

Abstract
The localization of the platelet glycoprotein GP Ib-IX complex (GP Ibα, GP Ibβ, and GP IX) to membrane lipid domain, also known as glycosphingolipid-enriched membranes (GEMs or raft) lipid domain, is essential for the GP Ib-IX complex mediated platelet adhesion to von Willebrand factor (vWf) and subsequent platelet activation. To date, the mechanism for the complex association with the GEMs remains unclear. Although the palmitate modifications of GP Ibβ and GP IX were thought to be critical for the complex presence in the GEMs, we found that the removal of the putative palmitoylation sites of GP Ibβ and GP IX had no effects on the localization of the GP Ib-IX complex to the GEMs. Instead, the disruption of GP Ibα disulfide linkage with GP Ibβ markedly decreased the amount of the GEM-associated GP Ibα without altering the GEM association of GP Ibβ and GP IX. Furthermore, partial dissociation with the GEMs greatly inhibited GP Ibα interaction with vWf at high shear instead of in static condition or under low shear stress. Thus, for the first time, we demonstrated that GP Ibβ/GP IX mediates the disulfide-linked GP Ibα localization to the GEMs, which is critical for vWf interaction at high shear.
Keywords: Blood, Cell Adhesion, Lipid Raft, Platelet, Thrombosis
Introduction
The glycoprotein (GP)2 Ib-IX complex (GP Ibα, GP Ibβ, and GP IX) expressed on the surface of platelets plays critical roles in hemostasis and thrombosis through its interaction with von Willebrand factor (vWf) (1–3). In the complex, GP IX associates noncovalently with GP Ibβ, and two GP Ibβ (Cys122) molecules disulfide-link with one GP Ibα (Cys484 and Cys485) through two disulfide bonds (Fig. 1). In humans and mice, lack of or dysfunction of the GP Ib-IX complex causes Bernard-Soulier syndrome (2) and abrogates thrombus formation in mouse models of thrombosis (4, 5). To date, it is not entirely understood how the GP Ib-IX/vWf interaction is regulated, but this process in GP Ib-IX complex has been reported to involve posttranslational modification (6), intracelluar phosphorylation at cytoplasmic regions (7), and ectodomain shedding by membrane-associated disintegrin and metalloproteinase (ADAM) metallopeptidase (8). In addition to these molecular modifications of the complex itself, it has been realized that spatial localization of the complex in the platelet membrane mediated by the platelet glycosphingolipid-enriched membranes (GEMs) plays critical roles in the GP Ib-IX/vWf interaction. We and others have reported that (i) dissociation of GP Ibα from the GEMs inhibited platelet adhesion and activation (9), and (ii) association with the GP Ib-IX complex of signaling molecules and their activation, such as Src and phospholipase Cγ2, occurs exclusively in the platelet GEMs (10).
FIGURE 1.

Schematic view of the platelet GP Ib-IX complex. The depicted polypeptide arrangement of GP Ibα, GP Ibβ, and GP IX is based on the recently published stoichiometry of 1:2:1 (3). Circles with S inside represent the extracellular disulfide linkage between GP Ibα and GP Ibβ. The intracellular membrane-proximal cysteines of GP Ibβ (Cys148) and GP IX (Cys154) were reported to be the potential sites for palmitate modification as shown (15). The dashed rectangle depicts either the GP Ibα ligand binding domain or the WM23 binding region (macroglycopeptide).
