

Summary
Strategically mutated neoceptors, e.g., with anionic residues in TMs 3 and 7 intended for pairing with positively charged amine-modified nucleosides, were derived from the antiinflammatory A2A adenosine receptor (AR). Adenosine derivatives functionalized at the 5′, 2, and N6 positions were synthesized. The T88D mutation selectively enhanced the binding of the chain-length-optimized 5′-(2-aminoethyl)uronamide but not 5′-(2-hydroxyethyl)uronamide, suggesting a critical role of the positively charged amine. Combination of this modification with the N6-(2-methylbenzyl) group enhanced affinity at the Q89D- and N181D- but not the T88D-A2AAR. Amino groups placed near the 2- or N6-position only slightly affected the binding to mutant receptors. The 5′-hydrazide MRS3412 was 670-and 161-fold enhanced, in binding and functionally, respectively, at the Q89D-A2AAR compared to the wild-type. Thus, we identified and modeled pairs of A2AAR-derived neoceptor-neoligand, which are pharmacologically orthogonal with respect to the native species.
Introduction
The clinical use of adenosine agonists as cytoprotective agents [1] has been limited by the widespread occurrence of adenosine receptors (ARs), thus leading to undesirable side effects of exogenously administered adenosine derivatives. For example, experimental A1AR agonists developed for the treatment of cerebral ischemia [2, 3] also tended to cause a bradycardiac effect through the same subtype of ARs present in the nodal conduction system of the heart. Compounds introduced as A2AAR agonists for the treatment of inflammatory conditions via action on immune cells [4–6] are at risk for also causing hypotension through vascular A2AARs. Prodrug schemes for overcoming the problem of actions of adenosine agonists in multiple tissues [7, 8] have not yet proven fruitful.
In order to overcome this inherent nonselectivity of activating the ARs and other native G protein-coupled receptors (GPCRs), we have introduced the concept of neoceptors [9, 10], a means of mutating GPCRs for eventual application to tissue-targeted gene therapy. By this strategy, the putative ligand binding site of a GPCR is reengineered with the aid of molecular modeling for unique activation by synthetic, small molecular agonists (neoligands) built to have a structural complementarity to the altered binding site. The synthetic agonist would be administered to activate exclusively the engineered receptor, i.e., neoceptor. The feasibility of neoceptors has been demonstrated for antiinflammatory A2A and cardioprotective A3ARs [4, 5, 11]. Certain positively charged amine-modified nucleosides have demonstrated enhanced affinity at these ARs that had been strategically mutated with anionic residues. These mutant receptors retained the ability to activate the usual second messenger systems. Specifically, amino groups introduced in the ribose moiety of adenosine have been found to interact favorably with receptors modified in the putative ribose binding regions associated with TMs (transmembrane helical domains) 3 and 7 [9, 10]. Rhodopsin-based molecular modeling of the receptors (residue identifiers of format X.YZ in parentheses refer to the TM X and residue YZ with respect to a highly conserved amino acid in each TM numbered 50) indicated that hydrophilic residues T (3.36) and H (7.43) were in proximity to the ribose moiety. 5′-(2-ami-noethyl)uronamidoadenosine (MRS 3366 1; Figure 1) displayed selective enhancement of affinity at the T88D (3.36) mutant A2AAR, at which the standard AR agonists were poorly recognized [10]. At the H272E (7.43) mutant A3AR, the 3′-amino-3′-deoxy derivative of adenosine 2 displayed selective affinity enhancement [9]. Amino groups placed at other positions on the nucleosides did not enhance the receptor binding. Amino groups placed close to the nucleoside pharmacophore generally tended to reduce affinity of agonists at the wild-type (WT) ARs, while certain amine substitutions selectively enhanced binding affinity at mutant ARs.
Figure 1.

Structures of Nonselective Adenosine Receptor Ligands (3, 4) and Several Amine Derivatives (1, 2) Previously Studied at A2AAR Neoceptors
In the present study, we have expanded the range of positions for concerted ligand derivatization and mutation of the human A2AAR. New mutant A2AARs have been expressed in COS-7 cells and initially examined using the prototypical nonselective AR agonist NECA 3 and antagonist CGS15943 4. We have systematically varied the chain length of the 5′-substituents found to interact putatively with the carboxylate group of T88D [10]. Furthermore, at the 2 and N6 positions we have incorporated substitutions [12–14], the effects of which on AR affinity have already been characterized. Thus, we are assembling SAR patterns for mutant A2AARs in an effort to achieve selective matching of neoceptor-neoligand pairs.
Other similar approaches have been reported for the orthogonal mutation of receptors and enzymes to recognize unnatural and structurally modified ligands and substrates [15–19]. Conklin and coworkers have introduced RASSLs (receptors activated solely by synthetic ligands) and demonstrated this approach for nonpep-tide opiate and other ligands. Shokat and coworkers [16, 20] have mutated kinases to accept structurally modified inhibitors for use as mechanistic probes. Schreiber and coworkers [17] have utilized uniquely matched proteins and ligands. By the “bump and hole” approach, a sterically enlarged ligand is designed to fit into a side-chain-truncated protein. The laboratories of Katzenellenbogen [18] and Koh [19] have matched mutant steroid and other hormone receptors with reciprocally altered ligands. In several cases, X-ray crystallographic data are available to guide the design of the ligands. For nearly all of the GPCRs, such data are lacking, and homology modeling guided by site-directed mutagenesis is the principal means of gaining structural insights.
Results
Design and Synthesis of Neoligands
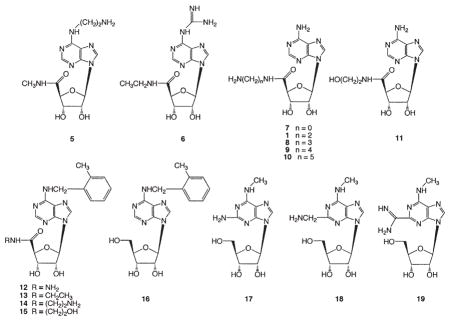
Various adenosine analogs functionalized at the 5′, 2, and N6 positions were prepared as potential neoligands (Table 1). The modifications included the addition of amino and various H-bonding groups to both 9-ribo-side-5′-uronamido 3–15 and 9-riboside 17–19 derivatives. Certain 5′-uronamido derivatives of adenosine, notably NECA 3, are known to display enhanced affinity at the A2AAR. Metrifudil 16, an adenosine agonist that was studied in humans [21], and its 5′-uronamide derivatives 12–15 were included due to the nonselective enhancement by the N6-(2-methylbenzyl) group of affinity at the ARs.
Table 1.
