

Abstract
The versatile optical and biological properties of a localized surface plasmon resonance (LSPR) sensor that responds to protein conformational changes are illustrated. The sensor detects conformational changes in a surface-bound construct of the calcium-sensitive protein calmodulin. Increases in calcium concentration induce a 0.96 nm red-shift in the spectral position of the LSPR extinction maximum (λmax). Addition of a calcium chelating agent forces the protein to return to its original conformation and is detected as a reversal of the λmax shift. As opposed to previous work, this work demonstrates that these conformational changes produce a detectable shift in λmax even in the absence of a protein label, with a signal:noise ratio near 500. In addition, the protein conformational changes reversibly switch both the wavelength and intensity of the resonance peak, representing an example of a bimodal plasmonic component that simultaneously relays two distinct forms of optical information. This highly versatile plasmonic device acts as a biological sensor, enabling the detection of calcium ions with a biologically-relevant limit of detection of 23 μM, as well as the detection of calmodulin-specific protein ligands.
Keywords: Nanoparticle, Localized Surface Plasmon Resonance, conformational change, ion sensor, calcium detection
The structure of a protein directly impacts its function. In a healthy cell, protein activity is precisely controlled through conformational changes induced by ligand binding, pH, or ionic strength. Protein misfolding or improper modulation of protein conformation can lead to the onset of several diseases, including Alzheimer’s, Huntington’s, Parkinson’s, and diabetes.1,2 Understanding the dynamic behavior of proteins is, therefore, extremely important in the development of disease diagnostics and therapeutics. Protein replacements or chaperones have been used to treat protein-related diseases;3 however, the efficacy of these treatments and development of new therapies is dependent upon the quality of the tools used to characterize structural variations in proteins.
X-ray crystallography is a frequently utilized tool for characterizing protein structure, and can reveal atomic-level details in the structure of a protein. However, because proteins must be crystallized for characterization, this method prohibits the observation of dynamic structural changes. A host of other methods are more suitable for gathering information about structural changes in real time. These can roughly be divided into two categories based on whether they characterize the secondary or tertiary structure of a protein. Secondary structure analytical tools, including circular dichroism, FT-IR and Raman spectroscopy, and NMR, focus on quantifying the degree of α-helix, β-sheet or unfolded conformation in a protein.4 NMR can be used to obtain detailed information about the positions of secondary structure components within a protein, but complexities involved with assigning chemical shifts hinder real-time analysis.5 Analysis of amide vibrational bands with FT-IR and Raman spectroscopy reveals information about protein helicity, and when performed at THz frequencies, can provide information about solvation and motion during protein folding.4,6 Circular dichroism is often used to monitor protein folding in real-time, though high acquisition averaging or long scan times are required to reduce noise in kinetic measurements.4 Tools for analyzing tertiary structure primarily provide information on global protein properties, such as radius of gyration or refractive index. Small-angle x-ray and neutron scattering (SAXS and SANS, respectively) provide information about the size and shape of solution-phase proteins with nanometer resolution.7,8 Fluorescence methods such as Fluorescence Resonance Energy Transfer (FRET)9,10 and Total Internal Reflection Fluorescence (TIRF)11 provide real-time detection of nanometer-scale conformational changes, but require the protein to be labeled in vitro with fluorescent molecules. Surface Plasmon Resonance (SPR) was purported to detect protein conformational changes based on refractive index,12,13 but pH effects in the matrix layer in which the protein was embedded were subsequently shown to dominate the sensor response.14 Vibrational techniques based on quartz crystal microbalance (QCM)15 and acoustic shear wave propagation16 have been used to monitor conformational changes in calmodulin by measuring changes in the resonant frequency of crystal oscillation. Although conformational changes were detected, the relatively low signal:noise ratio presents a serious limitation for this approach.
In this work we describe the application of Localized Surface Plasmon Resonance (LSPR) spectroscopy to the study of protein conformational changes. LSPR refers to the collective oscillation of conduction electrons that occurs when light impinges on nanoscale noble metal surfaces. This collective electron oscillation gives rise to wavelength-selective extinction and enhanced electromagnetic fields at the nanoparticle surface.17 The most commonly used nanoparticle materials (silver and gold) have extinction peaks in the visible and near IR region of the electromagnetic spectrum. The exact spectral position of the extinction peak (λmax) depends on nanoparticle size, shape, composition, and local refractive index.18 The dependence of λmax on refractive index is utilized in LSPR biosensing applications, where ligand-binding to a surface immobilized receptor results in refractive index increases and gives rise to shifts in the position of λmax.19 Unlike SPR, which utilizes planar metal films that support propagating plasmons with z-direction 1/e decay lengths of ~200 nm, the enhanced electromagnetic fields in LSPR are strongly localized, with 1/e decay lengths of ~ 5 – 10 nm in any direction normal to the nanoparticle surface.20,21 Further, in LSPR the decay length can be tuned by varying the nanoparticle size, shape, and composition. As a result, LSPR sensors are largely insensitive to bulk refractive index changes, and “selectively” respond to nanoscale analytes at the nanoparticle surface. This localized sensitivity has been utilized to detect structural changes in supported lipid bilayers,22 and was shown to detect conformational changes in a labeled protein.23 Improvements in LSPR instrumentation have reduced the standard deviation in λmax measurements to the order of 10−3 nm,23,24 providing a signal:noise ratio of 103 – 104 for typical ligand binding events. Here, we demonstrate for the first time that this extreme sensitivity to local refractive index allows the detection of reversible tertiary conformational changes in an unlabeled calmodulin protein, and describe the unique biodetection capabilities conferred by the resulting plasmon switching response.