In the presence of sterol, the GEMs on the cell membranes can be formed by a highly ordered packing of the saturated acyl chains of the enriched glycosphingolipids, which confers to the GEMs, and their protein constituents, a resistance to detergent solubilization. As a result, the GEMs can be extracted by non-ionic detergents (such as Triton X-100 and Brij series), isolated by high speed centrifugation over sucrose density gradient (floating in the low density upper fraction), and visualized by microscopy (size ranging from tens of nm to 100 nm). Because of the structural compatibility of the acyl chains of saturated long chain fatty acids (e.g. palmitate and myristate) (11) with that of glycosphingolipids, it has been speculated that acyl modification can cause proteins localization to the GEMs. However, a number of studies have suggested that lipid modifications are not prerequisites for receptor interaction with the GEMs because both palmitolyation-dependent (12) and -independent (11, 13) GEM associations have been reported. To date, there is no widely accepted mechanism for the GEM localization; rather, it is mediated by the specific targeting signals residing in the individual protein itself (14). In the case of the human GP Ib-IX complex, because GP Ibβ and GP IX are palmitoylated possibly through the intracellular membrane-proximal free cysteines (Cys148 in GP Ibβ and Cys154 in GP IX) (15), it has been speculated that palmitoylation of GP Ibβ and GP IX may play various roles in mediating the GP Ib-IX complex localization to the platelet GEMs (16); however, the experimental evidence has been lacking to support this notion. In this study, we intend to investigate how the GP Ib-IX complex associates with the GEMs.
EXPERIMENTAL PROCEDURES
Antibodies and Chemicals
Polyclonal antibodies against human GP Ibβ, GP IX, flotillin-1, and caveolin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The GP Ibα-specific monoclonal antibody WM23 binding within the macroglycopeptide (Fig. 1) was kindly provided by Dr. Michael Berndt at Monash University, Australia (17). The cholesterol-depriving chemicals, saponin and methyl-β-cyclodextrin (MβCD) were purchased from Sigma.
Generation of Chinese Hamster Ovary (CHO) Cell Lines Expressing Wild-type and Mutant GP Ib-IX Complex
Site-directed mutagenesis was performed by using a commercial PCR-based mutagenesis kit (QuikChange; Stratagene, La Jolla, CA). CHO cells expressing wild-type and mutant GP Ib-IX complex were generated and maintained as described previously (17–19). GP Ibα-positive cells were either sorted by WM23-conjugated magnetic beads or by Beckman-Coulter Altra Highpressure, a high speed cell sorter at the Flow Cytometry Core Facility of Baylor College of Medicine.
[3H]Palmitate Labeling
CHO cells expressing wild-type or mutant GP Ibα (2 × 106) were labeled with 250 μCi of [3H]palmitate (Amersham Biosciences) for 4 h in serum-free DMEM plus 2% dialyzed fetal bovine serum (FBS; Invitrogen). Labeled cells were harvested and washed, lysed with cold 1% Triton X-100, and immunoprecipitated with WM23. After extensive washing with lysis buffer, the samples were heated at 70 °C for 10 min and visualized using SDS-PAGE and fluorography.
Sucrose Density Floatation Assay
Resting platelets (9) or CHO cells (20) expressing the GP Ib-IX complex were lysed with 1.6 ml of ice-cold Triton X-100 or Brij 35 MES-buffered saline (25 mm MES, pH 6.5, 150 mm NaCl, 2 × proteinase inhibitor (Roche Diagnostics)) on ice for 1 h. The sample was then mixed with 1.6 ml of 80% sucrose in MES, transferred to the bottom of a 14 × 95-mm centrifuge tube (Seton Scientific), and gently overlaid with 4.8 ml of 30% and then 1.6 ml of 5% sucrose in MES. The gradient was then centrifuged at 34,000 rpm (∼146,000 × g) in a SW40 Ti rotor (Beckman) for 18 h at 4 °C followed by fractionation (from the top of the gradient) into 12 800-μl samples. An equal volume (15 μl) of each fraction was resolved in reducing SDS-PAGE and transferred to a PVDF membrane. GP Ibs were detected by Western blotting. The GEM fraction floats within sucrose gradient fractions 1–4, as identified by blotting flotillin-1 or caveolin (for CHO cells). The depletion of cholesterol by saponin was performed by pretreating the cell lysates in a ratio of 0.5% (g/v). Blots represent three independent experiments.