Binding Affinity of Ligands at WT and Mutant Human A2AARs Expressed in COS7 Cells
 | ||||
|---|---|---|---|---|
| Compound | Ki (nM)
|
|||
| Constructb | ||||
| WT | T88D | Q89D | N181D | |
| 3c | 21.4 ± 8.7 | >10,000 | 1.5 ± 0.4 | 9.3d |
| 4c | 0.84 ± 0.22 | 0.91 ± 0.09 | 0.35 ± 0.12 | 0.45d |
| 5 | >100,000 | >100,000 | >10,000 | >10,000 |
| 6 | 4,510 ± 150 | 5,680 ± 980 | 773 ± 129 | 1,140 ± 180 |
| 7 | 11,000 ± 2,900 | 32,800 ± 11,400 | 16.4 ± 2.1 | 1,180 ± 480 |
| 1 | 46,100 ± 4,200a | 4,400 ± 1,600a | 3,600 ± 1,000a | 493 ± 138 |
| 8 | 74,200 ± 24,800 | 57,600 ± 11,700 | 10,800 ± 1,500 | 4,140 ± 160 |
| 9 | >100,000 | >100,000 | 55,200 ± 8,400 | 48,100 ± 4,400 |
| 10 | >100,000 | >100,000 | 124,000 ± 36,000 | >100,000 |
| 11 | 2,110 ± 260 | 3,870 ± 1210 | 41.7 ± 7.3 | 13,300 ± 1,600 |
| 12 | >100,000 | >10,000 | 545 ± 145 | >100,000 |
| 13 | 2,590 ± 140 | >100,000 | 246 ± 36 | 3,470 ± 430 |
| 14 | >100,000 | >100,000 | 443 ± 16 | 320 ± 52 |
| 15 | >10,000 | >100,000 | 444 ± 127 | 6090 ± 380 |
| 17 | >10,000c | >100,000 | >10,000c | >100,000 |
| 18 | >100,000 | >100,000 | >100,000 | >100,000 |
| 19 | >100,000 | >100,000 | >100,000 | >100,000 |
Compound 4 is the only nonnucleoside (nonselective antagonist CGS15943) included. Membranes from COS-7 cells transfected with WT or mutant A2AAR cDNA were incubated with 2.0 nM [3H]ZM241385 in duplicate, together with increasing concentrations of the competing compounds, in a final volume of 0.2 ml Tris-HCl buffer (50 mM, [pH 7.4]) at 25°C for 120 min. The Ki values are expressed as mean ± standard error from three independent experiments, unless noted. The Kd values determined for saturation of binding of [3H]ZM241385 were (nM, mean ± SEM): WT, 2.4 ± 0.7; T88D, 4.2 ± 0.5; Q89D, 4.6 ± 1.8; and N181D, 4.1 ± 0.4.
From Kim et al. [10].
Binding affinities at the N253D-A2AAR could not be determined, since this construct lost the ability to bind either agonist or antagonist radioligand.
Ki values of the nonselective agonist NECA 3 and antagonist CGS15943 4 bound at the T88E-A2AAR were >10,000 and 0.67 ± 0.17 nM, respectively. Ki values for 17 of 63 and 17 μM determined at WT and Q89D-A2AARs, respectively (n = 1).
n = 1.
Synthetic methods for the adenosine analogs are shown in Figures 2 and 3. A N6-(2-aminoethyl) derivative 5 was synthesized (Figure 2A) from the protected 6-chloro 5′-uronamide precursor 20 [22].
Figure 2. Synthesis of N6-Derivatized Adenosine Analogs 5 and 6.

Reagents: (A) (i) ethylendiamine, rt., 24 hr; (ii) 1 N HCl, MeOH-H2O (1:1), 55°C, 36 hr, 65%.(B). (i) 2,2-dimethoxypropane, p-TsOH, acetone, 4 hr, 85%; (ii) TBDMS-Cl, imidazole, pyridine, 1 hr, 83%; (iii) guanidine solution, DABCO, 3 hr, 70%; (iv) Bu4N+F-, THF, 1hr, 95%; (v) KMnO4, KOH, H2O, 5 hr, 50%; (vi) H2SO4, 3 hr, 60°C, 84%; (vii) (a) ClCOOEt, N,N- i-Pr2NEt, DMF, 0°C; (b) EtNH2 in MeOH, 0°C 1 hr, RT 24 hr, 50%.
Figure 3. Synthetic Routes to Various Mono-and Disubstituted Adenosine-5′-Uronamide.

Derivatives (A) Synthesis of adenosine-5′-uronamide analogs 1 and 8–10.
(B) Synthesis of N6-(2-methylbenzyl)adeno-sine-5′-uronamide analogs 12–15. Reagents: (i) CrO3, AcOH, 18%; (ii) TMSCHN2, MeOH, 88%; (iii) RNH2, DMF, 60°C; (iv) 1N HCl MeOH-H2O (1:1) 55°C, 2 d; (v) RP-SiO2 C18, for 7, Amberlite CG-50, for 11 and 12–15; (vi) Boc-NH(CH2)nNH2, iPr2NEt, DMF, 60°C, 54%–97%; (vii) Amberlite CG-50, 13%–67%; (viii) 2-MeBnBr; (ix) ethylamine or ethanol-amine, MeOH, heat, Dimroth rearrangement and aminolysis of ester.
In the first step of the synthesis of the N6-guanidino derivative 6 of NECA, the readily accessible 6-chloro-9-β-D-ribofuranosyl-9H-purine [23] was converted into its 2′,3′-O-isopropylidene derivative [24] 21 (Figure 2B). The guanidinolysis of the 2′,3′-O-isopropylidene derivative 21 in the presence of DABCO afforded 24 contaminated with byproducts; therefore the tert-butyl-dimethylsilyl group (TBDMS) was used to protect the 5′-hydroxyl function in intermediate 22 [25]. The subsequent guanidinolysis of 22 afforded in good yield 23, which was then deprotected on treatment with tetrabu-tylammonium fluoride solution in tetrahydrofuran. Subsequent oxidation of the 5′-OH group with Ru(OH)Cl3 and NaIO4 afforded the carboxylic acid 25. However, to avoid contamination by colloidal metallic Ru, the oxidation was performed with KMnO4 in aqueous KOH. Subsequent deprotection of the isopropylidene group by acid hydrolysis in diluted aqueous H2SO4 gave 26. A mixed anhydride derived from acid 26 was treated without isolation with ethylamine in MeOH to give the desired 9-[N-ethyl(β-D-ribofuranosyluronamide)] 6-gua-nidinopurine 6 in a satisfactory yield.
Various 9-riboside-5′-uronamido derivatives 1, 7–11 were prepared as shown in Figure 3A. The 2′,3′-isopro-pylidene-protected derivative of adenosine 27 was oxidized using chromium trioxide in acetic acid to provide the carboxylic acid 28. Amide derivatives 1, 7–11 were synthesized by similar methods to those described by Gallo-Rodriguez et al. [22]. Initially, we attempted to couple 28 and the appropriate amines using 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide; however, low yields were obtained. Aminolysis of ester 29 with an amines or hydrazine (in the case of 7) improved yields. In the case of compounds 1, 8–11, a mono-N-Boc-protected diamine was used in this reaction. The protecting groups (isopropylidene and N-Boc) were removed under mild acidic conditions. The resulting nucleosides were purified by ion-exchange column chromatography or reverse-phase silica gel column chromatography, and their purity was confirmed by HPLC.
The N6-(2-methylbenzyl) derivatives 12–15 were synthesized as shown in Figure 3B by the Dimroth rearrangement. The intermediate 32 was alkylated at the N1 position by 2-methylbenzyl bromide, which rearranged to the N6 position under basic conditions to provide 41–44. Deprotection and purification procedures were the same as above. An alternate route used to prepare 13 and 15 was alkylation of the ester 29 followed by simultaneous Dimroth rearrangement and aminolysis of the methyl ester group and then acidic deprotection.
