

Abstract
Sodium 4-phenylbutyrate (4PBA) improves the intracellular trafficking of ΔF508-CFTR in cystic fibrosis (CF) epithelial cells. The underlying mechanism is uncertain, but 4PBA modulates the expression of some cytosolic molecular chaperones. To identify other 4PBA-regulated proteins that might regulate ΔF508-CFTR trafficking, we performed a differential display RT-PCR screen on IB3-1 CF bronchiolar epithelial cells exposed to 4PBA. One transcript up-regulated by 4PBA encoded ERp29, a luminal resident of the endoplasmic reticulum (ER) thought to be a novel molecular chaperone. We tested the hypothesis that ERp29 is a 4PBA-regulated ER chaperone that influences ΔF508-CFTR trafficking. ERp29 mRNA and protein expression was significantly increased (∼1.5-fold) in 4PBA-treated IB3-1 cells. In Xenopus oocytes, ERp29 overexpression increased the functional expression of both wild-type and ΔF508-CFTR over 3-fold and increased wild-type cystic fibrosis transmembrane conductance regulator (CFTR) plasma membrane expression. In CFBE41o− WT-CFTR cells, expression of and short circuit currents mediated by CFTR decreased upon depletion of ERp29 as did maturation of newly synthesized CFTR. In IB3-1 cells, ΔF508-CFTR co-immunoprecipitated with endogenous ERp29, and overexpression of ERp29 led to increased ΔF508-CFTR expression at the plasma membrane. These data suggest that ERp29 is a 4PBA-regulated ER chaperone that regulates WT-CFTR biogenesis and can promote ΔF508-CFTR trafficking in CF epithelial cells.
Keywords: Cystic Fibrosis, Endoplasmic Reticulum (ER), Epithelial Cell, Ion Channels, Trafficking, 4-Phenylbutyrate, CFTR, ERp29
Introduction
Cystic fibrosis (CF)3 is the most common lethal autosomal recessive disease among Caucasians and results from a paucity of functional cystic fibrosis transmembrane conductance regulator (CFTR). CFTR is a cAMP-activated chloride (Cl−) channel that is localized in the apical plasma membrane of epithelial cells where it has an integral role in regulating the transport of electrolytes and water. The most common mutation of CFTR, ΔF508-CFTR (deletion of a phenylalanine at position 508), is a temperature-sensitive trafficking mutant (1). ΔF508-CFTR is retained in the endoplasmic reticulum (ER) where it has prolonged associations with several cytosolic chaperones belonging to the heat shock protein (Hsp) family (2–5) and with the ER-resident chaperone calnexin (6–9). ΔF508-CFTR is targeted for rapid intracellular degradation (10) at least in part by the ubiquitin/proteasome system (11, 12) and so mostly fails to reach its appropriate subcellular location at the apical membrane (13, 14). Molecular chaperones are potential therapeutic targets to facilitate improvement of ΔF508-CFTR trafficking, but so far, little is known about the exact role of the chaperones in CFTR folding and trafficking. Of particular note, investigations of classical ER chaperones, including BiP/grp78, endoplasmin/grp94, and calreticulin, have failed to demonstrate a central role for these ER chaperones in promoting the proper folding, assembly, and trafficking of CFTR (2, 3, 6, 15).
Sodium 4-phenylbutyrate (4PBA) improves ΔF508-CFTR intracellular trafficking in CF epithelial cells such as the IB3-1 CF human bronchiolar epithelial cell line (genotype ΔF508/W1282X) as early as 4–8 h after exposure and restores CFTR function at the plasma membrane without altering CFTR mRNA expression (16). Because CFTR mRNA expression was not altered by 4PBA and because 4PBA and other butyrates are considered transcriptional regulators, we previously investigated whether 4PBA alters expression of molecular chaperones implicated in ΔF508-CFTR trafficking. We initially focused on 70-kDa heat shock cognate protein (Hsc70), a cytosolic chaperone involved in targeting a number of cellular proteins for ubiquitination and proteasome degradation (17). After 48 h of 4PBA treatment, both the expression of Hsc70 and complex formation between Hsc70 and ΔF508-CFTR decreased in IB3-1 cells. This Hsc70·ΔF508-CFTR complex may target ΔF508-CFTR for rapid intracellular degradation by the ubiquitin/proteasome pathway (4). Our group subsequently observed that 4PBA decreases the steady-state expression of Hsc70 by increasing the rate of Hsc70 mRNA turnover and that this effect requires new mRNA synthesis (18). These data suggested that IB3-1 cells undergo a complex response to 4PBA, a notion that has since been confirmed in genomics and proteomics profiling experiments (19, 20). Moreover, it appears that multiple elements of this 4PBA response (i.e. 4PBA-regulated genes or targets) could influence ΔF508-CFTR trafficking.
To identify candidate 4PBA targets that might improve ΔF508 trafficking, we performed differential display RT-PCR on 4PBA-treated IB3-1 cells. One cDNA species exhibiting a time-dependent increase in PCR product abundance encoded ERp29, which was of particular interest given the dearth of knowledge about ER chaperones involved in CFTR folding. ERp29 is a resident of the ER lumen (21) that, although expressed ubiquitously in mammalian tissues, is generally abundant in epithelia (22, 23). Several lines of evidence implicate ERp29 as a distinct type of molecular chaperone (24, 25), but direct evidence for a chaperone activity and identification of physiological client proteins (i.e. folding substrates) remain lacking. Notably, ERp29 lacks classical chaperone and oxidoreductase activities (26) but exhibits chaperone-like properties at both the biophysical and cellular levels (27–31). It has also been inferred that ERp29 prefers hydrophobic substrates such as membrane proteins (29, 32).
Recognizing parallels between the epithelial expression of ERp29 and CFTR, we tested the hypothesis that increased expression of ERp29 in response to 4PBA would contribute to improved ΔF508-CFTR trafficking. Our data establish that 4PBA leads to increased ERp29 expression and that ERp29 overexpression can improve trafficking of both wild-type (WT-) CFTR and ΔF508-CFTR. We also observed that reductions in ERp29 expression led to decreased WT-CFTR expression and maturation of newly synthesized channel as well as decreasing CFTR-mediated chloride transport in CFBE41o− cells that express WT-CFTR. These data therefore suggest that increased expression of ERp29 may contribute to improved ΔF508-CFTR trafficking in response to 4PBA and that ERp29 may indeed act as a chaperone of a medically important plasma membrane protein.
EXPERIMENTAL PROCEDURES
Cell Culture
IB3-1 CF epithelial cells (genotype ΔF508/W1282X) (68) were cultured as described previously (4, 18). Immortalized CFBE41o− bronchial epithelial cells that stably overexpress WT-CFTR (CFBE41o− WT) after lentiviral transduction and puromycin selection (33) were a generous gift of Dr. J. P. Clancy (University of Alabama at Birmingham). Cells were routinely cultured at 37 °C as described previously (33). Human colon adenocarcinoma T84 cells (cell line CCL248, American Type Culture Collection, Manassas, VA) were cultured in a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 medium (Invitrogen) supplemented with 100 units/ml penicillin, 0.1 mg/ml streptomycin, and 5% fetal bovine serum at 37 °C in 5% CO2. IB3-1 and T84 cells, which endogenously express ΔF508- or WT-CFTR, respectively, were studied alongside our other model systems that utilized heterologous overexpression to minimize potential artifacts confounding these data.
Antibodies
Rabbit antiserum to ERp29 (anti-ERp29) was as described previously (22) or obtained from GeneTex (San Antonio, TX). Rabbit anti-BiP/grp78 was purchased from Sigma-Aldrich. Mouse monoclonal anti-CFTR (clone CF3) was from Abcam (Cambridge, MA), and mouse monoclonal anti-CFTR-C terminus (clone 24-1) was from R&D Systems (Minneapolis, MN). Rabbit CFTR antiserum 169 directed against a human CFTR R-domain peptide was described previously (34). Mouse monoclonal anti-CFTR directed against NBD2 (clone 596) was obtained from Dr. John Riordan (University of North Carolina-Chapel Hill) via the CF Foundation Therapeutics CFTR antibody distribution program. Mouse monoclonal anti-CFTR direct against NBD1 (clone 3g11) was obtained from Dr. William Balch (Scripps Institute, San Diego, CA) via the CFTR Folding Consortium. Mouse monoclonal anti-GAPDH was from Millipore (Billerica, MA).
Differential Display RT-PCR
Differential display RT-PCR was performed using the RNAimageTM kit (GenHunter). Total RNA was isolated from IB3-1 cells treated with 1 mm 4PBA for 0, 4, 8, or 24 h using the RNAwiz reagent (Ambion, Austin, TX) according to the manufacturer's instructions. Reverse transcription used an oligo(dT)-anchored primer, and the subsequent PCR assay (40 cycles: 94 °C, 15 s; 40 °C, 2 min; 72 °C, 30 s; final cycle: 72 °C, 5 min) used the same anchored primer in combination with 16 different arbitrary primers (HAP-1 to HAP-16) in the presence of [α-33P]dATP (Amersham Biosciences). PCR products were resolved on an 8 m urea, 6% polyacrylamide gel and visualized by autoradiography. Differentially expressed cDNAs were extracted, reamplified by PCR, and cloned into pCRII (Invitrogen). The cloned inserts were sequenced by automated analysis (Nucleic Acid Research Core, Children's Hospital of Philadelphia) and identified by Blast search.
Cloning of Human ERp29 cDNA
A full-length ERp29 cDNA was isolated by PCR using ERp29-specific primers 5′-CCGGCGATATGGCTGCCG-3′ and 3′-AGGAGCTGTAAAAAGGCTGTCTGT-5′ and total RNA obtained from IB3-1 cells that underwent randomly primed reverse transcription using Superscript II reverse transcriptase (Invitrogen). The ERp29 cDNA was cloned into pCRII vector and then subcloned into pSK(−) (Stratagene) (pSK-ERp29) by digestion with HindIII/XhoI. The ERp29 cDNA was similarly subcloned into pcDNA4 (Invitrogen) by digestion with BamHI/NotI. All constructs were confirmed by sequencing.