Ristocetin-induced vWf Binding
CHO cells (2 × 105) expressing wild-type (CHOαWTβIX) or mutant (CHOαSSβIX) were plated on a 24-well plate coated with 0.01% poly-l-lysine. After 36 h, the cells were washed with PBS and 0.5% BSA and incubated for 20 min at room temperature with either WM23 (1 μg/ml) or increasing concentrations of human vWf (0–8 μg/ml; American Diagnostica) in the presence of 1.5 mg/ml ristocetin (Sigma). Nonspecific binding was determined in the absence of modulator or in the presence of a blocking GP Ibα antibody, AK2 (Research Diagnostics) (21). Unbound vWf was removed by washing with 0.1 m sodium acetate and 0.5 × PBS. Cell-bound vWf was then revealed by HRP-labeled rabbit anti-human vWf (Dako, Germany). To normalize for the differences in GP Ibα expression between the cell lines, the data are expressed as a ratio of vWf binding to WM23 binding (the mean value ± S.E. represent three different experiments).
Flow Chamber Assay
CHO cells expressing wild-type or mutant GP Ibα were incubated on immobilized vWf (20 μg/ml) for 1 min in a parallel-plate flow chamber and then perfused with TBS and 0.5% BSA at flow rates that generated wall shear stresses of 2.5, 10, or 20 dynes/cm2 (17). To deplete cholesterol from the plasma membrane, cells were pretreated for 1 h at 37 °C in TBS containing 10 mm MβCD and 0.5% fatty acid-free BSA (Sigma) (22), washed, and then perfused in the presence of TBS and 0.5% fatty acid-free BSA. The experiments were recorded in real time for 1 min, by a high speed digital camera (model Quantix; Photometrics, Tucson, AZ) connected to an inverted stage microscope (Eclipse TE300; Nikon, Garden City, NY).
RESULTS
Localization of the GP Ib-IX Complex in the Platelet and CHO GEMs
We have reported that a small portion of GP Ibα resides in the platelet GEMs (9). Because the preexisting heterogeneity of the GEMs has been recently recognized (23), we sought to determine whether the GP Ib-IX complex indeed localizes to the GEMs and in what quantity in two cell systems, platelets and the GP Ib-IX-expressing CHO cells. We isolated and washed human platelets as described previously (9). The platelets or CHO cells were lysed with different detergents, followed by the sucrose density gradient centrifugation. In agreement with our previous observation (9), <20% of GP Ibα as well as GP Ibβ and GP IX in the Triton X-100-treated platelets localized to the low density GEM fractions (fractions 1–4); however, much more (>50%) of the GP Ib-IX complex was detergent-resistant if Brij 35 was used (Fig. 2A). When the GP Ib-IX complex-expressing CHO cells (CHOαβIX) were analyzed by the same flotation assay, we observed that Triton X-100 treatment solubilized almost all of the GP Ib-IX complex into the high density non-GEM fractions, whereas Brij 35 treatment maintained the majority (>90%) of the GP Ib-IX complex in low density fractions (Fig. 2B). As controls for Brij 35 treatment in both platelets and CHO cells, the non-GEM marker CD71 was found in the high density fractions, and two GEM markers, flotillin and caveolin (only CHO cells), were found in low density fractions, demonstrating that there is indeed specificity.
FIGURE 2.