Calmodulin was chosen as the plasmonic response modulator because it is a ubiquitous and highly conserved intracellular protein whose activity is regulated by intracellular calcium concentration. This 17 kDa protein consists of N- and C-terminal globular domains connected by a flexible, predominantly α-helical linker.25-28 Each globular domain binds two Ca2+ ions via EF-hand helix-loop-helix motifs. Calcium binding occurs cooperatively29 and induces a conformational change that exposes N- and C-terminal hydrophobic pockets not present in the Ca-free (apo) form of the protein.30 These hydrophobic pockets allow calmodulin to bind to aromatic and aliphatic side chains on over 100 different target enzymes and proteins.31-33 In its Ca-bound state, calmodulin adopts a relatively rigid dumbbell-like structure 6.5 nm in length.25 NMR studies of apo-calmodulin revealed a more compact conformation with greater disorder in the helix linker regions, suggesting increased flexibility in the Ca-free state.30 SAXS studies also revealed differences in the hydrodynamic radii of the two conformers, with the Ca-bound state extending in radius by approximately 1 nm.34 Thus, the Ca-bound and Ca-free states of calmodulin are marked by distinct structural differences that we hypothesized could be distinguished based on LSPR λmax. We show that conformational changes in surface-immobilized calmodulin can be tracked in real-time based on LSPR spectral shifts, allowing the determination of kinetic rates for reorientation of the calmodulin monolayer. In addition, equilibrium LSPR shifts are used to determine the calcium affinity constant, and the LSPR sensor is shown to exhibit physiologically-relevant sensitivity to calcium concentration and calmodulin-specific ligands. The plasmonic switching behavior induced by calmodulin conformational changes exhibits an interesting bimodal character that could potentially be utilized in opto-electronic devices.
Nanosphere lithography (NSL)35 was used to create monodisperse, surface-confined Ag nanoprisms on glass according to a previously published procedure.36 For all experiments, 80 nm of Ag (D. F. Goldsmith) was evaporated over 390 nm polystyrene nanospheres in order to create nanoprisms with approximate in-plane widths of 100 nm and out of plane heights of 80 nm. Fabrication details can be found in the Supporting Information.
The Ag nanoparticle substrates were then functionalized with a self-assembled monolayer (SAM) prepared in-house according to a previously published procedure.37 Substrates were incubated in a 1 mM ethanolic solution of 96% hydroxyl-terminated triethylene glycol and 4% maleimide-terminated triethylene glycol for 24-48 hours, then rinsed with ethanol and dried in N2. The substrate was then immediately drop-coated with a 1 mM solution of a phosphonate capture ligand in DMSO for 30 minutes at 35° C. Finally, cutinase-calmodulin constructs were immobilized by exposing the phosphonate-terminated SAM surface to a 500 nM solution of the protein construct in 20 mM, pH 8.0 Tris buffer at room temperature. The reaction was followed in real-time until it reached completion, generally ~ 15 minutes. The reaction was considered complete when Δλmax/dt reached zero.
Cutinase-calmodulin is a fusion protein construct containing an N-terminal cutinase fused to a C-terminal calmodulin domain linked through a short, flexible GGGS peptide linker. In previous work23 we designed and cloned a similar construct, cutinase-calmodulin-cutinase, to achieve the three domain fusion protein. For this work, we used the truncated cutinase-calmodulin (CutCaM) fragment generated in the aforementioned work for cloning this gene fragment into a pET-21d (Novagen) vector between the N-terminal NcoI and C-terminal XhoI sites. A BamHI site separates the cutinase- and GGGS-calmodulin domains (see Supporting Information for complete details).
The recombinant CutCamCut construct has an overall mass of 63 kDa, with the calmodulin domain contributing 19 kDa and each cutinase domain contributing 22 kDa. The N-terminal cutinase on CutCamCut is rendered inactive by mutagenesis of an active site serine residue to alanine, ensuring that each construct is anchored to the nanoparticle surface by the C-terminal cutinase only.
Macroscale UV-vis extinction measurements were performed in standard transmission geometry with unpolarized light coupled into a photodiode array spectrometer (Model BRC711E, BWTek, Newark, DE) using lenses. The probe diameter was approximately 1 mm. A home-built flow cell was used to control the external environment of the Ag nanoparticle substrates. A program written in Labview (National Instruments, Austin, TX) was used to acquire spectra and provide real-time readout. The real-time response of CutCaMCut was recorded with 600 ms integration times and no averaging, with the maximum wavelength determined by fitting a 100 nm spectral region around the peak to a fourth order polynomial. The real-time response of CutCaM was recorded with a 300 ms integration time and averaged twice, with the maximum wavelength determined by fitting a 150 nm spectral region around the peak to a 20th order polynomial. To improve the signal:noise ratio for the CutCaM measurements, the intensity of the light source was maintained near the saturation level of the spectrophotometer (this required the light source intensity to be increased three-fold for the nanoparticle surface compared to the nanoparticle-free reference surface). All spectra and λmax time traces were recorded in an aqueous environment containing 20 mM Tris buffer at pH 8.
