation-negative cases with the exception that renal anomalies were more common in MLL2 mutation-positive cases. Similarly, we observed no significant phenotypic—including the severity of developmental delay—differences between individuals grouped by mutation type. However, the phenotypic data available to us for analysis was limited and, for many cases, we lacked specific information about each malformation present. Furthermore, the most typical phenotypic characteristic, the distinctive facial appearance, was not compared in detail between cases although it would be of interest to study facial images ‘blinded’ to mutation status to investigate its power to predict genotype. Analysis of genotype-phenotype relationships using both a larger set of Kabuki cases, and with access to more comprehensive phenotypic information would be valuable.
No MLL2 mutation could be identified in 26 of the cases referred to us with a diagnosis of Kabuki syndrome. In three of these cases, aCGH identified structural variants that could be of clinical significance although additional investigation is required. A fourth case had the classical deletion observed in individuals with Sotos syndrome, and in retrospect it appears that this case was included in the cohort erroneously. The 22 remaining cases, including one parent-offspring pair, represent individuals with fairly classic phenotypic features of Kabuki syndrome without a MLL2 mutation. This observation suggests that Kabuki syndrome is genetically heterogeneous. To this end, in these 22 cases, we sequenced the protein-coding exons of UTX, a gene that encodes a protein that directly interacts with MLL2 but no pathogenic changes were found (data not shown). Exome sequencing of a subset of these MLL2 mutation-negative cases to identify other candidate genes for Kabuki syndrome is underway.
Whether Kabuki syndrome is the most appropriate diagnosis for the MLL2 mutation-negative cases is unclear. Some of the MLL2 mutation-negative cases appear to have a facial phenotype that differs somewhat from that of the MLL2 mutation-positive cases. Whether these MLL2 mutation-negative cases diagnosed by expert clinicians should be considered Kabuki syndrome, a variant thereof, or a separate disorder remains to be determined. Our opinion is that there is simply not yet enough information to make an informed decision about this issue.
Most of the mutations in MLL2 are predicted to result in haploinsufficiency. However, it is unclear by what mechanism(s) haploinsufficiency of MLL2 could cause Kabuki syndrome. MLL2 encodes a histone 3 lysine 4 (H3K4) methyltransferase, one of at least 10 proteins (genes for which have not to our knowledge yet been screened in Kaubki cases in which MLL2 mutations were not found) that have been identified to specifically modify the lysine residue at the fourth amino acid position of the histone H3 protein [Kouzarides, 2007]. MLL2 has a SET domain near its C-terminus that is shared by yeast Set1, Drosophila Trithorax (TRX) and human MLL1 [FitzGerald and Diaz, 1999]. MLL2 appears to regulate gene transcription and chromatin structure in early development [Prasad et al., 1997]. In mice, loss of MLL2 results in embryonic lethality before E10.5, and while Mll2+/− mice are viable, they are smaller than wild type (Kai Ge, personal communication).
Kabuki syndrome is the most common of a small, but growing group of multiple malformation syndromes accompanied by developmental delay that are caused by mutations in genes that encode proteins involved in histone methylation [De Sario, 2009]. The most notable of these is CHARGE syndrome, which is one of the syndromes often considered in the differential diagnosis of children ultimately diagnosed with Kabuki syndrome. CHARGE syndrome is caused by mutations in CHD7, which encodes a chromodomain protein that recognizes the trimethylated H3K4 side chain [Vissers et al., 2004]. Other disorders caused by defects of histone methylation status include several intellectual disability syndromes, some of which are also characterized by malformations (e.g., cleft lip/palate) that overlap with those found in individuals with Kabuki syndrome.
Kabuki syndrome is one of the most common causes of heritable developmental delay. Discovery that mutations in MLL2 are the most common cause of Kabuki syndrome highlights the role that disrupted regulation of histone methylation plays as a cause of human birth defects. Characterizing the spectrum of mutations in MLL2 is a small but important first step toward understanding the mechanism(s) that underlies Kabuki syndrome.