A2AAR Modeling and Site-Directed Mutagenesis
Molecular modeling was used to make predictions of which complementary groups on the ligand and receptor may interact. The human A2AAR was modeled as described, based on a rhodopsin template [26], and representative adenosine derivatives were docked in a putative binding site [10]. Modeling has predicted the proximity of the amino groups of the adenosine analogs to specific residues of the human A2AAR. The derivatives that possess a positively charged nitrogen atom linked through the N6-position (5, 6) are hypothesized to interact with an anionic group placed in the vicinity of N253. The derivatives that bear an amino group linked through the 5′ position (7–10, 14) are hypothesized to interact with an anionic group placed in the vicinity of T88 or N181. The two-carbon homolog in this series 1 was already demonstrated to have enhanced affinity at the T88D mutant A2AAR [10]. The derivatives that bear an amino group linked through the 2 position are hypothesized to interact with an anionic group placed in the vicinity of Q89 or N181.
Several new mutant human A2AARs were prepared to test these hypotheses. The N181D- and N253D-A2AARs were constructed for the present study and expressed in COS-7 cells. The T88D and Q89D-A2AARs prepared previously [10, 27] were utilized to examine the new ligand structures. A summary of the affinities of 5′-N-ethyl-uronamidoadenosine (NECA) 3 and 5-amino-9-chloro-2-(2-furyl)-1,2,4-triazolo[1,5-c]quinazoline (CGS15943) 4, standard nonselective AR agonist and antagonist, respectively, at the mutant receptors transiently expressed in COS-7 cells is provided in Table 1. The affinity of NECA 3 was greatly reduced at both T88 mutant ARs. The N253D-A2AAR lost the ability to bind either agonist or antagonist radioligand with high affinity. The other mutant receptors, nevertheless, could be characterized using the conventional A2AAR antagonist radioligand [3H]ZM241,385 (4-2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a] [1, 3, 5]triazin-5-yl-amino]ethylphe-nol) [28].
Interaction of Adenosine Derivatives with the WT ARs
Initially, the adenosine derivatives were examined in assays at the native ARs. Binding assays were carried out at recombinant human A1, A2A, and A3ARs stably expressed in CHO cells (Table 2). In a functional assay at the WT A2AAR consisting of activation of adenylate cyclase [29], the adenosine derivatives were all agonists. The assay of cAMP was also used to examine activation of the Gs-coupled human A2BAR stably expressed in CHO cells. Nucleosides that were moderately selective in binding to the A1 AR were the 2-hydroxy- ethyluronamide 11 and the N6-aminoethyl derivative 5, with Ki values of 12.8 and 775 nM, respectively. The N6-guanidino derivative of NECA 6 was somewhat selective for the A1 and A3ARs and displayed strikingly high affinity at the A3AR, with a Ki value of 5 nM, and at the A2BAR, with a 3-fold enhancement over NECA 3. The hydrazide 12 was somewhat selective for the A3AR, and the N-ethyluronamide 13 had mixed selectivity for both A1 and A3ARs. Within the series of 5′-N-aminoal-kyluronamides 1, 8–10, the highest affinity at all subtypes was observed for the two-carbon homolog 1, which displayed an order of affinity of A1 > A2A, A2B ≫A3. The isoelectronic 2-hydroxyethyl analog 11 was of considerably higher affinity/potency than 1 at all four native AR subtypes. Thus, consistent with previous observations, amine functionalization in the ribose region disfavored binding to the native ARs. The hydrazide 7 was similar in affinity to 1 at A1, A2A, and A2BARs and more potent at the A3AR. Appending a 2-methylbenzyl group at the N6 position of 1 to provide 14 reduced affinity/potency at all four native AR subtypes. A comparison of the affinities of NECA 3 and its hydroxylated analog 11 showed a dramatic loss of affinity at A2A and A3ARs as a result of the 2-hydroxyl group.
Table 2.
Affinity or Potency of Various Adenosine Derivatives at Human Adenosine Receptors
| Compounda | hA1 Binding Ki(nM) b | hA2A Binding Ki(nM) c | hA2A Activation % c | hA2B Activation EC50d | hA3 Binding Ki(nM) c |
|---|---|---|---|---|---|
| 3 | 6.8 ± 2.4 | 2.2 ± 0.6 | 100 | 140 ± 19 (n = 4) | 16.0 ± 5.4 |
| 5 | 775 ± 235 | >100,000 | 32.9 ± 2.0 | >10,000 | 24,600 ± 8,200 |
| 6 | 7.0 ± 1.0 | 628 ± 39 | 102 ± 4 | 54.5 ± 13.3 | 5.1 ± 1.3 |
| 7 | 171 ± 15 | 2,230 ± 330 | 92.2 ± 1.0 | 12,000 ± 4200 | 378 ± 33 |
| 1 | 245 ± 48 | 3,120 ± 370 | 90.3 ± 1.4 | 15,600 ± 2,800 | >100,000 |
| 8 | >100,000 | >10,000 | 51.0 ± 7.6 | >10,000 | >10,000 |
| 9 | >100,000 | >100,000 | 11.6 ± 4.6 | >10,000 | >10,000 |
| 10 | >100,000 | >100,000 | 0 | >10,000 | >100,000 |
| 11 | 12.8 ± 3.1 | 505 ± 30 | 84.5 ± 3.4 | 942 ± 38 | 9450 ± 1760 |
| 12 | >10,000 | >10,000 | 78.9 ± 4.5 | >100,000 | 394 ± 74 |
| 13 | 106 ± 30 | 5430d | 108 ± 3 | >1000 | 78.8 ± 25.5 |
| 14 | >10,000 | 55,900 ± 4,600 | 91.4 ± 5.2 | >1,000 | >10,000 |
| 15 | 368 ± 147 | >10,000 | 102 ± 2 | 3,720d | 1,640 ± 400 |
| 16 | 39.3 ± 8.3 | 764 ± 105 | 112 ± 6 | ND | 47.2 ± 10.8 |
| 17 | 484 ± 22 | >10,000 | 93.3 ± 5.5 | >10,000 | 39.0 ± 2.4 |
| 18 | >10,000 | >10,000 | 42.2 ± 1.3 | >10,000 | 719 ± 37 |
| 19 | >10,000 | >10,000 | 2 | >10,000 | 2,730 ± 268 |
n = 3, unless noted. ND, not determined.
Structures shown in Table 1. 1, MRS3366; 3, NECA; 6, MRS3218; 7, MRS3412; 11, MRS3407; 14, MRS3417.
Binding experiments carried out for various subtypes of human recombinant adenosine receptors stably expressed in mammalian cells using the following radioligands (concentration): hA1 (CHO cells), [3H]R-PIA (2.0 nM); hA2A (HEK-293 cells), [3H]CGS21680 (15 nM); hA3 (CHO cells), [125I]I-AB-MECA (0.5 nM).
At hA2A and hA2B receptors expressed in CHO cells, cAMP was measured. For hA2A receptors, values are expressed as percent stimulation of cyclic AMP production at 100 μM (value for NECA is 100%).
n = 1.
The 2-position derivatives of N6-methyladenosine 17–19 recently reported by us [12] tended to be selective for the human A3AR and nearly inactive at A2A and A2BARs. This characteristic was also noted for other N6-methyladenosine derivatives [12, 30].