The GP Ib-IX complex associates with the GEMs of platelets and CHO cells. A, resting human platelets were lysed with cold 0.1% Triton X-100 or 1% Brij 35. Lysates were subjected to sucrose gradient density centrifugation. Equal volume aliquots of each fraction (total 12 fractions) were analyzed by reducing SDS-PAGE followed by Western blotting with anti-GP Ibα monoclonal antibody WM23, anti-GP Ibβ, and anti-GP IX polyclonal antibodies. Blots represent three independent experiments. The GEM fraction floats within sucrose gradient fractions 1–4, and the position of the GEMs was identified by blotting flotillin-1. B, CHO cells expressing the GP Ib-IX complex were lysed with 0.5% Brij 35 or 0.1% Triton X-100 and subjected to sucrose gradient density centrifugation. The localization of GP Ibs in the density gradient was revealed by the same antibodies as A. The CHO GEM markers used are flotillin and caveolin. The non-GEM marker used is CD71. C, the association of the GP Ib-IX complex with the GEMs on CHO cells is cholesterol-dependent because saponin (0.5%/g/v), a cholesterol-depriving chemical, depleted the majority of the GP Ib-IX complex and caveolin from the GEMs.
In addition, our data revealed an apparent difference in the distribution pattern of the GP Ib-IX complex between human platelets (Fig. 2A) and CHO cells (Fig. 2B) upon Brij 35 extraction, and we think that there could be two reasons for it. First, a platelet has more GP Ib-IX molecules than a GP Ib-IX-expressing CHO cell. It has been established that a platelet has ∼25,000 copies of GP Ib-IX (24), whereas a GP Ib-IX-expressing CHO cell has only at most 5–10% of as many copies as a platelet (25). Therefore, with a limited availability and capacity of the GEMs on cell membrane, the percentage of GP Ib-IX present in CHO cell GEMs will likely increase. Because of that, we initially chose 0.5% instead of 1% Brij 35 to extract the CHO GP Ib-IX complex, although later we found that the GP Ib distribution pattern showed minor changes in response to various concentrations of Brij 35 ranging from 0.5% to 1% (data not shown). Second, the two cell types have different lipid composition in their GEMs, which may respond differently to detergent solubilization. In support of this notion, we observed that the floating pattern of platelet GP Ibα showed a minor but detectable difference even when Brij series detergents were used, whereas that of CHO GP Ibα basically remained unchanged (data not shown).
Furthermore, we found that the GEM association of the GP Ib-IX complex expressed in CHO cells was decreased by the cholesterol-solubilizing chemical saponin (Fig. 2C), as evidenced by detectable signals of caveolin and the GP Ib-IX complex across almost of all the fractions starting with fraction 3 (instead of fraction 2 in nontreated cells; Fig. 2B). In addition, saponin treatment translocated the flotillin from fraction 3 to fraction 2 as well. These results suggested that cholesterol is one of the structural components of the complex-associated and partially of the flotillin-associated GEMs. In addition, because it has been shown that Brij series detergents are less effective in removing glycerophospholipids than Triton X-100 (26), the increasing amount of the GP Ib-IX complex localized at Brij 35-extracted GEMs than Triton X-100-extracted GEMs (Fig. 2, A and B) suggested that glycerophospholipids may be required for the GP Ib-IX complex association with the GEMs.
Thus, we demonstrated that the GP Ib-IX complex expressed in both platelets and CHO cells can associate with the GEMs, which are more sensitive to Triton X-100 solubilization than Brij 35. Our data also suggested that Brij series detergents are the optimal choice for isolating the GP Ib-IX complex-associated GEMs in both platelets and CHO cells, possibly because they can better maintain the integrity of the GEM structure during extraction.
GP Ibβ/GP IX Resides in the Same GEMs as the GP Ib-IX Complex
We subjected the GP Ibβ/GP IX-expressing CHO cells (CHOβIX) to the fractionation by sucrose density gradient centrifugation. To our surprise, we found a nearly identical distribution pattern of GP Ibβ and GP IX between CHOαβIX (Fig. 2B) and CHOβIX cells (Fig. 3A) upon extraction with Triton X-100 and Brij 35. Such striking similarity was also found in the distribution patterns of GP Ibα in CHOαβIX and GP Ibβ in CHOβIX when two concentrations of Brij 35, one concentration of Brij 58, Brij 78, Brij 98, one relatively strong detergent, Nonidet P-40, and also a weaker detergent, Tween 20, were used to extract the GP Ib-IX complex-associated GEMs (data not shown). Moreover, GP Ibβ and GP IX in CHOβIX cells were driven by saponin across all of the fractions starting from fraction 3 (instead of fraction 2) (Fig. 3B) in a similar way as they were in CHOαβIX cells, indicating a similar cholesterol dependence of GP Ibβ/GP IX GEM association in both cells. Thus, our data suggested that GP Ibβ/GP IX might mediate the entire complex, the GP Ib-IX complex, to associate with the GEMs.