Anthrax edema factor (EF) was prepared according to procedures described previously.38 Specific interaction with surface-bound calmodulin was assayed by exposing the CutCaMCut-functionalized sensor to 1 μM EF in the presence of 100 μM CaCl2. Protein binding proceeded over a 30 minute period. The LSPR λmax was monitored throughout the assay. Following binding, the sensor surface was rinsed with 20 mM Tris to remove physisorbed species.
This study aimed to demonstrate label-free LSPR-based detection of protein conformational changes. Our previous study using LSPR biosensors for the detection of protein conformational changes utilized a recombinant calmodulin construct with both C- and N-terminal cutinase labels.23 The C-terminal cutinase served to immobilize the construct, while the N-terminal moiety acted as an extra dielectric label to enhance the LSPR signal. In the presence of the N-terminal cutinase label, calmodulin conformational changes induced shifts in the LSPR λmax of 2.2 nm, amounting to a signal:noise ratio of over 500. Although the conformational changes were easily detectable, labels are undesirable because they can alter the structure and activity of a protein. We therefore sought to assess the sensitivity of LSPR biosensors to conformational changes in unlabeled calmodulin.
In this work, calmodulin was selectively immobilized on surface-confined Ag nanoprisms by utilizing a recombinant protein construct in which only the C-terminal end of calmodulin is fused to a cutinase domain.37 The cutinase moiety forms a covalent bond with self-assembled monolayers (SAMs) presenting terminal phosphonate groups, allowing label-free calmodulin to be flexibly and uniformly oriented at the nanoprism surface. Irreversible binding of the calmodulin construct (hereafter CutCaM) was verified by monitoring λmax as a function of time (Figure 1). Saturation of the surface-bound phosphonate ligands was ensured by allowing the reaction to proceed until dλmax/dt reached zero. Subsequent rinsing of the nanoprism surface removed any physisorbed species. A representative immobilization shows a λmax shift of 3.81 nm to the red upon CutCaM binding, indicating a refractive index (R.I.) increase at the nanoprism surface due to CutCaM immobilization. This red shift lies within the expected Δλmax range for adsorption of a monolayer of protein, which typically ranges from 3 to 15 nm depending on the size of the protein.
Figure 1.

Immobilization of CutCaM construct. (A) Schematic depicting covalent bond formation between cutinase and the phosphonate-terminated polyethylene glycol (PEG) SAM which serves to immobilize calmodulin in a uniform orientation at the nanoparticle surface. Protein construct and nanoparticle are not drawn to scale. (B) Time trace showing λmax changes upon exposure of the LSPR sensor to 500 nM CutCaM (1), followed by rinsing with 20 mM Tris (2). The λmax shifts a total of 3.81 nm in response to protein binding.
This work demonstrates the first reported detection of conformational changes in an unlabeled protein. To induce conformational changes in the surface-bound calmodulin, the CutCaM-functionalized nanoprism surface was exposed to sequential five-minute cycles of 2 mM CaCl2 and 2 mM EGTA. All solutions were buffered using saline-free Tris at pH 8 in order to minimize effects due to pH changes. Calcium binding to calmodulin triggers a transition to the “open” conformation, in which the length and radius of gyration of the protein increase. Chelation of calcium ions by EGTA triggers a reversion to the “closed” conformation (Figure 2A). The λmax of the LSPR sensor was monitored at 600 ms intervals over the entire CaCl2/EGTA cycle. Conformational transitions were detected by shifts in the λmax of the LSPR sensor (Figure 2B). The λmax changes were repeatable over several cycles and had an average magnitude of 0.96 ± .06 nm, with red shifts occurring in response to calmodulin opening and blue shifts occurring in response to calmodulin closing. As a comparison, these shifts are plotted next to the shifts previously observed from cutinase-labeled calmodulin (CutCaMCut). As expected, the response from label-free calmodulin is smaller due to the lower mass of the mobile portion of the protein construct (19 kDa) compared to the mass of the mobile portion of the CutCaMCut construct (41 kDa). The sensor response can be approximated by equation (1),
| (1) |
where m is the sensitivity factor in nm/RIU, Δn is the change in refractive index, d is the thickness of the adsorbed dielectric layer, and ld is the decay length of the electromagnetic field at the nanoparticle surface. Assuming a dielectric layer thickness that saturates the electromagnetic decay length, the response can be approximated by mΔn. Using the experimentally determined m value for this sensor of 241 nm/RIU (Figure S1), we calculate the R.I. change for label-free calmodulin to be 4 × 10−3 RIU, and the R.I. change for cutinase-labeled calmodulin to be 9 × 10−3 RIU. Thus, the R.I. change for CutCaMCut is roughly twice as large as the R.I. change for CutCaM, in keeping with the mass differences between the two constructs. The standard deviation (σ) in λmax for the CutCaM-functionalized sensor was further reduced from previous work down to 2 × 10−3 nm, providing a S/N of roughly 5 × 102 in response to the protein conformational change. Taking the smallest detectable shift to be 6 × 10−3 nm (3σ), this sensor is capable of detecting refractive index changes as small as 2.5 × 10−5 RIU.
Figure 2.