Ribonuclease (RNase) Protection Assay
Quantification of ERp29 mRNA by ribonuclease protection was performed essentially as described previously (4, 18) using the Direct Protect assay kit (Ambion). The ERp29-specific probe template comprised a 258-bp 3′-fragment obtained by digestion of pSK-ERp29 with SacI, and the probe was synthesized using a Maxiscript T3 kit (Ambion). Hybridization to 18 S rRNA using pTRI-18S (Ambion) as a template served as an internal control. The expression of ERp29 mRNA relative to that of 18 S rRNA was determined by fluorography and densitometry as described previously (4, 18) with statistical analysis as described below.
Quantitative PCR for CFTR mRNA Expression
Total mRNA was extracted with TRI Reagent® (Applied Biosystems, Ambion) from CFBE41o− WT cells. cDNA was synthesized from total RNA using oligo(dT) primers and reverse transcription reagents from Applied Biosystems. PCR for CFTR and GAPDH was performed using TaqMan Fast Master Mix and a predesigned primer-probe set for CFTR and GAPDH (Applied Biosystems) on a StepOne real time PCR machine (Applied Biosystem).
Depletion of ERp29 by siRNA
ERp29 expression was depleted using an ERp29 siRNA (Dharmacon/Thermo Fisher Scientific) that had been demonstrated previously to specifically decrease ERp29 expression in primary cultures of alveolar type II cells.4 ERp29 siRNA was delivered to CFBE41o− WT cells by transfection with Lipofectamine RNAimax reagent (Invitrogen) or to T84 cells by electroporation according to a commercially available optimized protocol and reagents (Amaxa, Lonza, Cologne, Germany). Control siRNA (Dharmacon/Thermo Fisher Scientific) was delivered under conditions identical to those of the ERp29 siRNA. Cells were either used for pulse-chase experiments (described below) or lysed for immunoblot (described below) or quantitative PCR analysis (described above) 48 h after electroporation or transfection.
Immunoblot
IB3-1, CFBE41o− WT, or T84 cell lysates were prepared in radioimmune precipitation assay buffer (150 mm NaCl, 50 mm Tris-HCl, pH 8, 1% Triton-X100, 1% sodium deoxycholate, 0.1% SDS) containing a protease inhibitor mixture (1:1000 dilution; Sigma). Lysates were passed through a 22-gauge needle and cleared by centrifugation (15,000 × g, 15 min, 4 °C). Protein content was determined with Bio-Rad DC reagents, and bovine γ-globulin was used as a standard. Equal amounts of protein were resolved by SDS-PAGE and transferred to nitrocellulose as described previously (16). Nonspecific binding was blocked by incubation with 5% nonfat milk in TBS (10 mm Tris-HCl, pH 8, 150 mm NaCl) and 0.05% Tween 20. Primary and horseradish peroxidase (HRP)-conjugated secondary antibodies were applied in TBS, 0.05% Tween 20, 1% BSA. Immunoreactivity was detected by chemiluminescence (ECL, Amersham Biosciences) and fluorography and was quantitated by densitometry (4, 18).
For experiments examining the presence of ERp29 in the cell culture media, IB3-1 cells were grown in T-25 tissue culture flasks. Cells were then incubated in fresh medium (5 ml) without (control) or with 1 mm 4PBA for 24 h or were placed in fresh medium (5 ml) without or with 1 mm 4PBA for 24 h after transfection with ERp29 cDNA (4 μg, 24 h). The conditioned media were then collected, and cell lysates (total volume, 0.5 ml) were prepared as above. Fresh, unconditioned medium served as a negative control. Proteins in the collected media were concentrated 10-fold (final volume, 0.5 ml) using an iCON Concentrator (molecular mass cutoff, 9 kDa; Thermo Scientific) according to the manufacturer's protocol. Equal amounts of cell lysate proteins were resolved by SDS-PAGE, and the corresponding volume of concentrated, conditioned media was similarly resolved by SDS-PAGE. ERp29 was then detected by immunoblot as described.
Immunoprecipitation
Lysates of IB3-1 cells were prepared as described above except that SDS was omitted from the radioimmune precipitation assay buffer. Antiserum directed against ERp29 (10 μl) (22) was incubated with cell lysates (200 μg of protein) at 4 °C overnight. Immune complexes were captured with protein A/G-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) and released by heating the samples at 65 °C for 20 min in 2× SDS-PAGE sample buffer. CFTR was detected by immunoblotting as above.
Pulse-Chase Analysis of CFTR Maturation
ERp29 expression was specifically depleted in CFBE41o− WT cells as described above. Forty-eight hours after siRNA delivery, cells were starved in Met/Cys-deficient growth media for 1 h, pulse-labeled with [35S]Met/Cys (400 μCi/ml; PerkinElmer Life Sciences), for 30 min and then chased with complete media containing an excess of unlabeled Met/Cys for 0–4 h prior to lysis in radioimmune precipitation assay buffer. CFTR was recovered from 500 μg of whole cell lysate protein by immunoprecipitation with protein A/G-Sepharose beads to which monoclonal anti-CFTR (clone 3G11) was covalently coupled (Crosslink IP kit, Thermo Scientific); covalent coupling was performed according to the manufacturer's instructions. Precipitated CFTR was resolved by SDS-PAGE and detected by fluorography. Depletion of ERp29 in these pulse-chase experiments was confirmed by immunoprecipitation with anti-ERp29, resolution of precipitated protein by SDS-PAGE, and fluorography (data not shown).
Transepithelial Ion Transport Measurements in Ussing Chambers
CFBE41o− WT cells were grown as polarized epithelial monolayers on SnapWells (Costar, Corning Life Sciences, Lowell, MA) and used when transepithelial resistance was >500 ohm·cm2 as assessed by an EVOM ohmmeter (World Precision Instruments, Sarasota, FL). After achieving this resistance, cells were transfected with control siRNA or ERp29 siRNA with Lipofectamine Plus (Invitrogen) 48 h prior to assay.
For short circuit current measurements, cells were mounted in a modified, vertical Ussing chamber, and the monolayers were continuously voltage-clamped to 0 mV after fluid resistance compensation using automatic voltage clamps (VCC 600, Physiologic Instruments, San Diego, CA). Filters were mounted in Bath Solution (115 mm NaCl, 25 mm NaHCO3, 2.4 mm KH2PO4, 1.2 mm K2HPO4, 1.2 mm MgCl2, 1.2 mm CaCl2, 10 mm glucose) warmed to 37 °C. The solution was pregassed and then continuously gas-lifted with a 95% O2, 5% CO2 mixture yielding a pH of 7.3–7.4. The short circuit current (Isc) was digitized at 0.05 Hz, and data were stored on a computer hard drive using Acquire and Analyze software build 2.2 (Physiologic Instruments). The data acquisition software measured transepithelial resistance automatically by passing a 5-mV, 0.2-s bipolar pulse across the monolayer and calculating resistance (R) by Ohm's law (V = IR); these resistance values remained stable throughout these experiments. A positive deflection in Isc is defined as the net movement of a cation toward the apical bath.
Amiloride (Sigma-Aldrich) was dissolved as 1000× stocks in Bath Solution. Forskolin and 3-isobutyl-1-methylxanthine (IBMX) (Sigma-Aldrich) were dissolved as 1000× stocks in DMSO. CFTRinh-172 (35) was obtained from the CFTR modulator library (Dr. Robert Bridges, Rosalind Franklin Chicago Medical School) and dissolved as a 1000× stock in DMSO. A basolateral to apical chloride gradient was imposed by replacing the apical bathing solution with a low chloride buffer containing 115 mm sodium gluconate, 25 mm NaHCO3, 2.4 mm KH2PO4, 1.2 mm K2HPO4, 1.2 mm MgCl2, 1.2 mm CaCl2, and 10 mm glucose. This buffer was also pH 7.3–7.4 when gassed with a 95% O2, 5% CO2 mixture. CFTR functional expression was determined in the presence of 10 μm amiloride and was defined as Isc that was inhibited by apical application of 10 μm CFTRinh-172 after treatment of the cells with 10 μm forskolin and 100 μm IBMX and imposition of the basolateral to apical chloride gradient.
Expression of Human CFTR and Human ERp29 in Xenopus Oocytes
Human CFTR (WT or ΔF508) and human ERp29 were expressed in Xenopus oocytes as described previously (35, 36). Briefly, WT-CFTR, ΔF508-CFTR, and ERp29 cRNAs were prepared using a cRNA synthesis kit (mMESSAGE mMACHINE, Ambion) according to the manufacturer's protocol. cRNA concentrations were determined spectroscopically. Oocytes were obtained from adult female Xenopus laevis (Nasco, Fort Atkinson, WI) under a protocol approved by the Institutional Animal Care and Use Committee of the Children's Hospital of Philadelphia. Harvested oocytes were enzymatically defolliculated and maintained at 18 °C in modified Barth's saline (88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 0.3 mm Ca(NO3)2, 0.41 mm CaCl2, 0.82 mm MgSO4, 15 mm Hepes, pH 7.6 supplemented with 10 μg/ml sodium penicillin, 10 μg/ml streptomycin sulfate, and 100 μg/ml gentamicin sulfate). Each batch of oocytes obtained from an individual frog was injected (50 nl/oocyte) using a Nanoject II microinjector (Drummond Scientific, Broomall, PA) with CFTR cRNA (WT or ΔF508; 10 ng) or a combination of ERp29 (1, 10, and 30 ng) and CFTR (10 ng) cRNAs dissolved in RNase-free water.
Electrophysiological Analyses in Xenopus Oocytes
Whole-cell current measurements were performed 24–48 h after injection using the two-electrode voltage clamp (TEV) method as described previously (35, 36). Oocytes were placed in a 1-ml chamber containing modified ND96 buffer (96 mm NaCl, 1 mm KCl, 0.2 mm CaCl2, 5.8 mm MgCl2, 10 mm Hepes, pH 7.4) and impaled with micropipettes of 0.5–5-megaohm resistance filled with 3 m KCl. The whole-cell currents were measured by voltage clamping the oocytes in 20-mV steps between −140 mV and +60 mV adjusted for resting transmembrane potential. Whole-cell currents were digitized at 200 Hz during the voltage steps, recorded directly onto a hard disk, and analyzed using pClamp software (version 8 or 8.1; Axon Instruments, Foster City, CA). To reduce the potential for error from series resistance, the voltage clamp (Axon Geneclamp 500B) was configured to clamp the bath potential to 0 mV. In this configuration, we independently monitored the oocyte membrane potential during our clamp protocol and routinely observed membrane potentials that were <5% depolarized from our target holding potentials.