Supplementary Material
Table I. MLL2 primer sequences and reaction conditions
Table II. Detailed summary of MLL2 mutation data in 110 cases of Kabuki syndrome
Acknowledgments
We thank the families for their participation and the Kabuki Syndrome Network for their support. Our work was supported in part by grants from the National Institutes of Health/National Heart Lung and Blood Institute (5R01HL094976 to D.A.N. and J.S.), the National Institutes of Health/National Human Genome Research Institute (5R21HG004749 to J.S., 1RC2HG005608 to M.J.B., D.A.N., and J.S.; and 5RO1HG004316 to H.K.T.), National Institute of Heath/National Institute of Environmental Health Sciences (HHSN273200800010C to D.N. and M.R.), National Institute of Neurological Disorders and Stroke (RO1NS35102 to C.S.M.) NIHR Manchester Biomedical Research Centre (D. D.), Ministry of Health, Labour and Welfare (K.Y., N.M., T.O., and N.N.), Ministry of Health, Labour and Welfare of Japan (N.M), Japan Science and Technology Agency (N.M.), Society for the Promotion of Science (N.M.), the Life Sciences Discovery Fund (2065508 and 0905001), the Washington Research Foundation, and the National Institutes of Health/National Institute of Child Health and Human Development (1R01HD048895 to M.J.B. and 5K23HD057331 to A.E.B.). S.B.N. is supported by the Agency for Science, Technology and Research, Singapore. A.W.B. is supported by a training fellowship from the National Institutes of Health/National Human Genome Research Institute (T32HG00035).
References
- Adam MP, Hudgins L. Kabuki syndrome: a review. Clin Genet. 2005;67(3):209–219. doi: 10.1111/j.1399-0004.2004.00348.x. [DOI] [PubMed] [Google Scholar]
- De Sario A. Clinical and molecular overview of inherited disorders resulting from epigenomic dysregulation. Eur J Med Genet. 2009;52(6):363–372. doi: 10.1016/j.ejmg.2009.07.004. [DOI] [PubMed] [Google Scholar]
- FitzGerald KT, Diaz MO. MLL2: A new mammalian member of the trx/MLL family of genes. Genomics. 1999;59(2):187–192. doi: 10.1006/geno.1999.5860. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99(4):570–573. doi: 10.1016/s0022-3476(81)80256-9. [DOI] [PubMed] [Google Scholar]
- Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, Sugimoto T, Sonoda T, Hasegawa T, Chinen Y, Tomita Ha HA, Kinoshita A, Mizuguchi T, Yoshiura Ki K, Ohta T, Kishino T, Fukushima Y, Niikawa N, Matsumoto N. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet. 2002;30(4):365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, Shendure J. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, Ishikawa N, Yamada Y, Fujita M, Umemoto H, et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31(3):565–589. doi: 10.1002/ajmg.1320310312. [DOI] [PubMed] [Google Scholar]
- Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99(4):565–569. doi: 10.1016/s0022-3476(81)80255-7. [DOI] [PubMed] [Google Scholar]
- Prasad R, Zhadanov AB, Sedkov Y, Bullrich F, Druck T, Rallapalli R, Yano T, Alder H, Croce CM, Huebner K, Mazo A, Canaani E. Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene. 1997;15(5):549–560. doi: 10.1038/sj.onc.1201211. [DOI] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- White SM, Thompson EM, Kidd A, Savarirayan R, Turner A, Amor D, Delatycki MB, Fahey M, Baxendale A, White S, Haan E, Gibson K, Halliday JL, Bankier A. Growth, behavior, and clinical findings in 27 patients with Kabuki (Niikawa-Kuroki) syndrome. Am J Med Genet A. 2004;127A(2):118–127. doi: 10.1002/ajmg.a.20674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table I. MLL2 primer sequences and reaction conditions
Table II. Detailed summary of MLL2 mutation data in 110 cases of Kabuki syndrome