FIGURE 3.

GP Ibβ/GP IX resides in the same density fractions as the GP Ib-IX complex in a cholesterol-dependent manner. A, CHO cells expressing only Ibβ and IX polypeptides (CHOβIX) were lysed with 0.5% Brij 35 or 0.1% Triton X-100 followed by a sucrose density gradient fractionation. B, GP Ibβ and GP IX resided in similar fractions as the GP Ib-IX complex in CHOαβIX cells (Fig. 2B), which is also sensitive to cholesterol deprivation.
GP Ibβ/GP IX-GEM Association Is Independent of Palmitoylation of GP Ibβ and GP IX
It has been reported that human GP Ibβ and GP IX each are palmitoylated possibly through their intracellular membrane-proximal free cysteines (15), an acyl modification thought to mediate the GP Ib-IX complex association with the GEMs (9, 16). However, there is no experimental evidence supporting this notion. To test it, we mutated the cysteine to serine in both GP Ibβ (βS) and GP IX (IXS) and incorporated the mutant cDNAs, along with wild-type GP Ibα cDNA, into CHO cells (Fig. 4A). After labeling with [3H]palmitate, we lysed the cells with cold 1% Triton X-100 and immunoprecipitated GP Ibα with WM23. Palmitoylated GP Ibβ and GP IX were then visualized using SDS-PAGE and fluorography. We found that GP Ibβ and GP IX in CHOαβWTIXWT were palmitoylated, whereas GP Ibβ in CHOαβSIXWT, GP IX in CHOαβWTIXS, or both GP Ibβ and GP IX in CHOαβSIXS were not modified by palmitate (Fig. 4B). Thus, our data provided the first experimental evidence showing that the intracellular membrane-proximal cysteines in both GP Ibβ and GP IX are the targets for palmitoylation (15). Next, we subjected our double mutant CHOβSIXS cells to fractionation by sucrose density gradient centrifugation. We found that the distribution patterns of the depalmitoylated GP Ibβ and GP IX (Fig. 4, C and D) are similar to the palmitoylated ones in either CHOαβIX (Fig. 2B) or CHOβIX cells (Fig. 3A) under the same detergent extraction conditions. These findings demonstrated that palmitoylation of GP Ibβ/GP IX is not essential for GP Ibβ/GP IX association with the GEMs and further suggested that other unidentified structural determinants residing in GP Ibβ/GP IX localize the GP Ib-IX complex to the GEMs.
FIGURE 4.

Palmitoylation of GP Ibβ and GP IX through intracellular membrane-proximal cysteines is not essential for GEM localization in CHO cells. Intracellular membrane-proximal cysteines of both GP Ibβ (βC148S) and GP IX (IXC154S) were mutated to the amino acid serine. A, stable CHO cells expressing mutant GP IbβS or GP IXS or both with wild-type GP Ibα were generated (WM23 staining). B, when cells were labeled with [3H]palmitate and visualized using SDS-PAGE and fluorography, no sign of palmitic modification was shown, indicating that the intracellular membrane-proximal cysteines of GP Ibβ and GP IX are the sites for palmitoylation in the GP Ib-IX complex. C and D, further sucrose density fractionation analysis showed that the depalmitoylated GP Ibs in the double mutant CHO cells distributed to the same sucrose gradient as their palmitoylated counterparts in the wild-type cells (Figs. 2B and 3A).