CFTR was activated by perfusion of the oocyte with buffer containing 10 μm forskolin and 100 μm IBMX for 25 min (35, 36). In all experiments, CFTR-mediated Cl− current (or functional expression) was defined as the difference between current measured before and 20 min after perfusion with forskolin/IBMX. Whole-cell currents were recorded at −100 mV for comparisons, and all measurements were performed at room temperature.
Surface Biotinylation of CFTR in Oocytes
Surface expression of CFTR in oocytes was determined by surface biotinylation using modifications of techniques we have used previously (37). cRNA for WT-CFTR (10 ng) was injected into Xenopus oocytes either alone or with ERp29 cRNA. After 48 h, oocytes were mechanically stripped of their vitelline membranes in hypertonic medium (300 mm sucrose in modified Barth's saline without penicillin, streptomycin, and gentamicin), and surface proteins were labeled with sulfo-NHS-biotin (Pierce). Oocytes (10 per group) were subsequently lysed in 0.15 m NaCl, 0.01 m Tris-HCl, pH 8.0, 0.01 m EDTA, 1.0% Nonidet P-40, 0.5% sodium deoxycholate, 1.0 mm phenylmethylsulfonyl fluoride, 0.1 mm N-p-tosyl-l-lysine chloromethyl ketone, 0.1 mm l-1-tosylamide-2-phenylethyl chloromethyl ketone, and 2 μg/ml aprotinin for 1 h at 4 °C and centrifuged at 13,000 × g for 15 min at 4 °C. CFTR was precipitated with CFTR antiserum 169 (34) overnight at 4 °C, recovered with protein A/G-Sepharose (Pierce), and subjected to SDS-PAGE. Biotinylated CFTR was detected on blots by probing with a streptavidin-HRP conjugate (Invitrogen).
Surface Biotinylation of ΔF508-CFTR and ERp29 in IB3-1 Cells
IB3-1 cells were incubated with or without 1 mm 4PBA for 24 h or transfected for 24 h with 4 μg of pCDNA4-ERp29 using the Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. Cells were rapidly cooled to 4 °C, washed in 1× PBS containing 0.1 mm CaCl2 and 1 mm MgCl2, and then incubated twice with 0.5 mg/ml sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate (sulfo-NHS-SS-biotin; Pierce) in 10 mm triethanolamine, 0.1 m borate buffer, pH 8 for 25 min at 4 °C. Nonreacted sulfo-NHS-SS-biotin was quenched by washing cells with 1× PBS, 192 mm glycine, 25 mm Tris-HCl, pH 8.3. Cells were then washed twice with 1× PBS containing 0.1 mm CaCl2 and 1 mm MgCl2 and lysed in radioimmune precipitation assay buffer. Biotinylated proteins were captured using neutravidin-agarose beads (Pierce), eluted into Laemmli 2× sample buffer, and resolved by SDS-PAGE. Biotinylated CFTR was detected by immunoblot analysis using a monoclonal antibody directed against residues 103–117 of CFTR (clone CF3, Abcam). Biotinylated ERp29 was detected using anti-ERp29 (GenTex).
Densitometry and Statistical Analyses
Densitometry of fluorograms was performed using an Alpha-Innotech 2200 Image Analysis System (Alpha-Innotech, San Leandro, CA) with two-dimensional integration of the selected band. The density of the lane surrounding the band was similarly determined by two-dimensional integration and used as a base-line density for background subtraction. For comparisons within an experiment, the density of the zero time or control was arbitrarily set to 1.0, and data are expressed relative to this control (mean ± S.E.). Statistical significance was then determined by Student's t test or one-way ANOVA as appropriate. For experiments examining the influence of ERp29 overexpression on WT- or ΔF508-CFTR functional expression in oocytes, data at −100-mV holding potential are presented as mean ± S.E. with significance determined by one-way ANOVA in comparison with oocytes injected with WT- or ΔF508-CFTR alone.
For all other data, statistical significance was determined by a two-tailed analysis using either Student's t test or a one-way ANOVA as appropriate. All statistical analyses were performed with SigmaStat version 2.03. A p value of ≤0.05 was considered significant.
RESULTS
4PBA Increases ERp29 Expression in IB3-1 CF Epithelial Cells
In IB3-1 cells, 4PBA decreases steady-state Hsc70 expression by increasing the rate of Hsc70 mRNA turnover (4, 18). Interestingly, the increase in Hsc70 mRNA turnover requires new mRNA synthesis (18), suggesting that 4-PBA causes a more global cellular adaptation. We performed differential display RT-PCR on RNA isolated from IB3-1 cells treated with 1 mm 4PBA to characterize alterations in gene expression associated with cellular adaptation to 4PBA. Among the mRNA species that were differentially expressed over the 24-h exposure, we identified ERp29 as exhibiting a time-dependent increase in abundance (data not shown). This finding was of particular interest because no ER chaperones have been clearly identified as 4PBA targets before. We confirmed and quantitated this 4PBA-induced increase in ERp29 mRNA abundance using a ribonuclease protection assay (Fig. 1). ERp29 mRNA expression increased significantly by ∼50% after 2 h and remained similarly elevated throughout the 24-h exposure to 4PBA.
FIGURE 1.

4PBA increases ERp29 mRNA expression in IB3-1 cells. A, ERp29 transcript was quantitated using a ribonuclease protection assay in IB3-1 cells treated for the indicated times with 1 mm 4PBA as described under “Experimental Procedures.” The hybridization of 18 S rRNA as an internal standard was constant under these conditions. These data are representative of n = 8 independent experiments. B, relative amount of ERp29 mRNA compared with the zero time point was determined by densitometry (n = 8 independent sample sets, each normalized by 18 S rRNA content). Means ± S.E. are shown with significance determined by one-way ANOVA in comparison with t = 0.
A corresponding increase in whole-cell expression of ERp29 protein was observed by immunoblot analysis of lysates from 4PBA-treated IB3-1 cells (Fig. 2A). Densitometric analysis suggested that ERp29 protein expression increased by a maximum of ∼50% after 4–8 h of 4PBA treatment (Fig. 2B). In contrast, using similar techniques, we found that Hsc70 expression decreased ∼40% in IB3-1 cells after treatment with 1 mm 4PBA for 24–48 h (4, 18).
FIGURE 2.

4PBA increases ERp29 protein expression in IB3-1 cells. IB3-1 cells were incubated at 37 °C with 1 mm 4PBA for the indicated times. Equal amounts of whole-cell lysate protein were resolved on 10% SDS-polyacrylamide gels. A, representative immunoblot performed with an ERp29-specific rabbit polyclonal antibody (22). B, densitometric analysis (mean ± S.E.) of 10 independent experiments normalized to zero time. Statistical significance was determined by ANOVA in comparison with the t = 0 sample. C, expression of BiP/grp78 relative to that of GAPDH was determined by densitometry of immunoblots equivalent to those in B (n = 4 independent experiments, all normalized to zero time). Immunoblots were probed with anti-BiP and anti-GAPDH, and each data set was normalized by GAPDH content (mean ± S.E.). By ANOVA, there were no significant differences for GAPDH or BiP/grp78 as a function of 4PBA exposure in these experiments.
Because ERp29 expression is reported to be induced by ER stress in some circumstances (25, 28, 38), we assessed whether 1 mm 4PBA was causing a generalized ER stress or an unfolded protein response. Increased expression of BiP/grp78, another ER-luminal chaperone, is a characteristic of ER stress or an unfolded protein response (39). As shown in Fig. 2C, BiP expression, normalized to the cytosolic marker GAPDH, did not change upon exposure to 1 mm 4PBA. Furthermore, IB3-1 cellular morphology was not affected by 1 mm 4PBA over the course of our experiments. These data as well as previous observations that 1 mm 4PBA does not alter expression of calnexin (16) and several other ER chaperones in IB3-1 cells (19) suggest that at such moderate exposures 4PBA causes neither a general alteration of ER chaperone expression nor a cellular stress response. However, BiP/grp78 and several other stress response proteins were reported to be up-regulated after harsher exposures to 4PBA (5 mm, 48 h; Ref. 20). We also observed changes in IB3-1 cell morphology (including modest rounding of cells) upon such harsher 4PBA exposures (data not shown). These observations further suggest that there are significant differences between the IB3-1 cellular responses to 1 versus 5 mm 4PBA with 5 mm 4PBA inducing a cellular stress response that was not evident upon exposure to 1 mm 4PBA.
Depletion of ERp29 Is Accompanied by Decreased Expression of WT-CFTR
Induction of ERp29 in 4PBA-treated bronchiolar epithelial cells led us to hypothesize that ERp29 is a chaperone for CFTR. Notably, ERp29, although ubiquitously expressed, is enriched in lung and has been implicated in membrane protein biogenesis (22, 23, 29, 32, 40). We initially tested this hypothesis using an RNA interference approach in CFBE41o− WT bronchial epithelial cells, which overexpress WT-CFTR, and in T84 colonic epithelial cells, which endogenously express WT-CFTR. As shown in Fig. 3 (A and B), delivery of ERp29-directed siRNA to CFBE41o− WT cells caused an ∼50% decrease in ERp29 and ∼40% decrease in CFTR whole-cell expression relative to cells transfected with control siRNA. Similarly, there was a 60% decrease in ERp29 and CFTR expression in T84 cells (Fig. 3, C and D). In neither cell line was expression of BiP/grp78 altered by delivery of ERp29 siRNA (Fig. 3), suggesting that the siRNA-mediated depletion of ERp29 caused neither a global alteration in ER chaperone expression nor an ER stress response. This diminution of CFTR expression upon ERp29 depletion was not due to decreased CFTR mRNA in the ERp29 siRNA-treated CFBE41o− WT cells; quantitative PCR showed that CFTR mRNA was apparent at 18–19 cycles in both control and ERp29 siRNA-transfected CFBE41o− WT cells lines (n = 3 experiments). These data provided initial support for the hypothesis that ERp29 is a chaperone for CFTR.