Interaction of Adenosine Derivatives with the Mutant A2AARs
Selective affinity enhancement was observed for some nucleoside derivatives at A2AAR mutant receptors expressed in COS-7 cells (Table 1). Amine-containing groups placed at the 2 position (17–19) on the nucleosides did not enhance the binding to mutant receptors, while the addition of the amine-containing groups to the N6 position in 5 and 6 caused a slight affinity enhancement. For example, a small enhancement was observed at the Q89D-A2AAR in comparison to WT for the 6-guanidino derivative 6 (6-fold). However, modification of the 5′ position provided many examples of selectively enhanced affinity at the mutant receptors.
A 5′-hydrazide analog (compound 7) displayed a 670-fold enhancement of affinity at the transiently ex pressed Q89D-A2AAR compared to the WT receptor and no enhancement at the T88D-A2AAR (Figure 4A). In contrast, the affinity at the T88D-A2AAR of compound 1, which has a 5′ chain elongated by two methylene units, was increased by ~10-fold over the WT. The 5′-(2-aminoethyl)uronamide derivative 1 [10] was preferred to its longer homologs 8–10 in binding enhancement at the T88D- and N181D-A2AARs. Thus, the length of the 5′ chain might be a key factor for enhancement. The affinity of compound 1 was also enhanced by 13-fold at the Q89D mutant receptor. The higher homolog 8 displayed a less pronounced enhancement at the Q89D-A2AAR compared with 1. Combination of the 5′-(2-aminoethyl)uronamide modification with the N6-(2-methylbenzyl) group in 14 provided enhancement at the Q89D- and N181D-A2AARs (Figure 4B) but not at the T88D-A2AAR. At the N181D-A2AAR, enhancement of affinity (fold over WT) was observed for 1 (94), 6 (4), 8 (16), 14 (>300), and 15 (>10).
Figure 4. Pharmacological Characterization of Two Neoagonists Indicates Selective Interaction with Neoceptors Derived from the A2AAR.

Binding (A and B) and functional (C and D) effects of two adenosine derivatives, the hydrazide derivative 7 (A) and the N6-(2-methyl-benzyl)-5′-aminoethyluronamide derivative 14 (B) at WT (■) and mutant A2AARs (T88D [▲], N181D [◆], and Q89D [▼]) transiently expressed in COS-7 cells. In the binding experiments, cell membranes (10–20 μg protein) were incubated with the radiolabeled antagonist [3H]ZM241385 (2.0 nM) in duplicate, together with increasing concentrations of the competing nucleoside, in a final volume of 0.4 ml Tris-HCl buffer (50 mM, pH 7.4) at 25°C for 120 min. Results were from a representative experiment performed in duplicate. The Ki values listed in Table 1 were from at least three separate experiments. In the functional experiments, cells expressing WT or mutant receptors were then treated with agonist 7 (C) or 14 (D) in the presence of rolipram (10 μM) and adenosine deaminase (3 units/ml) and incubated at 37°C for 1 hr. cAMP accumulation was determined using a competitive protein binding method [29]. The EC50 values (n = 3) determined for stimulation of cAMP formation were (nM, mean ± SEM): WT, 826 ± 138; T88D, 2970 ± 980; Q89D, 5.1 ± 0.8; N181D, 120 ± 22 for 7; and WT, 5800 ± 1230; T88D, 12,600 ± 3200; Q89D, 58 ± 12; N181D, 52 ± 6 for 14.
In order to characterize the functional properties of neoceptor-neoligand pairs, effects on adenylate cyclase by selected neoligands were studied in CHO cells transiently expressing WT and mutant A2AARs (T88D, Q89D, and N181D). Concentration-response curves for cAMP accumulation at the WT and mutant A2AARs (Figures 4C and 4D) were generated for two neoligands, 7 and 14. These nucleosides were demonstrated to be full agonists as compared with the known agonist NECA, and the potency orders were commensurate with the binding affinities. The potency compared to the WT A2AAR of the 5′-hydrazide analog 7 was 162-fold enhanced at the Q89D-A2AAR (EC50 5.1 ± 0.8 nM), and that of the aminoethyl derivative 14 was 112-fold enhanced at the N181D-A2AAR (EC50 52 ± 6 nM). Both nucleoside derivatives were less potent in activating the T88D mutant receptor than the WT receptor.
The Docking Result of Amine Derivatives with the Mutant A2AARs
Construction of molecular models of the WT and T88D-A2AARs and the docking of 1 have been described [10]. A similar approach was adopted to model interactions with the Q89D- and N181D-A2AARs. The modeling of the N181D/1 complex indicated a direct contact of the ligand with the newly added carboxylate group. At the Q89D-A2AAR, most of the nucleosides examined displayed enhanced affinity, and thus, these enhancements did not provide insights into specific interactions. Consistent with this observation, the modeling of the Q89D/7 complex did not indicate a direct contact with the carboxylate group.
For the side-chain refinement of Q89D- and N181D-A2AARs, after the mutation of each side chain the re-ceptor models were optimized through a molecular dynamics (MD) procedure. In the previous model of the T88D-A2AAR [10], the trans-rotamer of the χ1 angle of this side chain was preferred over the gauche+ conformation shown in the WT receptor because of the formation of H bonding of the new carboxylate group with the neighboring hydrophilic residue, N181 (5.42). This local conformational change toward the upper portion of TM5 was consistent with the binding profile of this mutant receptor; the extended 5′-aminoethyluronamide was enhanced in binding affinity, while 5′-amino-5′-deoxyadenosine 2 displayed poor binding affinity, similar to its binding affinity at the WT A2AAR. The MD result of the Q89D-A2AAR displayed the same χ1 angle as the WT receptor, i.e., gauche+. The side chain of Q89 formed a H bond with the carbonyl oxygen atom of C185 (5.46) in the resting WT receptor structure. However, in the Q89D-A2AAR, the carboxylate group did not display any H bonding with the surrounding residues. In the N181D-A2AAR, D181 (5.42) showed additional H bonding with the imidazole ring of H250 (6.52), keeping the same trans χ1 angle of N181 (5.42) in WT receptor without H bonding to H250 (6.52). Thus, both human A2A Q89D- and N181D-A2AARs showed the same preference of χ1 angle compared with the WT but with different intramolecular H-bonding character. The differences included the loss of H bonding with Q89D and the formation of H bonding in N181D-A2AAR.
In the case of the human A2AAR complex with the nonselective agonist NECA 3 [10], H bonds formed between the exocyclic amine and the side chain amide NH of N253 (6.55), between the purine N1 atom and the side chain of N181 (5.42), and between the purine N3 atom and the side chain of T88 (3.36). The 3′-OH group H bonded with the δ1 NH of H278 (7.43). The 5′-amide NH also formed a H bond with T88 (3.36), and the 5′-carbonyl group displayed H bonding with the hydroxyl group of S277 (7.42). NECA, which bound to the Q89D-and N181D-A2AARs, displayed similar binding orientations with respect to the same residues in proximity to NECA 3 bound in the WT human A2AAR.