Deficiency of α/β Disulfide Linkage Inhibits GP Ibα Instead GP Ibβ/GP IX Association with the GEMs
It has been established that the formation of a stable GP Ib-IX complex is through both noncovalent interaction of GP IX with GP Ibα/GP Ibβ (27) and covalent disulfide bonds between GP Ibα and GP Ibβ (3, 28). It is possible that the disulfide linkage between GP Ibα and GP Ibβ may be the driving force for the GP Ibα presence in the GP Ibβ/GP IX-associated GEMs. To test it, we mutated two of the extracellular membrane-proximal cysteines (Cys484 and Cys485) of GP Ibα (αSS) and extracted the GEMs by Brij 35. As shown in Fig. 5, more than half of mutant GP Ibα (∼60%) was solubilized to the high density fraction whereas other GP Ib components remained at low density (upper) fraction (∼100%). Thus, to the best of our knowledge, our data provide the first experimental evidence demonstrating that GP Ibα localization to the platelet GEMs is mediated primarily by a physical association of GP Ibβ/GP IX. Meanwhile, because there is still a certain amount of GP Ibα present in the GEMs (∼40%), other unidentified forces may facilitate GP Ibα localization to the GEMs.
FIGURE 5.

Disruption of α/β disulfide linkage inhibits GP Ibα association with the GEMs. CHO cells expressing mutant GP Ibα (with the C484S and C485S mutants) were analyzed by sucrose density gradient fractionation. A substantial amount (∼60%) of GP Ibα was solubilized to the high density fraction, whereas GP Ibβ and GP IX remained associated (∼100%).
Furthermore, because of the highly ordered packing of saturated glycosphingolipid acyl chains, the GEM membrane can resist the incorporation of detergents for solubilization whereas the non-GEM membrane cannot. Therefore, detergent solubilization can be used to isolate GEMs as well as to assess the affinity of proteins with the membrane lipids. In our case, we have tested a series of detergents to extract the GEMs from platelets and GP Ib-IX-expressing CHO cells, of which the Brij series can consistently isolate the GP Ib-IX-associated GEMs. When the α/β disulfide linkage was removed, GP Ibα appeared less in the GEMs. Thus, our data suggested that GP Ibβ/IX has much higher affinity to the GEMs lipids than GP Ibα, which one would expect can cause different membrane distribution of GP Ibα and GP Ibβ/IX if their disulfide linkage is absent. From this point of view, we think that biochemical analysis by detergent solubilization can, to some extent, reflect the real membrane organization of the GP Ib-IX complex in intact cells.
Partial Dissociation of GP Ibα from the GEMs Causes Dysfunction of GP Ibα in vWf Binding under High Fluid Shear Stresses
To investigate the potential effects of partial dissociation from the GEMs on GP Ibα function, we first investigated any effects that the mutations had on GP Ibα expression. We found that the mutant GP Ibα (CHOαSSβIX) was expressed at a level of 10–20% less than that of wild type (CHOαWTβIX), implicating that GP Ibβ/IX-mediated GEM localization plays a role in the efficient expression of GP Ibα in CHO cells. After sorting to achieve comparable expression levels of GP Ibα (Fig. 6A), we further evaluated the effects on the ristocetin-induced vWf binding in static condition, which we found was insignificant at all concentrations of vWf tested (Fig. 5B). Under flow, however, we found that, compared with wild-type, high shear stresses (10 or 20 dynes/cm2) caused much faster rolling of mutant GP Ibα-expressing CHO cells over a glass coverslip coated with 20 μg/ml vWf. In contrast, low shear stress (2.5 dynes/cm2) had minor effects (Fig. 6C). In addition, we also tested the effect of cholesterol-depriving chemical, MβCD on CHOαWTβIX interaction with immobilized vWf and found that consistent with our previous observation in platelets (9), cholesterol deprivation abolished the GP Ib-IX/vWf interaction only at high shear stress (data not shown). Thus, our data suggested that cholesterol-dependent localization of the GP Ibα within the GEMs plays a critical role in vWf interaction at high shear.