FIGURE 3.

Depletion of ERp29 is associated with decrease in WT-CFTR expression. ERp29-specific or control siRNA was delivered to CFBE41o− WT bronchial epithelial cells that overexpress WT-CFTR (A and B) or T84 colonic adenocarcinoma cells that endogenously express WT-CFTR (C and D) as described under “Experimental Procedures.” After 48 h, cells were lysed, and equal amounts of whole-cell lysate protein were resolved by SDS-PAGE. A, ERp29, CFTR, BiP/grp78, and GAPDH (as a loading control) were detected by immunoblot in lysates of CFBE41o− WT cells. Data representative of three independent experiments are shown. B, relative expression of ERp29 (gray bars) and CFTR (open bars) in CFBE41o− WT was quantified by densitometry of n = 3 independent experiments as described under “Experimental Procedures.” C, ERp29, CFTR, BiP/grp78, and GAPDH (as a loading control) were detected by immunoblot in T84 cells. Data representative of three independent experiments are shown. D, relative expression of ERp29 (gray bars) and CFTR (open bars) in CFBE41o− WT was quantified by densitometry of n = 3 independent experiments as described under “Experimental Procedures.” For B and D, data (mean ± S.E.) were normalized to control siRNA values, and expression of ERp29 and CFTR in control versus ERp29-specific siRNA-treated cells was compared by t test.
ERp29 Enhances Functional Expression of WT-CFTR and ΔF508-CFTR in Xenopus Oocytes
We initially used the Xenopus oocyte expression system and TEV technique to examine the influence of ERp29 overexpression on functional expression of WT- and ΔF508-CFTR. Whole-oocyte currents were measured 24–48 h after injection of cRNA encoding CFTR (WT or ΔF508) alone or co-injection of cRNAs encoding CFTR and ERp29.
Fig. 4A illustrates the whole-oocyte current/voltage (I/V) relationship for oocytes injected with both WT-CFTR and ERp29 prior to (closed circles) and after (open circles) 20 min of incubation with 10 μm forskolin/100 μm IBMX (which activate endogenous protein kinase A and subsequently CFTR). These data suggest that ERp29 overexpression does not alter the characteristic linear whole-cell I/V relationship that is typically observed for forskolin/IBMX-activated CFTR in oocytes (36). Fig. 4B shows the forskolin/IBMX-stimulated whole-cell currents measured at a holding potential of −100 mV in oocytes injected with CFTR alone (10 ng) compared with currents obtained in oocytes co-injected with CFTR (10 ng) and increasing amounts of ERp29 cRNA. In the corresponding immunoblot (Fig. 4B), no ERp29 expression was observed in oocytes injected with CFTR alone, whereas overexpressed ERp29 was readily detected in those injected with ERp29 cRNA.
FIGURE 4.

ERp29 enhances functional and surface expression of WT-CFTR in Xenopus oocytes. X. laevis oocytes were injected with cRNAs for WT-CFTR (10 ng) or ERp29 (10 ng) alone or co-injected with WT-CFTR (10 ng) and either 1, 10, or 30 ng of ERp29 cRNA. A, I/V relationship (adjusted for resting transmembrane potential) of whole-cell current was determined by TEV 24–48 h after co-injection with 10 ng of CFTR and 1 ng of ERp29 cRNAs before (closed circles) and after (open circles) stimulation with forskolin/IBMX as detailed under “Experimental Procedures.” Data are presented as mean ± S.E. for n = 29 oocytes. B, forskolin/IBMX-stimulated currents at −100-mV holding potential (adjusted for resting transmembrane potential) were determined for oocytes injected with cRNA for WT-CFTR alone (10 ng; closed bar) or ERp29 alone (10 ng; open bar that is not visible due to small magnitude) or co-injected with CFTR and either 1, 10, or 30 ng of ERp29 cRNA (gray bars). Data are presented as mean ± S.E. for the indicated number of oocytes, and statistical significance was determined by ANOVA in comparison with oocytes injected with WT-CFTR alone. Immunoblot detection of ERp29 in similarly injected oocytes was performed as described under “Experimental Procedures.” Groups of 10 oocytes were lysed, and 10% of this lysate (i.e. one oocyte equivalent) was loaded per lane. C, detection of CFTR at the oocyte surface by surface biotinylation was performed 48 h after injection as described under “Experimental Procedures.” Biotinylated CFTR was revealed with a streptavidin-HRP conjugate after precipitation with CFTR antiserum 169. Equivalent results were obtained in three independent experiments.
With regard to CFTR functional expression, oocytes co-injected with CFTR and 1 ng of ERp29 had forskolin/IBMX-stimulated whole-cell currents that were ∼3.2-fold greater than oocytes injected with CFTR cRNA alone (−5.4 ± 1.1 μA, n = 29 versus −1.7 ± 0.5 μA, n = 35; mean ± S.E.; p < 0.05 by ANOVA). Interestingly, greater amounts of ERp29 cRNA were less effective at enhancing CFTR functional expression. Specifically, in oocytes co-injected with WT-CFTR and 10 ng of ERp29 cRNA, the forskolin/IBMX-stimulated currents (−3.3 ± 0.5 μA, n = 27; p < 0.05) were only ∼1.9-fold greater than the control, whereas in oocytes injected with 30 ng of ERp29 cRNA (−2.7 ± 0.3 μA, n = 13; p not significant), the forskolin/IBMX-stimulated currents were not significantly different from oocytes injected with CFTR alone. Oocytes injected with ERp29 cRNA alone had forskolin/IBMX-stimulated currents that were not significantly different from uninjected oocytes, confirming that the observed forskolin/IBMX-stimulated currents were indeed CFTR-mediated.
To assess the mechanism by which ERp29 overexpression increased CFTR functional expression, we determined the amount of CFTR in the plasma membrane by surface biotinylation (Fig. 4C). Oocytes were co-injected with cRNAs encoding CFTR and increasing amounts of ERp29 (0, 1, 10, and 30 ng), and CFTR surface expression was determined 48 h after injection. Surface expression of WT-CFTR was readily detected in oocytes co-injected with CFTR and 1 ng of ERp29 cRNAs, less robust in oocytes co-injected with CFTR and 10 ng of ERp29, and not detected in oocytes injected with CFTR alone or in oocytes co-injected with CFTR and 30 ng of ERp29. We suspect that our inability to detect CFTR in the latter two cases (oocytes injected with CFTR alone or co-injected with CFTR and 30 ng of ERp29 cRNA) was due to surface expression levels being below the limit of detection; this has been a consistent finding in other experiments in our laboratory (not shown). Nevertheless, the observed surface expression pattern of CFTR parallels that of CFTR activity as measured by TEV (Fig. 4B) with co-injection of 1 ng of ERp29 cRNA causing the greatest CFTR surface and functional expression. These data are therefore consistent with ERp29 acting as a chaperone that promotes WT-CFTR expression at the plasma membrane.
Next we examined whether ERp29 overexpression also influenced the functional expression of ΔF508-CFTR. Although ΔF508-CFTR is a “trafficking-defective” mutant, in oocytes, ΔF508-CFTR is delivered to the plasma membrane (41) because oocytes are maintained at 18 °C, and temperatures ≤27 °C are permissive for ΔF508-CFTR trafficking (1, 42). As shown in Fig. 5A, ERp29 overexpression did not alter the characteristic linear I/V relationship of ΔF508-CFTR (36). Similarly, Fig. 5B shows whole-cell currents measured at a holding potential of −100 mV in oocytes co-injected with ΔF508-CFTR and increasing amounts of ERp29 cRNA. Oocytes co-injected with ΔF508-CFTR and 1 ng of ERp29 cRNA had ∼2.3-fold greater forskolin/IBMX-stimulated currents (−0.9 ± 0.3 μA, n = 22) than did controls injected with ΔF508-CFTR cRNA alone (−0.4 ± 0.0 μA, n = 27; p < 0.05 by ANOVA; Fig. 5B). Currents obtained with co-injection of 10 ng of ERp29 (−0.9 ± 0.2 μA, n = 19) were similarly ∼2.25-fold greater than controls (p < 0.05). In contrast, currents in oocytes co-injected with 30 ng of ERp29 were not significantly higher than controls (−0.6 ± 0.2 μA, n = 14 versus −0.4 ± 0.0 μA, n = 27; p not significant). Again, oocytes injected with ERp29 cRNA alone had forskolin/IBMX-stimulated currents that were not significantly different from uninjected oocytes, indicating that the forskolin/IBMX-stimulated currents in these experiments were mediated by ΔF508-CFTR. Together, these data suggest that ERp29 increases ΔF508-CFTR functional expression in oocytes in a manner similar to its effect on WT-CFTR. Consistent with the relatively low current values observed for ΔF508-CFTR (versus WT-CFTR; Fig. 4), we were unable to detect ΔF508-CFTR at the plasma membrane by surface biotinylation (not shown). These data further support the hypothesis that ERp29 is a chaperone for CFTR.
FIGURE 5.

ERp29 enhances functional expression of ΔF508-CFTR in Xenopus oocytes. X. laevis oocytes were injected with cRNA for ΔF508-CFTR (10 ng) or ERp29 alone (10 ng) or co-injected with cRNAs for ΔF508-CFTR (10 ng) and either 1, 10, or 30 ng of ERp29. A, I/V relationship (adjusted for resting transmembrane potential) of whole-cell current was determined by TEV for oocytes co-injected with 10 ng of ΔF508-CFTR cRNA and 1 ng of ERp29 cRNA before (closed circles) and after (open circles) stimulation with forskolin/IBMX. Data are presented as mean ± S.E. for n = 22 oocytes. B, forskolin/IBMX-stimulated current at −100-mV holding potential (adjusted for resting transmembrane potential) of oocytes injected with ΔF508-CFTR alone (10 ng; closed bar) or ERp29 alone (10 ng; open bar) or co-injected with ΔF508-CFTR and either 1, 10, or 30 ng of ERp29 cRNA (gray bars). Data are presented as mean ± S.E. for the indicated numbers of oocytes, and the significance of differences from oocytes injected with ΔF508-CFTR alone was determined by ANOVA.