CutCaM conformational changes. (A) Schematic representation of the reversible conformational changes calmodulin undergoes in response to changing calcium concentration. In the presence of calcium (green circles) calmodulin adopts a rigid, extended structure. (B) LSPR λmax changes plotted over time as calmodulin undergoes conformational changes. LSPR response to CutCaM is plotted in red; LSPR response to the larger CutCaMCut construct described in reference 21 is plotted in black as a comparison. (C) First order kinetic fit to the CutCaM-induced λmax changes occurring between 40 and 50 minutes from the plot in 2B. Rate constants for the opening and closing transitions were found to be 0.057 s−1 and 0.13 s−1, respectively.
Monitoring λmax changes in real-time enabled the determination of the kinetics of the conformational change. The conformational transition can be approximated by a two-step process according to scheme 1:
where k1/−1 are the forward and reverse rates for calcium ion binding to calmodulin, and k2/−2 are the forward and reverse rates for the calcium-induced conformational change. Because the binding of calcium ions involves a negligible mass change that cannot be detected by LSPR, and because calcium binding occurs on a timescale 104 times faster than the conformational change,39 only the rate k2 is observed during the forward process. During the reverse process, calcium release occurs on a similar timescale to the conformational change,39 and the observed rate is a convolution of k−2 and k−1. For simplicity, we designate the forward and reverse rates by kopen and kclose, respectively. Data was fit to a first-order kinetic model according to equation (2),
| (2) |
where A is Δλmax at the final time t, k is the rate in s−1, and B is a correction for linear drift in the baseline over time due to solvent annealing effects (previously described elsewhere).40 These fits revealed opening and closing rate constants of 0.059 ± 0.01 s−1 and 0.13 ± 0.06 s−1, respectively, where the error is the calculated standard deviation from five repeat cycles on a single sample (Figure 2C). These opening and closing rate constants agree well with the rate constants previously observed for the N-terminally labeled calmodulin construct (CutCaMCut) of 0.033 ± 0.003 s−1 and 0.125 ± .02 s−1, respectively, indicating that the additional cutinase label had a modest impact on the kinetics of the conformational transition to the open state, and no impact on the transition to the closed state. This observation suggests that protein labels do not always necessarily disrupt the kinetics of conformational changes, and may be advantageous in formats where sensitivity is a concern. However, the rates observed for surface-immobilized calmodulin deviate significantly from conformational transition rates previously observed in solution. Previous studies demonstrated that calmodulin opening and closing rates occur on a microsecond to millisecond timescale,39,41,42 in agreement with the consensus that the physiological role of calmodulin is to act as a fast responder to transient calcium signals. The slower kinetic rates observed in this work may indicate that the transition being observed is a packing rearrangement in the calmodulin monolayer. Steric interactions between adjacent proteins may create an energetic penalty that slows this process. The observed rates are also limited by mixing and diffusion processes, and should more closely agree with rates observed in solution once these mixing limitations are overcome. Future work will aim to elucidate possible fast and slow processes that occur during conformational transitions in surface-confined protein layers through the incorporation of rapid-mixing devices or photolabile calcium compounds, and by varying the density of the immobilized Calmodulin construct.
In order to understand how the observed LSPR shifts in response to calmodulin conformational changes correspond to the surface orientation of the protein, we modeled the LSPR response to an adsorbed dielectric layer with changing thickness and refractive index. In the presence of a dielectric layer, the LSPR response can be approximated by a modified version of equation (1):
| (3) |
where neff is the effective refractive index over the entire enhanced electromagnetic field at the nanoparticle surface. The effective refractive index is a weighted average of the refractive index at each distance z from the nanoparticle surface, as described by equation (4):43
| (4) |
where n(z) is the refractive index at height z. Because the EM field strength decays exponentially away from the nanoparticle surface, the refractive index closest to the nanoparticle surface contributes most heavily to the LSPR response. The effective refractive index and resulting LSPR response are therefore dominated by the refractive index within the first few nanometers of the surface. In a traditional LSPR bioassay, the binding of proteins or antibodies to surface-bound receptors creates an additional dielectric layer, increasing both z and n(z) and producing a red shift in λmax. When detecting conformational changes, however, the total mass of the adsorbed protein layer remains constant. The LSPR response in this case is determined by changes in the density and refractive index of the adsorbed protein layer that occur as a result of the conformational change. For example, extension of calmodulin away from the nanoparticle surface in response to calcium binding will result in both an increase in the height and a decrease in the density of the protein monolayer. Referring to equation (4), we see that height increases (z) lead to larger neff and produce red shifts in the LSPR, while density decreases lead to a lower refractive index (n(z)) and produce blue shifts in the LSPR. The result of these competing effects on the overall LSPR response was modeled using equations 3 and 4, where neff for the dielectric layer ranges between 1.33 (water) and 1.6 (protein) and the EM decay length ld is taken to be 5 nm (Figure 3). This modeling revealed that, for a fixed amount of protein, the LSPR response is dominated by density rather than height changes in the protein monolayer. Thus, denser and shorter monolayers induce red-shifted extinction maxima, and taller, less dense monolayers induce blue-shifted extinction maxima. In this study we observed a red shift in response to calcium binding, indicating that calmodulin adopts a more densely packed conformation close to the nanoparticle surface in the presence of calcium. This suggests that, in response to calcium binding, calmodulin extends in a direction that moves the center of mass of the protein closer to the nanoparticle surface. The direction of this extension and the preference for calmodulin to create a dense monolayer on the surface may be influenced by hydrophobic interactions between adjacent proteins: upon calcium binding, the conformational change in calmodulin exposes surface hydrophobic pockets on the N- and C-terminal domains that enable binding of protein targets.30 In the absence of protein targets, inter-protein interactions via these hydrophobic pockets may force calmodulin into a denser packing formation. To quantitatively assess how these LSPR results compare to the expected SPR response, we also used equations (3) and (4) to model the response for a surface with an EM decay length of 200 nm. From the modeled SPR response curve it is apparent that the longer EM decay length dramatically reduces the sensitivity to small refractive index changes occurring close to the sensor surface. A schematic representation of the relative EM decay lengths for LSPR and SPR sensors, depicting the volume of the sensing region occupied by a roughly ~10 nm diameter protein in each case, is shown in Figure 3B. These modeling results demonstrate why SPR sensors have previously failed to detect conformational changes in protein monolayers, and why LSPR sensors provide an advantage in this application.