FIGURE 6.
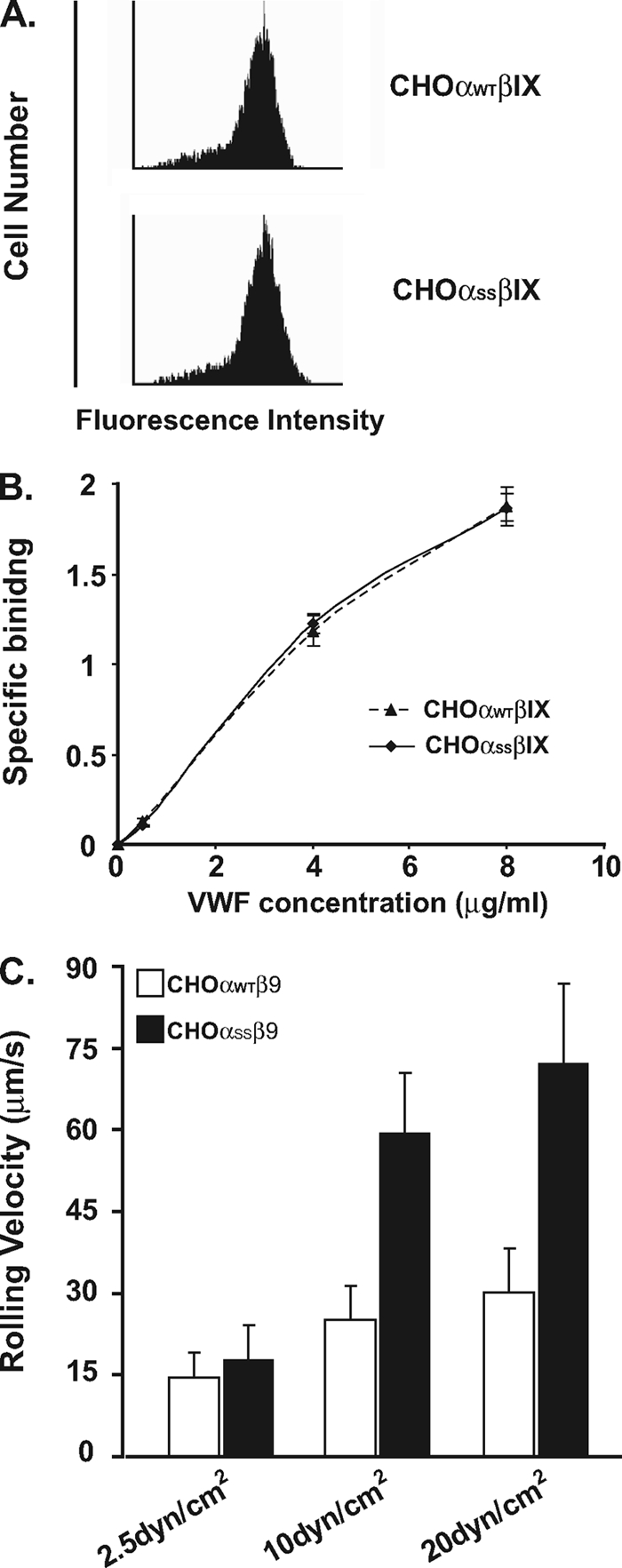
Partial dissociation of GP Ibα from the GEMs inhibits the interaction of the GP Ib-IX complex with vWf under high shear stresses. A, mutant GP Ibα (αSS) was transfected into CHO cells expressing wild-type Ibβ and IX (CHOαSSβIX). The sorted stable cell lines expressed comparable levels of either wild-type or mutant GP Ibα, detected by WM23 staining. B, ristocetin-induced vWf binding was determined as described under “Experimental Procedures.” In the presence of 1.5 mg/ml ristocetin, wild-type or mutant GP Ibα-expressing CHO cells bound to vWf equally at all concentrations of vWf tested. C, the cells expressing wild-type or mutant GP Ibα were incubated on immobilized vWf for 1 min in a parallel-plate flow chamber and perfused with TBS/0.5% BSA at flow rates that generated wall shear stresses of 2.5, 10 or 20 dynes/cm2. The apparent difference in the rolling velocity between CHOαWTβIX and CHOαSSβIX was only seen under high shear stresses (10 and 20 dynes/cm2), indicating that the association with the GEMs is critical for GP Ibα interaction with vWf at high shear.