Overexpression of ERp29 Increases ΔF508-CFTR Plasma Membrane Expression in IB3-1 Cells
We next addressed whether altered ERp29 expression modifies ΔF508-CFTR trafficking to the plasma membrane in CF epithelial cells. Surface biotinylation experiments were performed in IB3-1 cells that overexpressed ERp29 or had been treated with 4PBA as a positive control (Fig. 6). ΔF508-CFTR was not detected at the surface of IB3-1 cells grown under control conditions as expected but was clearly present at the surface of these cells after treatment with 1 mm 4PBA. When ERp29 was overexpressed by transient transfection, surface expression of ΔF508-CFTR was even more robust than in cells treated with 4PBA for 24 h, commensurate with greater ERp29 overexpression revealed by immunoblotting (Fig. 6A). Consistent with the data of Fig. 2, densitometric analysis indicated that cells treated with 4PBA had ∼40% increased ERp29 whole-cell expression of ERp29 (Fig. 6B, gray bars; #, p = 0.01 versus control; n = 3), whereas ERp29-transfected cells had ∼4-fold increased expression. Densitometry also indicated that ERp29-transfected cells had 4-fold greater ΔF508-CFTR surface expression than 4PBA-treated cells, which in turn had ∼5-fold greater ΔF508-CFTR surface expression than control cells (Fig. 6B, open bars; *, p < 0.05 versus 1 mm 4PBA; n = 4). As a control for integrity of the plasma membrane during these biotinylation experiments, we assessed the recovery of biotinylated GAPDH in concurrent experiments. In contrast to CFTR, biotinylated GAPDH was undetectable in the neutravidin-precipitated proteins despite strong detection of GAPDH immunoreactivity in whole-cell lysate, confirming that our protocol specifically labeled proteins at the cell surface (Fig. 6A, middle panel).
FIGURE 6.

Overexpression of ERp29 increases surface expression of ΔF508-CFTR in IB3-1 cells. IB3-1 cells were incubated without (Control) or with 1 mm 4PBA for 24 h or were transfected with ERp29 cDNA (4 μg, 24 h). Surface proteins were then biotinylated, captured with neutravidin beads, and resolved by SDS-PAGE as described under “Experimental Procedures.” The first lane (Input) contains 20 μg of IB3-1 whole-cell lysate proteins, whereas lanes with captured biotinylated samples (Biotinylation) contained 200 μg of total protein. A, top panel, CFTR was detected in the captured biotinylated fraction using monoclonal anti-CFTR (clone CF3, Abcam). Middle panel, anti-GAPDH immunoblot demonstrating the presence of GAPDH in IB3-1 whole-cell lysate but not in the captured biotinylated fractions. Lower panel, immunoblot detection of ERp29 in the biotinylated samples (20 μg of total protein) prior to neutravidin capture. These data are representative of three to four independent experiments. B, densitometric analysis of ERp29 whole-cell expression (gray bars; expressed relative to control; n = 3) and ΔF508-CFTR surface expression (open bars; expressed relative to 1 mm 4PBA; n = 4). Means ± S.E. are shown. #, p = 0.01 versus control (t test); *, p < 0.05 versus 1 mm 4PBA (ANOVA).
These data suggest that, in human CF bronchiolar epithelial cells, overexpression of ERp29 promotes ΔF508-CFTR trafficking to and expression at the cell surface. Furthermore, these data imply that a mechanism of action for 4PBA on CFTR trafficking may be via the induction of ERp29 expression. Finally, these data imply that ERp29 increases ΔF508-CFTR functional expression in oocytes (Fig. 4) by increasing channel number at the oocyte plasma membrane, similar to its mechanism of action on WT-CFTR.
ERp29 Is Detectable at Surface of IB3-1 Cells and in IB3-1 Cell Growth Media
Previous data have suggested that ERp29 may move through the secretory pathway and be found extracellularly in some situations (milk and cultured thyrocytes; Refs. 27 and 43) but not others (tooth enamel proteins and plasma; Refs. 22 and 44). Therefore, we examined whether ERp29 could be detected either at the surface or in the culture media of IB3-1 cells (Fig. 7).
FIGURE 7.

Detection of ERp29 in growth medium and at surface of IB3-1 cells. IB3-1 cells were grown under control conditions (−), in the presence of 1 mm 4PBA (+), transfected with 2 μg of ERp29, or transfected with ERp29 in the presence of 1 mm 4PBA for 24 h as indicated. A, whole-cell lysates were prepared, and ERp29 and GAPDH (as a loading control) were detected by immunoblot. Data are representative of n = 4 independent experiments. B, growth medium was collected and concentrated 10-fold as described under “Experimental Procedures.” ERp29 was detected in the conditioned medium by immunoblot but not in a similarly concentrated fresh culture medium control. Smaller or larger immunoreactive species were not detected. Immunoblots representative of n = 4 independent experiments are shown. C, surface proteins were biotinylated, captured with neutravidin beads, and resolved by SDS-PAGE as described under “Experimental Procedures.” ERp29 was detected in the biotinylated fraction. The “Input” lane represents the immunoreactivity of 10% of the total protein subjected to neutravidin capture in the Control lane. GAPDH immunoreactivity was absent from the biotinylated fraction (not shown), suggesting that intracellular proteins were not labeled by biotin in this experiment. These data are representative of n = 4 independent experiments. IP, immunoprecipitation; WB, Western blot.
As expected, whole cell expression of ERp29 was moderately increased after treatment with 1 mm 4PBA and more so upon transfection with ERp29 either in the absence or presence of 4PBA (Fig. 7A). By immunoblotting, ERp29 was readily detected in the culture medium of IB3-1 cells under control conditions and upon ERp29 transfection (Fig. 7B). In contrast, ERp29 was not found in the culture medium of 4PBA-treated IB3-1 cells, and 4PBA decreased ERp29 secretion from ERp29-transfected cells. In control experiments, ERp29 was not detected in fresh culture medium (Fig. 7B). Similarly, GAPDH was undetectable in the conditioned culture medium (data not shown), indicating that our detection of ERp29 there was not an artifact of cell lysis.
In surface biotinylation experiments (Fig. 7C), ERp29 was undetectable at the surface of IB3-1 cells under control conditions. However, treatment with 1 mm 4PBA, overexpression of ERp29 via transfection, or the combination of ERp29 transfection and 4PBA treatment all led to ERp29 being detected at the IB3-1 cell surface. GAPDH was not detected in the neutravidin-captured fraction (data not shown), again indicating that ERp29 surface expression was not an experimental artifact. These data suggest that, in IB3-1 cells, ERp29 can move beyond the ER and be externalized. Interestingly, 4PBA treatment increased the surface expression of ERp29 both in transfected and untransfected cells (Fig. 7C) while decreasing the amount of ERp29 secreted into the culture media (Fig. 7B). These data suggest that 4PBA promotes the association of ERp29 with the IB3-1 cell surface in addition to elevating ERp29 expression.
ERp29 Co-immunoprecipitates with ΔF508-CFTR in IB3-1 Cells
As the above data supported ERp29 being a chaperone for CFTR, we investigated whether ERp29 interacts with WT-CFTR and ΔF508-CFTR in epithelial cells using a co-immunoprecipitation approach. ERp29 appears generally unable to maintain stable interactions with clients following cell lysis (22, 27, 40, 45). However, as shown in Fig. 8, immunoprecipitation of endogenous ERp29 resulted in modest recovery of ΔF508-CFTR from IB3-1 whole-cell lysate. Because these experiments were performed in the absence of 4PBA, this likely reflects intracellular ERp29 and ΔF508-CFTR because IB3-1 cells exhibit no surface expression of ΔF508-CFTR under these conditions (Fig. 7A). Control immunoprecipitations lacking anti-ERp29 were negative for ΔF508-CFTR. In contrast, immunoprecipitation of endogenous ERp29 from lysates of T84 cells did not result in recovery of endogenously expressed WT-CFTR under identical experimental conditions. These results are consistent with reports suggesting that, in comparison with WT-CFTR, immature ΔF508-CFTR has more robust interactions with a variety of chaperones at or within the ER (2, 4, 6).
FIGURE 8.
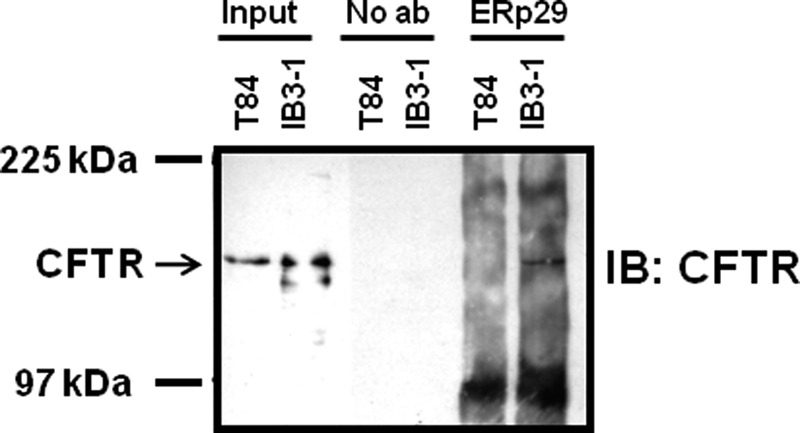
ERp29 co-immunoprecipitates with ΔF508-CFTR in IB3-1 cells. Whole-cell lysates of IB3-1 and T84 cells were prepared as described under “Experimental Procedures.” ERp29 and its stably interacting partners were then immunoprecipitated from equal sample amounts (200 μg of protein) using anti-ERp29 and resolved by SDS-PAGE. Input, 20 μg of whole-cell lysate; No ab, immunoprecipitation protocol performed with omission of anti-ERp29 as a control for nonspecific precipitation. CFTR (arrows) was detected with monoclonal anti-CFTR (clone CF3). These data are representative of three independent experiments. IB, immunoblot.