Figure 3.

Predicted LSPR and SPR response. (A) Response of LSPR and SPR sensors based on a sensing volume defined by an electromagnetic (EM) field with an intensity that decays exponentially away from the nanoparticle surface. The decay length (ld) of the EM field for the LSPR sensor is 5 nm; the decay length of the SPR sensor is 200 nm. The protein length at a given conformation is defined as d; the length of the closed conformation protein is designated as d0. (B) Schematic representation of the sensing volume (red) of the SPR and LSPR sensor, where the sensing volume is taken to be 5ld.
One of the unique consequences of LSPR-based conformational change detection is the presence of bimodal switching behavior in the LSPR plasmon peak. Although LSPR detection is typically based only on the magnitude and direction of spectral shifts in λmax, valuable information can be obtained from monitoring changes in the extinction intensity as well. Mie theory describes a linear dependence of the extinction intensity on the refractive index at the nanoparticle surface. In addition to observing reversible λmax wavelength shifts in response to calmodulin conformational changes, we also observed reversible changes in the extinction intensity (Figure 4). These intensity changes had an average value of .002 (a.u.) with a standard deviation in the intensity value of 1.5 × 10−5, providing a S/N ratio of 130. Although the higher S/N ratio for λmax wavelength measurements makes λmax a more attractive parameter for the measurement of conformational changes, the presence of bimodal switching could provide unique advantage in sensing and opto-electronic applications. Plasmonic devices offer a means to overcome the size and speed limitations of conventional electronic circuits by providing a means to transmit digital information using light at sub-wavelength dimensions.44 Tunable plasmonic devices capable of concurrently transmitting two forms of optical information could be a valuable asset in these devices.
Figure 4.
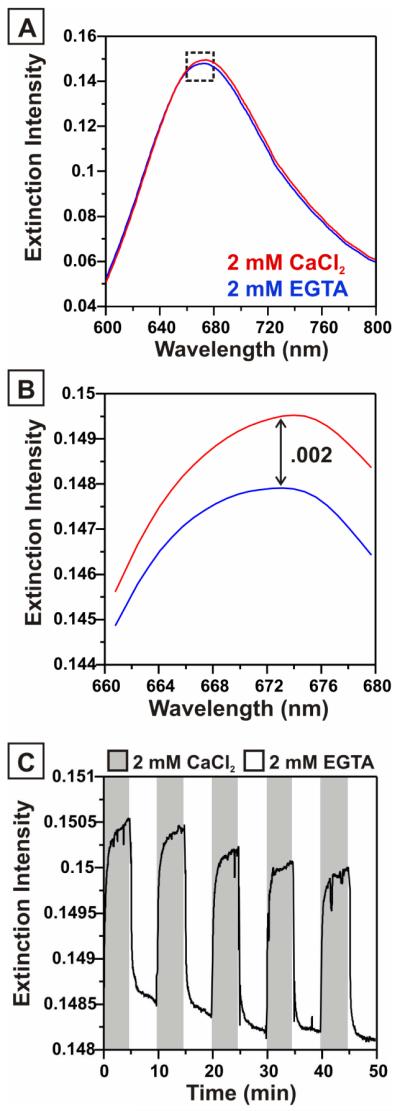
Extinction intensity changes in response to calmodulin conformational changes. (A) Extinction spectra from CutCaM-functionalized LSPR sensor collected in 2 mM CaCl2 (red) and 2 mM EGTA (blue). (B) Magnification of the dashed square region of the spectra shown in 4A, depicting a difference in extinction intensity of 0.002 (A.U.). (C) Time trace showing extinction intensity changes in response to calmodulin conformational transitions.
In addition to demonstrating the detection of protein conformational changes, we also aimed to determine the utility of the LSPR sensor for alternative biodetection applications. In order to maximize the sensitivity of the sensor for these applications we chose to utilize the labeled CutCaMCut construct as the surface receptor. With this construct we demonstrated the detection of calcium and calmodulin-specific ligands. Calcium detection is typically achieved using fluorescent dyes or electronic meters with dynamic ranges in the nanomolar to millimolar regime. However, the detection of small molecules (<100 Da) has previously posed a challenge for plasmonic sensors. By taking advantage of calmodulin conformational changes that occur in response to calcium, we were able to detect the binding of only 600 fmol Ca2+/cm2 at full calmodulin saturation. By measuring the LSPR response to calcium concentrations ranging from nanomolar to millimolar and fitting the resulting curve to the langmuir equation,
| (5) |
we calculated a dissociation constant (Kd = 1/Ka) for the calcium-calmodulin interaction of 52 μM (Figure 5A). At low concentrations, a small non-specific response due to changes in the bulk refractive index gives rise to a baseline Δλmax of 0.1 nm ± 0.012 nm, as indicated by the dashed line in Figure 5A. Taking the minimum detectable shift to be 0.124 nm (three standard deviations above the maximum baseline), and using the calculated Kd, the limit of detection of the LSPR calcium sensor is 23 μM. It is significant that this LOD lies at physiologically relevant calcium concentrations. This feature could be utilized for the detection of calcium transients capable of triggering cellular signaling cascades. As an important intracellular calcium messenger, calmodulin is uniquely poised to detect only calcium transients that lie above a biologically significant threshold.