In the Q89D-A2AAR/NECA 3 complex, which did not show direct interaction of the ligand with D89 (3.37), there were additional H bonds of the N6-amine with the side chain of N181 and of the ribose ring oxygen with the hydroxyl group of T88 (3.36). This was consistent with a 14-fold increase of binding affinity for NECA. The docking result of the human A2A N181D-A2AAR/NECA 3 complex displayed alternative H bonding between the exocyclic amine and the side chain of D181 (5.42). The nonbonding energy components of the three NECA 3 complexes with the WT, Q89D-, and N181D-A2AARs in dicated a correlation between the experimental binding affinity (-logKi) and the calculated nonbonding energy of complex (r2 value was 0.99). This implied an inverse relationship between the nonbonding van der Waals and the electrostatic energy.
For the docking studies of neoceptor and neoligand, the thermodynamically stable conformations of several ligands were calculated. Conformational searching was performed using MOPAC PM3 calculations. The resulting low-energy conformers of 1, 7, 8, 9, 10, and 11 displayed intramolecular hydrogen bonding of the terminal ammonium or the hydroxyl group with the 5′-carbonyl oxygen atom. In the case of longer aminoalkyl homologs 8, 9, and 10, the interaction of the terminal ammonium group and the N7 atom in the adenine ring was indicated, due to the length of the 5′ substituent.
The lowest-energy conformer of 1 was superimposed onto the N181D-A2AAR using an atom-by-atom fitting from the human A2AAR/NECA 3 complex. The overlay indicated that the 5′-ammonium group of 1 was unable to interact with the side chain of D181 (5.42). However, a different binding mode that was more energetically favorable than in the A2AAR/NECA 3 complex was suggested using the automatic docking program. This mode featured a direct interaction of the terminal positively charged ammonium ion and the negatively charged carboxylate ion of D181 (5.42). Figure 5A shows the docking result of the N181D-A2AAR/1 complex. The exocyclic amino group interacted with the hydroxyl group of S277 (7.43) and the imidazole ring of H278 (7.43) through H bonding. The 3′-OH group formed a H bond with the backbone carbonyl group of T88 (3.36). In the binding site of the 5′ substituent, the NH group formed a H bond with the imidazole ring of H250 (6.52). The terminal NH3+ group interacted with the negatively charged side chain of D181 (5.42) through a salt bridge (3.0 Å between the N of the terminal NH3+ in 1 and the O of the carboxylate ion) as well as through H bonding (2.1–2.8 Å). In addition, there were additional H bonds (2.5–2.7 Å) with the backbone carbonyl of D181 (5.42) and C185 (5.46). However, in the complex of 2-hydroxyethyluronamideadenosine 11, there was only H bonding (2.3 Å) with the side chain of D181 (5.42), consistent with its 27-fold decrease of the binding affinity compared with 2-aminoethyluronam-ideadenosine 1. In the case of the docking result of 8, having one more methylene unit, the complex still showed a salt bridge (2.8 and 3.3 Å between the N of the terminal NH3+ in 7 and the O of the carboxylate ion). The H bonds with the side chain of D181 of length 2.2 and 2.5 Å were weakened, and H bonding with the backbone carbonyl groups of D181 and C185, detected in the docking complex of 1, was absent. Correlating with its experimental data, the complex of 9 displayed just one H bond (2.7 Å) with D181 (5.42), and the complex of 10 showed a distortion of the α-helical structure of TM5. The r2 value of correlation between the binding affinity and the nonbonding energy of the complex was 0.96. Compound 14 docked with the same binding mode as compound 8, displaying additional bonding at the N6 position in upper TM7. In Figure 5A, the terminal NH3+ group reached the side chain of Q89 (3.37) due to the flexibility of 5′-terminal group, possibly explaining the increase of binding affinity at both Q89D- and N181D-A2AARs.
Figure 5. Proposed Modes of Docking of Two Neoagonists in Rhodopsin-Based Homology Models of Neoceptors Derived from the A2AAR.
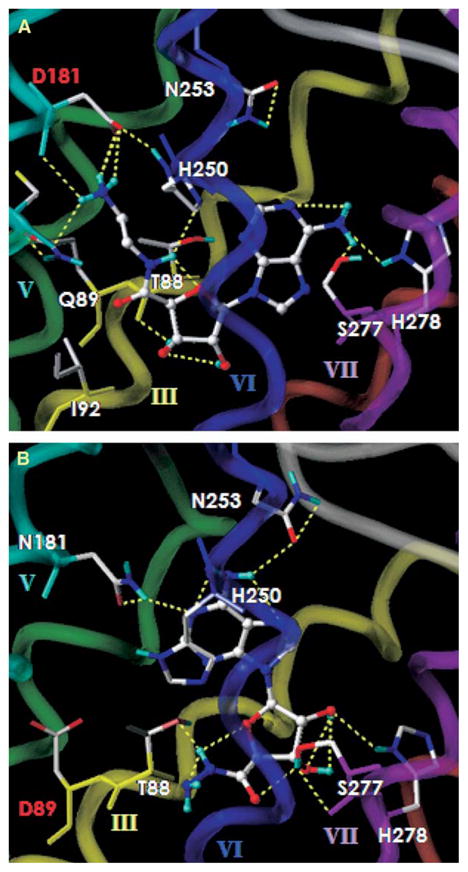
The binding site of (A) the N181D-A2AAR/1 and (B) the Q89D-A2AAR/7 complex. All ligands are displayed as ball-and-stick models in the atom-by-atom color, and the side chains of human A2AAR are shown as stick models. The H bonding between ligand and the mutant receptor is displayed in yellow. The A2AAR is represented by a tube model with a different color for each TM domain (TM3 in yellow, TM5 in green, TM6 in cyan, and TM7 in purple).
In the docking complex of the Q89D-A2AAR with 1, 7, and 11, energetically favorable binding modes followed a similar binding geometry of the ribose ring to that shown in the A2AAR/NECA 3 complex. In that complex, there was no interaction between the 5′-terminal group and the side chain of D89 (3.37). As illustrated in Figure 5B, the exocyclic amino group, the purine N1 atom, and the 5′-NH group of 7 interacted through H bonds with the N253 (6.55), N181 (542), and the T88 (3.36), respectively, as shown by the A2AAR/NECA 3 complex. The 2′-OH group instead of the 3′-OH group formed a H bond with the imidazole ring of H278 (7.43). Compounds 1 and 11 showed intramolecular H bonding of the 5′-terminal NH3+ or the hydroxyl group with the 5′-carbonyl oxygen in the bound conformation as well as in the unbound state. Because of the limitation of space at the 5′ binding site, a bulkier group decreased the binding affinity to the Q89D-A2AAR. The binding modes of several 5′-ammonium analogs with various lengths of the methylene chain in the N181D-A2AAR were different from those in the Q89D-A2AAR. This was consistent with two different modes observed for binding of 3′- and 5′-amino analogs in the T88D-A2AAR [10]. Thus, the results of molecular modeling explained the experimental data for some neoligands.
The loss of binding affinity at the WT and mutant A2AARs of 2-position derivatives, 17–19, seems to be characteristic of N6-methyl adenosine derivatives, which tend to have increased selectivity for the hA3 AR [12].