The observed co-immunoprecipitation of ΔF508-CFTR and ERp29 is consistent with ERp29 influencing ΔF508-CFTR trafficking via a direct or close interaction. Although such a stable interaction was not evident for WT-CFTR, we feel a transient interaction could still occur given the above data demonstrating that depletion of ERp29 decreases WT-CFTR expression and that ERp29 overexpression can promote increased WT-CFTR functional and surface expression in oocytes. These data also provide provisional identification of CFTR as a putative disease-relevant membrane protein substrate for ERp29. Further work is required to establish whether the observed interaction between ERp29 and ΔF508-CFTR is direct or occurs via intermediaries.
ERp29 Depletion Impedes CFTR Maturation and Decreases CFTR Functional Expression
Because of its predominant ER localization and its apparent traversal of the secretory pathway, we hypothesized that ERp29 would influence the trafficking and maturation of newly synthesized CFTR. Accordingly, we performed a pulse-chase experiment in CFBE41o− WT cells that had been treated with either ERp29-specific or non-targeting (control) siRNA (as in Fig. 3 above). As shown in Fig. 9, under control conditions, newly synthesized CFTR progressed over 1–2 h of chase from the lower molecular weight, ER-localized form (Fig. 9, arrow B) to the higher molecular weight, mature form that results from glycosyl processing in the Golgi (Fig. 9, arrow C). The mature form predominated after 4 h of chase. In contrast, after siRNA-mediated depletion of ERp29, less immature CFTR (Band B) was present after pulse-labeling even though, as described above, CFTR mRNA expression was unchanged as assessed by quantitative PCR. Also, the appearance of mature CFTR when ERp29 is depleted was at least delayed if not frankly prevented. The rate at which Band B disappeared (∼20% over the 1st h of chase) when ERp29 was depleted was similar to that under control conditions (∼30% over the 1st h). Together, these data suggest that ERp29 may facilitate or promote both assembly of nascent CFTR in the ER and its trafficking to and eventual maturation in the Golgi. Because ERp29 appears to traverse the secretory pathway, this may occur by ERp29 accompanying or “escorting” newly synthesized CFTR throughout its trafficking itinerary.
FIGURE 9.

ERp29 depletion impedes WT-CFTR maturation. CFBE41o− WT bronchial epithelial cells that overexpressed WT-CFTR were transfected with control (non-target) or ERp29-specific siRNA as described under “Experimental Procedures.” After 48 h, cells were pulse-labeled with [35S]Met/Cys and chased in the presence of excess unlabeled Met/Cys. Cells were lysed at the indicated times of chase, and CFTR was recovered from equal amounts of whole-cell lysate protein by immunoprecipitation using monoclonal anti-CFTR (clone 3G11). Immunoprecipitated CFTR was detected by SDS-PAGE and fluorography. A, representative fluorograms (equal time exposure; n = 4 independent experiments). Arrows B and C depict immature and mature CFTR as described in the text. B, relative expression of CFTR B form in CFBE41o− WT in cells transfected with control siRNA (gray bars) or ERp29 siRNA (open bars) was quantified by densitometry of n = 4 independent experiments after 0 and 1 h of chase. Data (mean ± S.E.) were normalized to values in control siRNA-transfected cells at t = 0.
We then performed Ussing chamber experiments to assess the CFTR-mediated chloride transport in control and ERp29 siRNA-treated CFBE41o− WT polarized monolayers (Fig. 10). As shown in Fig. 10A, control siRNA- or ERp29 siRNA-transfected CFBE41o− WT had similar base-line short circuit currents (−2.6 ± 0.7 μA/cm2 for Isc control versus −5.9 ± 1.9 μA/cm2; p not significant) as well as negligible amiloride-sensitive Isc. In the presence of a basolateral to apical chloride gradient, there was a significant ∼40% reduction in CFTR-mediated Isc (i.e. Isc activated by forskolin/IBMX and inhibited by CFTRinh-172) in the ERp29 siRNA-treated cells (157.7 ± 20.1 μA/cm2, n = 12) versus the control siRNA-treated cells (262.2 ± 40.8 μA/cm2, n = 12; p = 0.03; Fig. 10B). Together, these data indicate that specific depletion of ERp29 decreases the maturation and function of CFTR in the CFBE41o− model of bronchial epithelia.
FIGURE 10.

Depletion of ERp29 decreases CFTR-mediated chloride transport in CFBE41o− WT cells. CFBE41o− WT cells were grown as polarized monolayers and then transfected with control or ERp29 siRNA as described under “Experimental Procedures.” Assessment of CFTR-mediated currents was performed in Ussing chambers 48 h after transfection also as described under “Experimental Procedures.” A, representative Isc trace for cells transfected with control (closed circles) or ERp29 siRNA (open circles). Addition of 10 μm amiloride, addition of 10 μm forskolin and 100 μm IBMX, imposition of a basolateral to apical chloride gradient (Apical low Cl−), and addition of 10 μm CFTRinh-172 (Inh-172) are indicated. In these experiments, anion secretion results in a downward deflection in Isc. B, summary data for CFTR-mediated current, defined as Isc inhibited by CFTRinh-172 after stimulation with forskolin/IBMX and imposition of the basolateral to apical chloride gradient. Dark gray bar, control siRNA; light gray bar, ERp29 siRNA. Means ± S.E. for n individual experiments are shown. *, p = 0.03 by two-tailed t test.
DISCUSSION
The development of novel, mechanism-based therapies for CF is founded on the hypothesis that repair of mutant CFTR dysfunction will result in improved clinical outcomes (46). For ΔF508-CFTR, repair of function requires “correction” of its aberrant intracellular trafficking. 4PBA is one of the prototype ΔF508-CFTR “correctors” with demonstrated positive effects both in vitro (16) and in pilot human studies (47, 48). However, the mechanism by which 4PBA corrects ΔF508-CFTR remains unclear.
We (4, 18) and others (5) have suggested that 4PBA may improve ΔF508-CFTR trafficking by altering expression of the cytoplasmic 70-kDa heat shock proteins Hsc70 and Hsp70. We have also shown that such alterations in Hsc70 and Hsp70 expression can modulate the intracellular trafficking of the epithelial sodium channel ENaC (49). However, it is also clear that the cellular response to 4PBA in IB3-1 CF epithelial cells is quite complex (19, 20).
Here, we sought to identify additional 4PBA-regulated species in IB3-1 CF epithelial cells that could contribute to improved ΔF508-CFTR trafficking and identified ERp29 as a potential 4PBA-regulated chaperone of ΔF508-CFTR. We found that ERp29, a resident of the ER lumen, has increased mRNA and protein abundance in IB3-1 cells after treatment with 4PBA and that ERp29 interacts with ΔF508-CFTR in these cells. We also demonstrate that ERp29 overexpression promotes WT- and ΔF508-CFTR functional expression in Xenopus oocytes; for WT-CFTR, this effect correlates with increased CFTR abundance at the plasma membrane. Overexpression of ERp29 similarly led to increased ΔF508-CFTR surface expression in IB3-1 cells, and siRNA-mediated depletion of ERp29 led to decreased expression and maturation of WT-CFTR as well as decreased CFTR-mediated chloride current in CFBE41o− WT cells. Together, these data suggest that ERp29 is a target of 4PBA and that ERp29 can indeed function as a bona fide molecular chaperone or escort for an integral membrane protein.
The ER utilizes two execution pathways to control the biogenesis and trafficking of membrane or soluble secretory proteins. In one, correctly folded and assembled proteins are packaged into membrane carriers to be transported from the ER to the Golgi complex on the way to various cellular or extracellular destinations. These carriers are formed by the activity of cytosolic protein complexes, which for many proteins, including CFTR, involve COP II (coat complex II) machinery (50). In contrast, proteins that are recognized as inappropriately folded or assembled are diverted to proteasome-mediated degradation in the cytosol, a process known as ER-associated degradation (51).
Folding of newly synthesized integral membrane proteins like CFTR is likely facilitated by chaperone machines residing both in the cytosol and in the ER. Some central chaperone components of these machines are functionally modulated by co-chaperones with the objective of balancing the kinetic and thermodynamic constraints of client proteins to create an environment that will optimize their correct folding (50). Many components of this chaperone machinery also recognize misfolded proteins like ΔF508-CFTR and facilitate their degradation via ER-associated degradation.
A number of cytoplasmic and ER-resident chaperones and co-chaperones appear to be involved in regulating the biogenesis of WT- and/or ΔF508-CFTR. For example, cytoplasmic proteins such as Hsp90, Hsp40s, and the Hsp70 nucleotide exchange factor HspBP1 facilitate the Hsp70-dependent folding of CFTR (3–5, 52–54). In contrast, the cytosolic Hsc70-interacting protein CHIP is an E3 ubiquitin ligase that promotes CFTR degradation (55). Furthermore, data in yeast suggest that ER-associated degradation of CFTR is predominantly controlled by cytosolic rather than ER-resident chaperones (56), although a recently characterized ER membrane protein, Derlin-1, does appear to be pivotally involved (57, 58).
Relatively little is known about the influence of ER-resident chaperones on the biogenesis of WT- or ΔF508-CFTR. Calnexin, an ER membrane protein, interacts with immature forms of CFTR and ΔF508-CFTR by binding to a luminal loop (6, 8, 9, 59). However, calnexin has a more prolonged interaction with ΔF508- than WT-CFTR (6), and calnexin overexpression leads to enhanced retention of ΔF508 in the ER (8). Intriguingly, it appears that the classical ER chaperones BiP/grp78 and endoplasmin/grp94 do not interact with CFTR stably (2, 3, 6), and neither BiP nor calnexin appear to have central roles in ER-associated degradation of CFTR (8, 9, 56, 59). These findings raised the prospect that other more recently discovered ER residents could play a key role in the regulation of CFTR biogenesis.