Figure 5.

LSPR detection of calcium ions and calmodulin-specific ligands. (A) Binding curve describing λmax response of the CutCaMCut functionalized LSPR sensor to varying calcium concentration. Data (black diamonds) is fit to a langmuir isotherm (red line) in order to determine Kd. The non-specific response of 0.1 nm due to bulk refractive index changes is depicted by the dashed black line. (B) Time trace depicting LSPR detection of anthrax edema factor (EF) binding to calmodulin. Dashed lines indicate exposure of the LSPR sensor to (1) 500 nM CutCaMCut, (2) 20 mM Tris, (3) 100 μM CaCl2, (4) 1 μM EF, and (5) 20 mM Tris.
Finally, we demonstrated the detection of anthrax edema factor (EF), a calmodulin-specific ligand that, in conjunction with two other protein factors, is responsible for anthrax toxicity.45,46 Calmodulin binding to EF induces a structural transition in EF that activates the enzyme for catalysis.47 This activation allows EF to catalyze the conversion of ATP to cAMP, disrupting ATP stores and causing fluid loss in affected cells. It has been shown that the EF enzyme binds to calmodulin in a calcium-sensitive manner, with optimal binding affinity occurring at calcium concentrations above 10 μM.48 We therefore monitored the binding of 1 μM EF to surface-bound CutCaMCut in the presence of 100 μM CaCl2. The LSPR λmax shifted 6.6 nm in response to EF binding (Figure 5B). The shift due to EF binding was larger than the shift due to CutCaMCut immobilization, as expected based on the size of EF (80 kDa) compared to CutCaMCut (63 kDa). A non-specific binding assay in which the CutCaMCut surface was exposed to bovine serum albumin demonstrated no changes in λmax in reponse to the non-calmodulin specific ligand (Figure S2), indicating that the EF-CutCaMCut interaction was specific. Because of its promiscuous interactions with a wide variety of ligands, calmodulin can act as a useful surface receptor for over one hundred proteins of interest.
Perhaps the most striking conclusion from this study is the versatility of this calmodulin-based LSPR sensor. We demonstrated initially that the LSPR λmax is sensitive to protein conformation, and by monitoring λmax as a function of time we can study the rates of conformational transitions. In addition, the direction of the observed λmax shift provides information about the surface orientation and density of the protein monolayer. In this case, calmodulin extends in a way that creates a more densely-packed monolayer in response to calcium binding. Although we could not draw any conclusions about the precise secondary or tertiary structural changes that occurred as a result of the conformational transition, the LSPR sensor is nonetheless a valuable tool for studies in which the goal is to ascertain whether or not a structural change occurs in response to an external stimulus. The LSPR sensor could also be used to detect protein folding processes, for example by immobilizing a denatured protein on the LSPR sensor, then altering environmental conditions to induce folding. Protein folding should be accompanied by large changes in refractive index due to the changes in density that occur in transitioning from the unfolded to folded state, and should be detectable with even larger S/N ratios than reported in this work.
In addition to monitoring changes in protein structure, the LSPR sensor utilizes calmodulin conformational changes to enable the detection of small molecules that are otherwise “invisible” to plasmonic sensors. There is a strong motivation to develop sensors capable of detecting small molecules for applications ranging from water quality analysis to drug screening. This type of detection is challenging using conventional ligand interaction assays because of the large size differences between the receptor and the small molecule analyte. Previous work49,50 demonstrated that changes in the absorption properties of chromophoric proteins in response to small molecule binding can alter resonant interactions between the protein and nanoparticle, inducing a significantly enhanced LSPR response. Thus, chromophoric proteins offer a unique signal transduction method for the detection of small molecules. This study illustrates how conformational changes in proteins can likewise induce much larger LSPR shifts than expected based on small molecule binding alone. Taking advantage of the unique structural and optical properties of proteins will open up avenues for detecting a variety of molecular ligands that were previously undetectable using plasmonic sensors. Furthermore, the small (~ 1 cm) sensor format and easy spectroscopic readout of LSPR sensors presents the possibility of incorporating LSPR detection into portable sensors for field use.
This study demonstrated the detection of conformational changes in the intracellular protein calmodulin, in what is the first reported detection of conformational changes in an unlabeled protein using a plasmonic sensor. Calmodulin conformational transitions in response to calcium were detected by shifts of approximately 1 nm in the LSPR λmax. Although these shifts are significantly smaller than typical Δλmax values reported for protein binding assays, the extremely low noise level of 2 × 10−3 nm in our spectroscopic measurements provides a S/N of nearly 500 for the conformational change measurement. The relatively small size of calmodulin (19 kDa) suggests that LSPR sensors can also be utilized to detect conformational changes in a host of other proteins with masses as small as only a few kDa. Monitoring the λmax response to conformational changes in real time enabled the determination of the kinetics of the transition, and could also be utilized to measure the rates of other protein structural changes such as folding and unfolding.