Discussion
We have used both rational and empirical design processes to identify pairs of neoceptor-neoligand that are pharmacologically orthogonal with respect to the native species. In principle, it could be possible to redesign the binding site for recognition of a radically different structure, i.e., use the parent receptor as template only.The T88D-A2AAR is truly a “neoceptor,” since its recognition profile is distinct from that of the parent A2AAR. Only compound 1 was enhanced in affinity at this construct in comparison to the WT A2AAR. At the N181D-A2AAR, the degree of enhancement was higher than at the T88D-A2AAR. Compounds 1 and 14 were most highly enhanced in affinity at the N181D-A2AAR.
Orthogonality of selective interaction with mutant A2AARs has been achieved for two nucleoside derivatives, 7 and 14. The selectivity ratios for compound 7 (MRS3412) in binding to the mutant Q89D-A2AAR (transiently expressed) in comparison to the stably expressed WT A1-, A2A-, and A3ARs were 10, 136, and 23-fold, respectively. Comparable selectivity ratios for compound 14 (MRS3417) in binding to the mutant N181D-A2AAR (transiently expressed) were >31, 175, and >31-fold, respectively. The T88D, Q89D, and N181D mutations within the putative ribose binding region of the A2AAR agonist binding site [10] did not impair signaling properties of the receptor. In the functional assays of adenylate cyclase stimulation, compound 7 acting at the Q89D-A2AAR was more potent than 14 acting at the N181D-A2AAR, consistent with the binding results. Potency at the A2BAR was very weak for both agonists, so the presence of this subtype will not likely be a complicating factor in envisioned in vivo use of these nucleosides.
We have used molecular modeling to provide hypotheses for the selective enhancement of affinity observed for certain pairs of nucleoside and mutated receptor. At the T88D-A2AAR, most of the nucleosides examined bound weakly or not at all, and therefore the dramatic gain of affinity of 1 has been interpreted to imply a direct contact between the ligand and the D88 side chain. In contrast, at the Q89D-A2AAR, NECA 3 and most other agonists also displayed enhanced affinity (Table 1). The enhancements of the adenosine derivatives could not readily be ascribed to a specific electrostatic or H-bonding interaction with the newly introduced carboxylic acid group. This is supported by the observation that a variety of single amino acid replacements of Q89 enhanced agonist affinity, regardless of the chemical nature of the side chain. Thus, a specific conformational hypothesis to explain the 670-fold affinity enhancement of 7 was lacking. However, at the N181D-A2AAR, the affinity of NECA was only minimally increased. Thus, we have used molecular modeling to propose a direct interaction between 7 and the D88 side chain.
In addition to introducing novel molecules that may now be explored pharmacologically in the context of therapeutics, at the same time the neoceptor approach has validated key contact regions in the receptor models. We interpret the results in the context of formation of new electrostatic and other polar interactions to enhance affinity, i.e., a gain of function. Since the sequence of rhodopsin has only <20% homology to the AR sequences, this is an important aspect of the study.
The present study also emphasizes the limitations of the current GPCR modeling, since several predicted interactions (e.g., enhancement of the affinity of 2-amino derivatives at the Q89D-A2AAR) were not explicable. There are several limitations to the neoceptor approach as a rational approach: (1) the homology modeling method is imprecise (the limitations of using a rhodopsin template, especially for modeling agonist binding have been discussed [10]), and (2) the side chains may rearrange upon mutation to lose predictability of interaction with the ligand. It is evident that electrostatic interactions, e.g., from an amine-bearing ligand and carboxylate-modified protein, are not the only basis on which a selective enhancement may be achieved. Other principles of enhancement might be based on more extensive H-bonding ability or on hydrophobic interactions.
In the projected therapeutic application of neoceptors, i.e., by introducing the gene for the mutant receptor using an organ- or tissue-targeted vector for in vivo delivery [9, 31–33], the protective role of native ARs would not be affected. Ideally, the native ligand and neoligand would exclusively activate their respective receptors. This orthogonal approach is predicated on the neoceptor interacting with the necessary second messenger systems. Thus, the addition of the neoceptor to the functioning of the tissue would be a parallel process, to be invoked only as needed clinically, and not to interfere with critical functions of endogenous adenosine. Therefore, the drug action would be site specific and would avoid side effects of activation of subtypes of native ARs.
Significance
GPCRs constitute the target mechanism of roughly half of the pharmaceutical substances in commercial use. The therapeutic use of agonists of GPCRs is prone to side effects, due to frequently widespread distribution of a given receptor subtype. For example, in the adenosine system, no potent and selective agonists of any of the four receptor subtypes have yet been approved for therapeutic use, in spite of the thousands of agonists synthesized and selectivity achieved. The A2AAR in the immune system is involved in suppressing prolonged inflammatory responses. It also has several cardiovascular actions, such as vasodilation [1, 2], which may be considered side effects in this context. One means of overcoming side effects of the A2A receptor and of other GPCRs is to establish a new basis for selectivity of uniquely tailored small molecular agonists for engineered receptors, which could potentially be delivered by genetic means to a target organ.
The neoceptor approach to engineering GPCRs for unique activation is based on the use of molecular modeling to identify and later alter the recognition elements of a GPCR binding site, such that only neoligands will activate the mutant receptor [9, 10]. The present study initially attempted to enhance A2A receptor affinity in the orthogonal pairs based on electrostatic attraction of a cationic nucleoside agonist and anionic TM of the receptor protein. This study revealed additional possibilities not predicted using receptor modeling, such as enhanced H-bonding ability. Selective enhancements of several hundred-fold were achieved. Also, other combinations not explored here are possible, e.g., placing a negative charge on the ligand and a positively charged side chain on the receptor.
Given the identification of unique neoligand/neceptor pairs, it is now appropriate to perform more extensive pharmacological studies, including functional cell systems and transgenic mice.
Experimental Procedures
Human A2AAR cDNA (expression vector pSVL-A2A) was kindly provided by Dr. M. Jacobson (Merck Research Labs, West Point, PA). Oligonucleotides used were synthesized by Bioserve Biotechnologies (Laurel, MD). [3H]ZM241385 (4-(2-[7-amino-2-(2-furyl)1,2,4]tri-azolo [2,3a] [1,3,5]triazin-5-ylamino]ethyl)phenol, 17 Ci/mmol) was from Tocris Cookson Ltd. (Bristol, United Kingdom). Adenosine de-aminase, CGS15943, and NECA were obtained from Sigma (Saint Louis, MO). All other compounds were obtained from standard commercial sources and were of analytical grade. Detailed methods used for chemical synthesis [22–25] and along with the coordinates of the hypothetical neoceptor-neoligand complex shown in Figure 5A, derived using molecular modeling [34–38], are provided in the Supplemental Data.
Site-Directed Mutagenesis
The protocols used were as described in the QuikChange Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA). Mutations were confirmed by DNA sequencing. Lipofectamine 2000 (Invitro-gen Life Technologies) was used for transfection of WT and mutant receptor cDNA to COS-7 cells.
Membrane Preparation
After 48 hr of transfection, COS-7 cells were harvested and homogenized with a Polytron homogenizer. The homogenates were centrifuged at 20,000 × g for 20 min, and the resulting pellet was resuspended in the 50 mM Tris-HCl buffer (pH 7.4) and stored at –80°C in aliquots. The protein concentration was determined by using the method of Bradford [39].