Here, we provide evidence that WT- and ΔF508-CFTR trafficking can indeed be influenced by the novel ER-luminal resident ERp29 and that ERp29 likely acts upon newly synthesized CFTR. These data provide strong functional evidence that ERp29 is, in fact, a bona fide molecular chaperone or co-chaperone, extending recent reports by us and others (31, 40). The presumed function of ERp29 as a chaperone was supported previously by cellular studies of thyroglobulin, a soluble secretory protein (27, 28, 30). Moreover, ERp29 was found to induce a conformational change in the polyoma virus VP1 protein, which in turn facilitated passage of the virus across the ER membrane and successful infection (60). Biophysical characterization and activity assays also suggested that ERp29 is functionally distinct from the classical ER chaperones (26, 29).
It has also been suggested that ERp29, like its Drosophila paralogue Windbeutel, could act to escort secretory proteins through post-ER compartments in addition to or instead of the ER chaperone functionality (25, 27, 40). Windbeutel has a crucial role escorting a glycosaminoglycan-modifying enzyme (Pipe) to the Golgi. In the Golgi, Pipe directs the ventral activation of an extracellular serine proteolytic cascade, which leads to determination of the ventral side of the embryo (61, 62). The findings of ERp29 in conditioned cell media (Fig. 7 and Ref. 27) and in milk (43) are consistent with an escort role from the ER to the cell surface. However, because ERp29 was not found extracellularly in other physiological contexts such as in tooth enamel matrix (22) and plasma (44), it remains unclear whether extracellular ERp29 reflects an active escort role or passive egress.
Our present data (Fig. 7) demonstrate that ERp29 is secreted into the growth medium under control conditions and more so when ERp29 is overexpressed. Under control conditions, ERp29 was not detected at the surface of IB3-1 cells, but interestingly, 4PBA treatment increased the amount of ERp29 at the cell surface while decreasing that found in the culture media. In ERp29-overexpressing IB3-1 cells, whereas some ERp29 was detectable at the surface, a similar shift in ERp29 from the culture medium to the cell surface was observed with 4PBA treatment. These data associate improved ΔF508-CFTR trafficking after 4PBA treatment or ERp29 overexpression with increased cell surface expression of ERp29 in IB3-1 cells.
Our data also suggest that depletion of ERp29 decreases the amount of newly synthesized immature CFTR (Fig. 9). Although we cannot rule out that ERp29 depletion slows the initiation of CFTR synthesis, we feel this is less likely as ERp29 depletion does not alter CFTR mRNA expression. Instead, we posit that ERp29 may promote completion of synthesis and/or stabilization of newly formed WT-CFTR as well as delivery of CFTR to the Golgi or later compartments (Fig. 9). Noting evidence for a similar role with connexin43 (40), it seems reasonable to speculate that ERp29 could be accompanying or escorting ΔF508- or WT-CFTR through the exocytic pathway and also that ERp29 may remain associated with ΔF508- or WT-CFTR at the cell surface. Whether ERp29 facilitates WT-CFTR and ΔF508-CFTR trafficking by acting as a chaperone in the ER or as a post-ER escort or by both means warrants further investigation.
Recent data suggest that the ΔF508 mutation in the cytosolic first nucleotide binding domain (NBD1) of CFTR can alter the interaction of NBD1 with the cytoplasmic face of the transmembrane domains of CFTR (63). Although it is reasonable to hypothesize that such ΔF508-related structural alterations at the cytoplasmic face of the transmembrane domains may portend altered structure and interactions of the luminal transmembrane domain face, this does not appear to be the case for the functional interaction of ERp29 with WT- versus ΔF508-CFTR. In oocytes, ERp29 overexpression induced by injection of moderate amounts of cRNA increased the functional expression of both WT- and ΔF508-CFTR by ∼3-fold. These data suggest that the luminal/extracellular loops of WT- and ΔF508-CFTR are similarly recognized by ERp29.
That ERp29 overexpression induced by injection of moderate amounts of cRNA was more effective at promoting ΔF508- or WT-CFTR functional expression than that with higher amounts was not surprising. We have seen and extensively discussed potential mechanisms underlying similar effects for chaperone regulation of ENaC trafficking in oocytes (49), and there are other such reports for ER chaperones (15, 64).
Our data suggest that association of ΔF508-CFTR with ERp29 is detectable by co-immunoprecipitation in IB3-1 cell extracts but that such an association of WT-CFTR with ERp29 is not readily detected in T84 colonic epithelial cells. Similar findings were made for the interaction of ERp29 with misfolded viral protein (45) and for calnexin with ΔF508-CFTR (8). Interestingly, association of ERp29 with thyroglobulin was only demonstrable by co-immunoprecipitation after cross-linking, suggesting a weak or transient interaction (27). Here, we found that ERp29 co-immunoprecipitated with ΔF508-CFTR in the absence of cross-linking, suggesting a more stable or robust interaction; this is consistent with previous suggestions that ERp29 could favor hydrophobic substrates such as integral membrane proteins (29, 32). That ERp29 appears critical for the appropriate processing and assembly of connexin43 hemichannels (40) as well as the expression of Surfactant Protein B, which has significant hydrophobic character, in alveolar type II cells4 supports this notion. Similarly, we have observed that ERp29 overexpression increases functional expression of the epithelial sodium channel ENaC in the oocyte system.5 Collectively, these data enrich the hypothesis that ERp29 may function as a general chaperone or escort for membrane and/or hydrophobic secretory proteins.
In summary, our findings from siRNA-mediated depletion, overexpression, and 4PBA treatment are consistent with increased expression of ERp29 contributing to improved ΔF508-CFTR intracellular trafficking in CF epithelial cells. These data establish ERp29 as a functional target of the prototype ΔF508-CFTR corrector 4PBA. These data may also implicate ERp29 as a critical element in the mechanism by which 4PBA improves the trafficking and secretion of other mutant proteins such as ABCA3 (65), Surfactant Protein C (66), and α1-antitrypsin (67). Furthermore, our results support ERp29 functioning as a molecular chaperone of integral membrane proteins and are the first data demonstrating that overexpression of an ER resident can facilitate trafficking of WT-CFTR and ΔF508-CFTR. ER-luminal chaperones such as ERp29 can now be regarded as candidate pharmaceutical targets for novel CF therapies. Further investigation of the role of ERp29 in the trafficking of ΔF508-CFTR is warranted to inform the potential utility of this novel therapeutic strategy for CF.
Acknowledgment
We thank Dr. Zsuzsa Bebok, University of Alabama at Birmingham, for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK058046 and R01 DK073185 from the NIDDK (to R. C. R.), R01 HL059959 (to S. G.) and K08 HL081080 from the NHLBI (to J. L. K.), and T35 HD0744 (a student research fellowship to L. A. through the American Pediatric Society-Society for Pediatric Research). This work was also supported by grants from the Pennsylvania-Delaware Chapter of the American Heart Association (to L. S.) and the Cystic Fibrosis Foundation (to R. C. R.).
S. Guttentag, personal communication.
T. Chang, L. Suaud, and R. C. Rubenstein, unpublished observations.
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane conductance regulator
- Hsc70
- 70-kDa heat shock cognate protein
- ER
- endoplasmic reticulum
- IBMX
- 3-isobutyl-1-methylxanthine
- 4PBA
- sodium 4-phenylbutyrate
- TEV
- two-electrode voltage clamp
- I/V
- current/voltage
- Hsp
- heat shock protein
- Isc
- short circuit current
- sulfo-NHS-SS-biotin
- sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate
- NHS
- N-hydroxysuccinimide
- ANOVA
- analysis of variance
- cRNA
- complementary RNA
- NBD
- nucleotide binding domain
- BiP
- immunoglobulin heavy chain binding protein.