In addition, the wide range of applications for the LSPR sensor described in this work illustrates the diverse functionality that can be conferred upon biosensors by taking advantage of the unique properties of biomolecules. The calmodulin-based LSPR sensor performs four distinct functions: 1) detection of protein conformational changes, 2) detection of protein-ligand interactions, 3) detection of small molecules such as calcium, and 4) bimodal plasmonic switching. These functions can be utilized for a wide array of applications, including the study of protein structure and function, quantification of aqueous calcium concentrations, and development of tunable and switchable plasmonic devices. Utilizing other proteins with distinct conformational triggers—light, pH, metal ions, etc—will further expand the range of functions of LSPR sensors. This work represents one example of how the properties of biomolecules can be harnessed in order to improve and diversify detection capabilities.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the National Science Foundation (Grants EEC-0647560, CHE-0911145, DMR-0520513, and BES-0507036), the National Cancer Institute (1 U54 CA119341-01), a Ruth L. Kirschstein National Research Service Award (5 F32 GM077020) to J.N.A., a George W. Beadle Postdoctoral Fellowship to Y.L., and a Ryan Fellowship to W.P.H. The authors thank the Wei-Jen Tang group from the University of Chicago for the donation of the anthrax edema factor used in this work.
Footnotes
SUPPORTING INFORMATION Data not included in the manuscript, including the SAXS structure of the CutCaMCut construct, refractive index sensitivity calculations and non-specific binding studies, are available on the World Wide Web at http://pubs.acs.org.
References
- (1).Dobson CM. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- (2).Dobson CM. Seminars in Cell and Developmental Biology. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- (3).Cohen FE, Kelly JW. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- (4).Pelton JT, McLean LR. Analytical Biochemistry. 2000;277:167–176. doi: 10.1006/abio.1999.4320. [DOI] [PubMed] [Google Scholar]
- (5).Ishima R, Torchia DA. Nature Structural Biology. 2000;7:740–743. doi: 10.1038/78963. [DOI] [PubMed] [Google Scholar]
- (6).Leitner DM, Havenith M, Gruebele M. Internation Reviews in Physical Chemistry. 2006;25:553–582. [Google Scholar]
- (7).Uzawa T, Akiyama S, Kimura T, Takahashi S, Ishimori K, Morishima I, Fujisawa T. PNAS. 2004;101:1171–1176. doi: 10.1073/pnas.0305376101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lipfert J, Doniach S. Annual Reviews of Biophysics and Biomolecular Structure. 2007;36:307–327. doi: 10.1146/annurev.biophys.36.040306.132655. [DOI] [PubMed] [Google Scholar]
- (9).Truong K, Ikura M. Current Opinion in Structural Biology. 2001;11:573–578. doi: 10.1016/s0959-440x(00)00249-9. [DOI] [PubMed] [Google Scholar]
- (10).Heyduk T. Current Opinion in Biotechnology. 2002;13:292–296. doi: 10.1016/s0958-1669(02)00332-4. [DOI] [PubMed] [Google Scholar]
- (11).Axelrod D. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- (12).Sota H, Hasegawa Y. Analytical Chemistry. 1998;70:2019–2024. doi: 10.1021/ac9713666. [DOI] [PubMed] [Google Scholar]
- (13).Gestwicki JE, Hsieh HV, Pitner JB. Analytical Chemistry. 2001;73:5732–5737. doi: 10.1021/ac0105888. [DOI] [PubMed] [Google Scholar]
- (14).Paynter S, Russell DA. Analytical Biochemistry. 2002;309:85–95. doi: 10.1016/s0003-2697(02)00255-5. [DOI] [PubMed] [Google Scholar]
- (15).Furusawa H, Komatsu M, Okahata Y. Analytical Chemistry. 2009;81:1841–1847. doi: 10.1021/ac8022229. [DOI] [PubMed] [Google Scholar]
- (16).Wang X, Ellis JS, Lyle E-L, Sundaram P, Thompson M. Molecular Biosystems. 2006;2:184–192. doi: 10.1039/b600186f. [DOI] [PubMed] [Google Scholar]
- (17).Willets KA. Annual Reviews in Physical Chemistry. 2007;58:267–297. doi: 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
- (18).Kreibig U, Vollmer M, editors. Optical Properties of Metal Clusters. Vol. 25. Springer-Verlag; Heidelberg, Germany: 1995. [Google Scholar]
- (19).Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Nature Materials. 2009;7:442–453. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- (20).Haes AJ, Zou S, Schatz GC, Van Duyne RP. Journal of Physical Chemistry B. 2004;108:109–116. doi: 10.1021/jp051178g. [DOI] [PubMed] [Google Scholar]