Radioligand Binding and Functional cAMP Assay
The procedures of [3H]ZM241385 binding to WT and mutant human A2AARs was as previously described [28]. For saturation experiments, membranes (20–40 μg of protein) were incubated with increasing concentrations (0.5–16 nM) of [3H]ZM241385 in duplicate, in a final volume of 0.4 ml of Tris-HCl buffer (50 mM, pH 7.4 at 20°), at 25° for 120 min. Nonspecific binding was defined as the binding retained on the filter and membranes in the presence of 1 μM CGS15943. For competition experiments, the membranes (20 μg of protein) were incubated with 2.0 nM [3H]ZM241385 in duplicate, with increasing concentrations of the competing compounds, in a final volume of 0.2 ml Tris-HCl buffer (50 mM, pH 7.4) at 25°C for 120 min. Binding reactions were terminated by filtration through Whatman GF/B glass-fiber filters under reduced pressure with an MT-24 cell harvester (Brandel, Gaithersburg, MD). The filters were washed three times with ice-cold buffer and placed in scintillation vials containing 5 ml scintillation fluid, and bound radioactivity was determined by using a liquid scintillation counter.
Intracellular cyclic AMP levels were measured with a competitive protein binding method [29]. Briefly, CHO cells expressing WT and mutant ARs were harvested by trypsinization. After resuspension in the medium, cells were plated in 24-well plates in 0.5 ml medium/ well. After 24 hr, the medium was removed and cells were washed three times with 1 ml/well of DMEM, containing 50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.4). Cells were then treated with agonists and/or test compounds in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL) and incubated at 37°C for 1 hr. The reaction was terminated upon removal of the medium, and the cells were lysed with 200 μl/well of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at –20°C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μl of the cell lysate, and 30 μl 0.1 M HCl. Bound radioactivity, separated by rapid filtration through Whatman GF/C filters under reduced pressure and washed once with cold buffer, was measured by liquid scintillation spectrometry.
Statistical Analysis
Binding and functional parameters were estimated with GraphPad Prism software (GraphPad, San Diego, CA). IC50 values obtained from competition curves were converted to Ki values by using the Cheng-Prusoff equation [40]. Data were expressed as mean ± standard error.
Molecular Modeling
All calculations were performed using the SYBYL program [34] version 6.9 on a Silicon Graphics Octane workstation (300 MHz MIPS R12000 [IP30] processor). All ligand structures were derived using the “Sketch Molecule” and were performed by random search for all rotatable bonds. The options of the random search consisted of 3000 iterations, 3 kcal energy cutoffs, and chirality checking. MMFF force field [35] and charge were applied using distance-dependent dielectric constants and the conjugate gradient method until the gradient reached 0.05 kcal/mol/Å. After clustering the low energy conformers from the result of the conformational search, the representative conformers from all groups were reoptimized by semi- empirical molecular orbital calculations using the PM3 method in the MOPAC 6.0 package [36].
The previously published human A2AAR model (PDB code: 1upe) built by homology modeling [10] from the X-ray structure of bovine rhodopsin with 2.8 Å resolution [26] was used for the docking study. For the side-chain refinement of the Q89D and N181D neo-ceptors, the optimized structures were then used as the starting point for subsequent 50 ps MD, during which the protein backbone atoms in the secondary structures were constrained. The options of MD at 300 K with a 0.2 ps coupling constant were a time step of 1 fs and a nonbonded update every 25 fs. The SHAKE algorithm [37] was employed to fix the lengths of bonds with hydrogen atoms. The average structure from the last 10 ps trajectory of MD was optimized with backbone constraints in the secondary structure and then the unconstrained structure was minimized as described above.
Compounds 1, 7, 8, 9, 10, and 11 were docked within the human A2AAR and neoceptors. Flexible docking was facilitated through the FlexiDock utility in the Biopolymer module of SYBYL 6.9. During flexible docking, the rotatable bonds in the 5′-terminal group of the neoligand and the side chains of acidic residues were defined with flexible bonds. After the hydrogen atoms were added to the receptor, atomic charges were recalculated by using Kollman all-atom for the protein and Gasteiger-Hückel for the ligand. H-bonding sites were identified for acidic residues of neoceptor and for the positively charged groups of the neoligands that were able to act as an H-bond donor or acceptor. The lowest energy conformer of neoli-gand was variously prepositioned in the putative binding cavity as a starting point for FlexiDock. Default FlexiDock parameters were set at 3000 generation for genetic algorithms. To increase the binding interaction, the torsion angles of the side chains that directly interacted within 5 Å of the ligands, according to the results of FlexiDock, were manually adjusted. The atom types of all ligands were manually assigned with an Amber all-atom force field [38]. Finally, the complex structure was minimized by using an Amber force field with a fixed dielectric constant (4.0), until the conjugate gradient reached 0.1 kcal · mol–1 · Å–1.
Supplementary Material
Acknowledgments
We thank A. Gross (NIDDK) for assistance in binding assays. We thank Dr. B. Li, Dr. A. Chen, and Dr. Z. Pittel (NIDDK) for assistance in preparing the mutant constructs. M.O. is on sabbatical from Toray Industries (Kamakura, Japan) and thanks them for financial support. S.T. thanks the Cystic Fibrosis Foundation (Bethesda, MD) for financial support.
Footnotes
Supplemental Data for this article is available online at http://www.chembiol.com/cgi/content/full/12/2/237/DC1/.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 2.Knutsen LJ, Lau J, Petersen H, Thomsen C, Weis JU, Shalmi M, Judge ME, Hansen AJ, Sheardown MJ. N-substituted adenosines as novel neuroprotective A1 agonists with diminished hypotensive effects. J Med Chem. 1999;42:3463–3477. doi: 10.1021/jm960682u. [DOI] [PubMed] [Google Scholar]
- 3.von Lubitz DKJE, Lin RC, Paul IA, Beenhakker M, Boyd M, Bischofberger N, Jacobson KA. Post-ischemic administration of adenosine amine congener (ADAC): analysis of recovery in gerbils. Eur J Pharmacol. 1996;316:171–179. doi: 10.1016/s0014-2999(96)00667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okusa MD, Linden J, Huang L, Rieger JM, Macdonald TL, Huynh LP. A2A adenosine receptor-mediated inhibition of renal injury and neutrophil adhesion. Am J Physiol Renal Physiol. 2000;279:F809–F818. doi: 10.1152/ajprenal.2000.279.5.F809. [DOI] [PubMed] [Google Scholar]
- 5.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 6.Cassada DC, Tribble CG, Long SM, Laubach VE, Kaza AK, Linden J, Nguyen BN, Rieger JM, Fiser SM, Kron IL, et al. Adenosine A2A analogue ATL-146e reduces systemic tumor necrosing factor-alpha and spinal cord capillary platelet-endothelial cell adhesion molecule-1 expression after spinal cord ischemia. J Vasc Surg. 2002;35:994–998. doi: 10.1067/mva.2002.123091. [DOI] [PubMed] [Google Scholar]