REFERENCES
- 1. Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. (1992) Nature 358, 761–764 [DOI] [PubMed] [Google Scholar]
- 2. Yang Y., Janich S., Cohn J. A., Wilson J. M. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 9480–9484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Loo M. A., Jensen T. J., Cui L., Hou Y., Chang X. B., Riordan J. R. (1998) EMBO J. 17, 6879–6887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rubenstein R. C., Zeitlin P. L. (2000) Am. J. Physiol. Cell Physiol. 278, C259–C267 [DOI] [PubMed] [Google Scholar]
- 5. Choo-Kang L. R., Zeitlin P. L. (2001) Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L58–L68 [DOI] [PubMed] [Google Scholar]
- 6. Pind S., Riordan J. R., Williams D. B. (1994) J. Biol. Chem. 269, 12784–12788 [PubMed] [Google Scholar]
- 7. Okiyoneda T., Wada I., Jono H., Shuto T., Yoshitake K., Nakano N., Nagayama S., Harada K., Isohama Y., Miyata T., Kai H. (2002) FEBS Lett. 526, 87–92 [DOI] [PubMed] [Google Scholar]
- 8. Okiyoneda T., Harada K., Takeya M., Yamahira K., Wada I., Shuto T., Suico M. A., Hashimoto Y., Kai H. (2004) Mol. Biol. Cell 15, 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rosser M. F., Grove D. E., Chen L., Cyr D. M. (2008) Mol. Biol. Cell 19, 4570–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ward C. L., Kopito R. R. (1994) J. Biol. Chem. 269, 25710–25718 [PubMed] [Google Scholar]
- 11. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., Riordan J. R. (1995) Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 12. Ward C. L., Omura S., Kopito R. R. (1995) Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 13. Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. (1990) Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 14. Kartner N., Augustinas O., Jensen T. J., Naismith A. L., Riordan J. R. (1992) Nat. Genet. 1, 321–327 [DOI] [PubMed] [Google Scholar]
- 15. Harada K., Okiyoneda T., Hashimoto Y., Ueno K., Nakamura K., Yamahira K., Sugahara T., Shuto T., Wada I., Suico M. A., Kai H. (2006) J. Biol. Chem. 281, 12841–12848 [DOI] [PubMed] [Google Scholar]
- 16. Rubenstein R. C., Egan M. E., Zeitlin P. L. (1997) J. Clin. Investig. 100, 2457–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bercovich B., Stancovski I., Mayer A., Blumenfeld N., Laszlo A., Schwartz A. L., Ciechanover A. (1997) J. Biol. Chem. 272, 9002–9010 [DOI] [PubMed] [Google Scholar]
- 18. Rubenstein R. C., Lyons B. M. (2001) Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L43–L51 [DOI] [PubMed] [Google Scholar]
- 19. Wright J. M., Zeitlin P. L., Cebotaru L., Guggino S. E., Guggino W. B. (2004) Physiol. Genomics 16, 204–211 [DOI] [PubMed] [Google Scholar]
- 20. Singh O. V., Vij N., Mogayzel P. J., Jr., Jozwik C., Pollard H. B., Zeitlin P. L. (2006) J. Proteome Res. 5, 562–571 [DOI] [PubMed] [Google Scholar]
- 21. Demmer J., Zhou C., Hubbard M. J. (1997) FEBS Lett. 402, 145–150 [DOI] [PubMed] [Google Scholar]
- 22. Hubbard M. J., McHugh N. J., Carne D. L. (2000) Eur. J. Biochem. 267, 1945–1957 [DOI] [PubMed] [Google Scholar]
- 23. Shnyder S. D., Hubbard M. J. (2002) J. Histochem. Cytochem. 50, 557–566 [DOI] [PubMed] [Google Scholar]
- 24. Hubbard M. J. (2002) Proteomics 2, 1069–1078 [DOI] [PubMed] [Google Scholar]
- 25. Mkrtchian S., Sandalova T. (2006) Antioxid. Redox. Signal. 8, 325–337 [DOI] [PubMed] [Google Scholar]
- 26. Hubbard M. J., Mangum J. E., McHugh N. J. (2004) Biochem. J. 383, 589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sargsyan E., Baryshev M., Szekely L., Sharipo A., Mkrtchian S. (2002) J. Biol. Chem. 277, 17009–17015 [DOI] [PubMed] [Google Scholar]
- 28. Park S., You K. H., Shong M., Goo T. W., Yun E. Y., Kang S. W., Kwon O. Y. (2005) Mol. Biol. Rep. 32, 7–13 [DOI] [PubMed] [Google Scholar]
- 29. Hermann V. M., Cutfield J. F., Hubbard M. J. (2005) J. Biol. Chem. 280, 13529–13537 [DOI] [PubMed] [Google Scholar]
- 30. Baryshev M., Sargsyan E., Mkrtchian S. (2006) Biochem. Biophys. Res. Commun. 340, 617–624 [DOI] [PubMed] [Google Scholar]
- 31. Barak N. N., Neumann P., Sevvana M., Schutkowski M., Naumann K., Malesević M., Reichardt H., Fischer G., Stubbs M. T., Ferrari D. M. (2009) J. Mol. Biol. 385, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 32. MacLeod J. C., Sayer R. J., Lucocq J. M., Hubbard M. J. (2004) J. Comp. Neurol. 477, 29–42 [DOI] [PubMed] [Google Scholar]
- 33. Bebok Z., Collawn J. F., Wakefield J., Parker W., Li Y., Varga K., Sorscher E. J., Clancy J. P. (2005) J. Physiol. 569, 601–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McGrath S. A., Basu A., Zeitlin P. L. (1993) Am. J. Respir. Cell Mol. Biol. 8, 201–208 [DOI] [PubMed] [Google Scholar]
- 35. Jiang Q., Li J., Dubroff R., Ahn Y. J., Foskett J. K., Engelhardt J., Kleyman T. R. (2000) J. Biol. Chem. 275, 13266–13274 [DOI] [PubMed] [Google Scholar]
- 36. Suaud L., Li J., Jiang Q., Rubenstein R. C., Kleyman T. R. (2002) J. Biol. Chem. 277, 8928–8933 [DOI] [PubMed] [Google Scholar]
- 37. Samaha F. F., Rubenstein R. C., Yan W., Ramkumar M., Levy D. I., Ahn Y. J., Sheng S., Kleyman T. R. (2004) J. Biol. Chem. 279, 23900–23907 [DOI] [PubMed] [Google Scholar]
- 38. Mkrtchian S., Fang C., Hellman U., Ingelman-Sundberg M. (1998) Eur. J. Biochem. 251, 304–313 [DOI] [PubMed] [Google Scholar]
- 39. Kaufman R. J. (2002) J. Clin. Investig. 110, 1389–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Das S., Smith T. D., Sarma J. D., Ritzenthaler J. D., Maza J., Kaplan B. E., Cunningham L. A., Suaud L., Hubbard M. J., Rubenstein R. C., Koval M. (2009) Mol. Biol. Cell 20, 2593–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Drumm M. L., Wilkinson D. J., Smit L. S., Worrell R. T., Strong T. V., Frizzell R. A., Dawson D. C., Collins F. S. (1991) Science 254, 1797–1799 [DOI] [PubMed] [Google Scholar]
- 42. French P. J., van Doorninck J. H., Peters R. H., Verbeek E., Ameen N. A., Marino C. R., de Jonge H. R., Bijman J., Scholte B. J. (1996) J. Clin. Investig. 98, 1304–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu C. C., Howell K. E., Neville M. C., Yates J. R., 3rd, McManaman J. L. (2000) Electrophoresis 21, 3470–3482 [DOI] [PubMed] [Google Scholar]
- 44. Muthusamy B., Hanumanthu G., Suresh S., Rekha B., Srinivas D., Karthick L., Vrushabendra B. M., Sharma S., Mishra G., Chatterjee P., Mangala K. S., Shivashankar H. N., Chandrika K. N., Deshpande N., Suresh M., Kannabiran N., Niranjan V., Nalli A., Prasad T. S., Arun K. S., Reddy R., Chandran S., Jadhav T., Julie D., Mahesh M., John S. L., Palvankar K., Sudhir D., Bala P., Rashmi N. S., Vishnupriya G., Dhar K., Reshma S., Chaerkady R., Gandhi T. K., Harsha H. C., Mohan S. S., Deshpande K. S., Sarker M., Pandey A. (2005) Proteomics 5, 3531–3536 [DOI] [PubMed] [Google Scholar]
- 45. Ferrari D. M., Nguyen Van P., Kratzin H. D., Söling H. D. (1998) Eur. J. Biochem. 255, 570–579 [DOI] [PubMed] [Google Scholar]
- 46. Rubenstein R. C. (2005) Curr. Opin. Pediatr. 17, 385–392 [DOI] [PubMed] [Google Scholar]
- 47. Rubenstein R. C., Zeitlin P. L. (1998) Am. J. Respir. Crit. Care Med. 157, 484–490 [DOI] [PubMed] [Google Scholar]
- 48. Zeitlin P. L., Diener-West M., Rubenstein R. C., Boyle M. P., Lee C. K., Brass-Ernst L. (2002) Mol. Ther. 6, 119–126 [DOI] [PubMed] [Google Scholar]
- 49. Goldfarb S. B., Kashlan O. B., Watkins J. N., Suaud L., Yan W., Kleyman T. R., Rubenstein R. C. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 5817–5822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kelly J. W., Balch W. E. (2006) Nat. Chem. Biol. 2, 224–227 [DOI] [PubMed] [Google Scholar]
- 51. Aridor M. (2007) Adv. Drug Deliv. Rev. 59, 759–781 [DOI] [PubMed] [Google Scholar]
- 52. Strickland E., Qu B. H., Millen L., Thomas P. J. (1997) J. Biol. Chem. 272, 25421–25424 [DOI] [PubMed] [Google Scholar]
- 53. Meacham G. C., Lu Z., King S., Sorscher E., Tousson A., Cyr D. M. (1999) EMBO J. 18, 1492–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang H., Schmidt B. Z., Sun F., Condliffe S. B., Butterworth M. B., Youker R. T., Brodsky J. L., Aridor M., Frizzell R. A. (2006) J. Biol. Chem. 281, 11312–11321 [DOI] [PubMed] [Google Scholar]
- 55. Jiang J., Ballinger C. A., Wu Y., Dai Q., Cyr D. M., Höhfeld J., Patterson C. (2001) J. Biol. Chem. 276, 42938–42944 [DOI] [PubMed] [Google Scholar]
- 56. Zhang Y., Nijbroek G., Sullivan M. L., McCracken A. A., Watkins S. C., Michaelis S., Brodsky J. L. (2001) Mol. Biol. Cell 12, 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun F., Zhang R., Gong X., Geng X., Drain P. F., Frizzell R. A. (2006) J. Biol. Chem. 281, 36856–36863 [DOI] [PubMed] [Google Scholar]
- 58. Ahner A., Nakatsukasa K., Zhang H., Frizzell R. A., Brodsky J. L. (2007) Mol. Biol. Cell 18, 806–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Farinha C. M., Amaral M. D. (2005) Mol. Cell. Biol. 25, 5242–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Magnuson B., Rainey E. K., Benjamin T., Baryshev M., Mkrtchian S., Tsai B. (2005) Mol. Cell 20, 289–300 [DOI] [PubMed] [Google Scholar]
- 61. Konsolaki M., Schüpbach T. (1998) Genes Dev. 12, 120–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sen J., Goltz J. S., Konsolaki M., Schüpbach T., Stein D. (2000) Development 127, 5541–5550 [DOI] [PubMed] [Google Scholar]
- 63. Loo T. W., Bartlett M. C., Clarke D. M. (2008) J. Biol. Chem. 283, 28190–28197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dorner A. J., Wasley L. C., Kaufman R. J. (1992) EMBO J. 11, 1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cheong N., Madesh M., Gonzales L. W., Zhao M., Yu K., Ballard P. L., Shuman H. (2006) J. Biol. Chem. 281, 9791–9800 [DOI] [PubMed] [Google Scholar]
- 66. Wang W. J., Mulugeta S., Russo S. J., Beers M. F. (2003) J. Cell Sci. 116, 683–692 [DOI] [PubMed] [Google Scholar]
- 67. Burrows J. A., Willis L. K., Perlmutter D. H. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 1796–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zeitlin P. L., Lu L., Rhim J., Cutting G., Stetten G., Kieffer K. A., Craig R., Guggino W. B. (1991) Am. J. Respir. Cell Mol. Biol. 4, 313–319 [DOI] [PubMed] [Google Scholar]