- (21).Whitney AV, Elam JW, Zou S, Zinovev AV, Stair PC, Schatz GC, Van Duyne RP. Journal of Physical Chemistry B. 2005;109:20522–20528. doi: 10.1021/jp0540656. [DOI] [PubMed] [Google Scholar]
- (22).Jonsson MP, Jonsson P, Dahlin AB, Hook F. Nano Letters. 2007;7:3462–3468. doi: 10.1021/nl072006t. [DOI] [PubMed] [Google Scholar]
- (23).Hall WP, Anker JN, Lin Y, Modica J, Mrksich M, Van Duyne RP. Journal of the American Chemical Society. 2008;130:5836–5837. doi: 10.1021/ja7109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dahlin AB, Tegenfeldt JO, Hook F. Analytical Chemistry. 2006;78:4416–4423. doi: 10.1021/ac0601967. [DOI] [PubMed] [Google Scholar]
- (25).Babu YS, Sack JS, Greenhough TJ, Bugg CE, Means AR, Cook WJ. Nature. 1985;315:37–40. doi: 10.1038/315037a0. [DOI] [PubMed] [Google Scholar]
- (26).Kretsinger RH, Rudnick SE, Weissman LJ. Journal of Inorganic Biochemistry. 1986;28:289–302. doi: 10.1016/0162-0134(86)80093-9. [DOI] [PubMed] [Google Scholar]
- (27).Babu YS, Bugg CE, Cook WJ. Journal of Molecular Biology. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- (28).Wilson MA, Brunger AT. Journal of Molecular Biology. 2000;301:1237–1256. doi: 10.1006/jmbi.2000.4029. [DOI] [PubMed] [Google Scholar]
- (29).Linse S, Helmersson A, Forsen S. Journal of Biological Chemistry. 1991;266:8050–8054. [PubMed] [Google Scholar]
- (30).Zhang M, Tanaka T, Ikura M. Nature Structural Biology. 1995;2:758–767. doi: 10.1038/nsb0995-758. [DOI] [PubMed] [Google Scholar]
- (31).Hoeflich KP, Ikura M. Cell. 2002;108:739–742. doi: 10.1016/s0092-8674(02)00682-7. [DOI] [PubMed] [Google Scholar]
- (32).Cohen P, Klee CB, editors. Calmodulin. Elsevier; New York: 1988. [Google Scholar]
- (33).Van Eldik LJ, Watterson DM, editors. Calmodulin and Signal Transduction. Academic Press; San Diego: 1998. [Google Scholar]
- (34).Seaton BA, Head JF, Engelman DM, Richards FM. Biochemistry. 1985;24:6740–6743. doi: 10.1021/bi00345a002. [DOI] [PubMed] [Google Scholar]
- (35).Hulteen JC, Van Duyne RP. Journal of Vacuum Science and Technology A. 1995;13:1553–1558. [Google Scholar]
- (36).Haes AJ, Van Duyne RP. Journal of the American Chemical Society. 2002;124:10596–10604. doi: 10.1021/ja020393x. [DOI] [PubMed] [Google Scholar]
- (37).Hodneland CD, Lee Y-S, Min D-H, Mrksich M. PNAS. 2002;99:5048–5052. doi: 10.1073/pnas.072685299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Drum CL, Shen Y, Rice PA, Bohm A, Tang W-J. Acta Crystollographica D. 2001;D57:1881–1884. doi: 10.1107/s0907444901014937. [DOI] [PubMed] [Google Scholar]
- (39).Evenas J, Malmendal A, Akke M. Structure. 2001;9:185–195. doi: 10.1016/s0969-2126(01)00575-5. [DOI] [PubMed] [Google Scholar]
- (40).Malinsky MD, Kelly KL, Schatz GC, Van Duyne RP. Journal of the American Chemical Society. 2001;1223:1471–1482. [Google Scholar]
- (41).Tsuruta H, Sano T. Biophysical Chemistry. 1990;35:75–84. doi: 10.1016/0301-4622(90)80062-c. [DOI] [PubMed] [Google Scholar]
- (42).Evenas J, Forsen S, Malmendal A, Akke M. Journal of Molecular Biology. 1999;289:603–617. doi: 10.1006/jmbi.1999.2770. [DOI] [PubMed] [Google Scholar]
- (43).Jung LS, Campbell CT, Chinowsky TM, Mar MN, Yee SS. Langmuir. 1998;14:5636–5648. [Google Scholar]
- (44).Ozbay E. Science. 2006;311:189–193. doi: 10.1126/science.1114849. [DOI] [PubMed] [Google Scholar]
- (45).Leppla SH. PNAS. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Leppla SH. Advanced Cyclic Nucleotide and Protein Phosphorylation Research. 1984;17:189–198. [PubMed] [Google Scholar]
- (47).Drum CL, Yan S-Z, Bard J, Shen Y-Q, Lu D, Soelaiman S, Grabarek Z, Bohm A, Tang W-J. Nature. 2002;415:396–402. doi: 10.1038/415396a. [DOI] [PubMed] [Google Scholar]
- (48).Shen Y, Lee Y-S, Soelaiman S, Bergson P, Lu D, Chen A, Beckingham K, Grabarek Z, Mrksich M, Tang W-J. European Molecular Biology Organization. 2002;21:6721–6732. doi: 10.1093/emboj/cdf681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhao J, Das A, Zhang X, Schatz GC, Sligar SG, Van Duyne RP. Journal of the American Chemical Society. 2006;128:11004–11005. doi: 10.1021/ja0636082. [DOI] [PubMed] [Google Scholar]
- (50).Das A, Zhao J, Schatz GC, Sligar SG, Van Duyne RP. Analytical Chemistry. 2009;81:3754–3759. doi: 10.1021/ac802612z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.