- 7.Maillard M, Nikodijevic O, LaNoue KF, Berkich DA, Ji XD, Bartus R, Jacobson KA. Adenosine receptor prodrugs: Synthesis and biological activity of derivatives of potent, A1-selective agonists. J Pharm Pharmacol. 1994;83:46–53. doi: 10.1002/jps.2600830112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalpiaz A, Scatturin A, Menegatti E, Bortolotti F, Pavan B, Biondi C, Durini E, Manfredini S. Synthesis and study of 5′-ester prodrugs of N6-cyclopentyladenosine, a selective A1 receptor agonist. Pharm Res. 2001;18:531–536. doi: 10.1023/a:1011018730459. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson KA, Gao ZG, Chen A, Barak D, Kim SA, Lee K, Link A, van Rompaey P, van Calenbergh S, Liang BT. Neoceptor concept based on molecular complementarity in GPCRs: A mutant adenosine A3 receptor with selectively enhanced affinity for amine-modified nucleosides. J Med Chem. 2001;44:4125–4136. doi: 10.1021/jm010232o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SK, Gao ZG, Van Rompaey P, Gross AS, Chen A, Van Calenbergh S, Jacobson KA. Modeling the adenosine receptors: Comparison of binding domains of A2A agonist and antagonist. J Med Chem. 2003;46:4847–4859. doi: 10.1021/jm0300431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parsons M, Young L, Lee J-E, Jacobson KA, Liang BT. Distinct cardioprotective effects of adenosine mediated by differential coupling of receptor subtypes to phospholipases C and D. FASEB J. 2000;14:1423–1431. doi: 10.1096/fj.14.10.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohno M, Gao ZG, Van Rompaey P, Tchilibon S, Kim SK, Harris BA, Blaustein J, Gross AS, Duong HT, Van Calenbergh S, et al. Modulation of adenosine receptor affinity and intrinsic efficacy in nucleosides substituted at the 2-position. Bioorg Med Chem. 2004;12:2995–3007. doi: 10.1016/j.bmc.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hocek M. Syntheses of purines bearing carbon substituents in positions 2, 6 or 8 by metal- or organometal-mediated C C bond-forming reactions. Eur J Org Chem. 2003;12:245–254. [Google Scholar]
- 14.Gao ZG, Blaustein J, Gross AS, Melman N, Jacob-son KA. N6-Substituted adenosine derivatives: Selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coward P, Wada HG, Falk MS, Chan SD, Meng F, Akil H, Conklin BR. Controlling signaling with a specifically designed Gi-coupled receptor. Proc Natl Acad Sci USA. 1998;95:352–357. doi: 10.1073/pnas.95.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Shah K, Yang F, Witucki L, Shokat KM. Engineering Src family protein kinases with unnatural nucleotide specificity. Chem Biol. 1998;5:91–101. doi: 10.1016/s1074-5521(98)90143-0. [DOI] [PubMed] [Google Scholar]
- 17.Belshaw PJ, Schreiber SL. Cell-specific calcineurin inhibition by a modifed cyclosporin. J Am Chem Soc. 1997;119:1805–1806. [Google Scholar]
- 18.Tedesco R, Thomas JA, Katzenellenbogen BS, Kat-zenellenbogen JA. The estrogen receptor: a structure- based approach to the design of new specific hormone-receptor combinations. Chem Biol. 2001;8:277–287. doi: 10.1016/s1074-5521(01)00006-0. [DOI] [PubMed] [Google Scholar]
- 19.Koh JT. Designing selectivity and discrimination into re-engineered ligand-receptor interfaces; beyond bumps and holes. Chem Biol. 2002;9:17–23. doi: 10.1016/s1074-5521(02)00087-x. [DOI] [PubMed] [Google Scholar]
- 20.Niswender CM, Ishihara RW, Judge LM, Zhang C, Shokat KM, McKnight GS. Protein engineering of protein kinase A catalytic subunits results in the acquisition of novel inhibitor sensitivity. J Biol Chem. 2002;277:28916–28922. doi: 10.1074/jbc.M203327200. [DOI] [PubMed] [Google Scholar]
- 21.Wildbrandt R, Frotscher U, Freyland M, Messerschmidt W, Richter R, Schulte-Lippern M, Zschaege B. Treatment of glomerulonephritis with metrifudil. Preliminary report Med Klin (Munich) 1972;67:1138–1140. [PubMed] [Google Scholar]
- 22.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q, Olah ME, van Galen PJM, et al. Structure-activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J Med Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Žemlic ka J, Šorm F. The reaction of dimethylchl-oromethylene ammonium chloride with 2′,3′,5′-tri-O-acetyl-inosine: a new synthesis of 6-chloro-9-(beta-D-ribofuranosyl)-purine. Collect Czech Chem Commun. 1965;30:1880–1885. [Google Scholar]
- 24.Kappler F, Hampton A. Approaches to isozyme-specific inhibitors. 17 Attachment of a selectivity-inducing substituent to a multisubstrate adduct Implications for facilitated design of potent, isozyme-selective inhibitors. J Med Chem. 1990;33:2545–2551. doi: 10.1021/jm00171a032. [DOI] [PubMed] [Google Scholar]
- 25.Camp D, Li Y, McCluskey A, Moni RW, Quinn RJ. Diimidazo[1,2-c:4′,5′-e]pyrimidines: N6-N1 conforma-tionally restricted adenosines. Bioorg Med Chem Lett. 1998;8:695–698. doi: 10.1016/s0960-894x(98)00101-2. [DOI] [PubMed] [Google Scholar]
- 26.Palczewski K, Kumasaka T, Hori T, Behnke CA, Moto-shima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp TE, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Q, Lee BX, Glashofer M, van Rhee AM, Jacobson KA. Mutagenesis reveals structure-function parallels between human A2A-adenosine receptors and the biogenic amine family. J Med Chem. 1997;40:2588–2595. doi: 10.1021/jm970084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao ZG, Jiang Q, Jacobson KA, IJzerman AP. Site-directed mutagenesis studies of human A2A adenosine receptors: involvement of Glu13 and His278 in ligand binding and sodium modulation. Biochem Pharmacol. 2000;60:661–668. doi: 10.1016/s0006-2952(00)00357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 30.Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G. N6-alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2B ligand. J Med Chem. 2002;45:3271–3279. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- 31.Yang A, Sonin D, Jones L, Barry WH, Liang BT. A beneficial role of cardiac P2X4 receptors in heart failure: rescue of the calsequestrin-overexpression model of cardiomyopathy. Am J Physiol Heart Circ Physiol. 2004;287:1096–1103. doi: 10.1152/ajpheart.00079.2004. [DOI] [PubMed] [Google Scholar]
- 32.Brooks AD, Ng B, Liu D, Brownlee M, Burt M, Federoff HJ, Fong Y. Specific organ gene transfer in vivo by regional organ perfusion with herpes viral amplicon vectors: implications for local gene therapy. Surgery. 2001;129:324–334. doi: 10.1067/msy.2001.111697. [DOI] [PubMed] [Google Scholar]
- 33.Slavin AJ, Tarner IH, Nakajima A, Urbanek-Ruiz I, McBride J, Contag CH, Fathman CG. Adoptive cellular gene therapy of autoimmune disease. Autoimmun Rev. 2002;1:213–219. doi: 10.1016/s1568-9972(02)00051-4. [DOI] [PubMed] [Google Scholar]
- 34.SYBYL Molecular Modeling System, version 6.9 (computer program) Tripos Inc; St. Louis, Missouri: [Google Scholar]
- 35.Halgren TA. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J Comput Chem. 1999;20:730–748. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 36.Stewart JJP. MOPAC: a semiempirical molecular orbital program. J Comput Aided Mol Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 37.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion for a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 38.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 39.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.