DISCUSSION
We have reported that the GP Ib-IX complex localization to the platelet GEMs is essential for platelet adhesion and activation (9); however, we do not know what the targeting signals for its localization to the GEMs are. In this study, we demonstrated that the association with the GEMs of GP Ibα is mediated by two elements, one is GP Ibβ/GP IX harboring an unidentified targeting structure; the other is the α/β interchain disulfide linkage.
To the former, we found that the lipid modification of GP Ibβ and GP IX is not required, other structural determinants in GP Ibβ/GP IX mediate the GEM localization. Because the transmembrane domain (TMD) is the major segment mediating membrane lipid/receptor interaction (13, 29–31) which is highly conserved between human and mouse GP Ibβ/GP IX, it is most likely that the GP Ib-IX GEM-targeting signals reside in the TMDs of GP Ibβ and GP IX. Furthermore, previous reports suggested that (i) the length of the TMD can determine the localization of proteins to the GEMs such that the addition or removal of residues from the TMD could alter the ability of the protein localization to the GEMs (32), and (ii) membranes containing cholesterol tend to be thicker than membranes that do not contain the sterol (33). Therefore, in single TMD proteins, a longer TMD may be required for transmembrane protein localization to cholesterol-rich GEMs. Because it is predicted that the GP Ibβ TMD (25 amino acids) is significantly longer than both GP Ibα (21 amino acids) and GP IX (21 amino acids), considering the cholesterol dependence of the GP Ib-IX complex-associated GEMs (Figs. 2C and 3B), it is possible that GP Ibβ and its TMD play a more important role than GP IX in mediating GP Ibα localization to the GEMs, which is also supported by the finding that GP Ibβ is twice as many as GP IX in the GP Ib-IX complex (3). Nevertheless, our data do not rule out the possibility that other non-TMD segments of GP Ibβ and GP IX as well as their modifications (e.g. glycosylation and phosphorylation) play roles in the GEM localization of the GP Ib-IX complex.
To the latter, several lines of evidence showed that when specific antibodies were used to pull down the GP Ib-IX complex, the major dimeric form is α/β, instead of α/α or β/β (3, 34). Because our data indicated that the α/β disulfide linkage is the major force mediating GP Ibα into the GEMs, an interaction being essential for platelet adhesion and platelet activation upon vWf engagement (9), an enzymatically controlled preference for α/β disulfide bond formation, as opposed to other combinations (e.g. α/α or β/β), is most likely. Investigations have shown that platelet function could be regulated by a redox switch, a thiol-disulfide interchange catalyzed by enzymes, such as the protein disulfide isomerase (35) and other thiol isomerases (36). Several major platelet surface receptors have been reported to utilize this mechanism to regulate the affinity to their ligands (37), including fibrinogen receptor αIIbβ3, collagen receptor α2β1, and ADP receptor P2Y12. For a potential enzyme/substrate reaction to occur, a spatial closeness would be necessary. Interestingly, it has been reported that protein disulfide isomerase is membrane-proximate to the GP Ib-IX complex on platelet surface with a measurable fluorescence resonance energy transfer (FRET) efficiency of 14.8 ± 4.9% (38), which is comparable with that of GP Ib/FcRγIIA (16.3 ± 3.9%) (39). Even though these were two different experiments with a different antibody set for detection of the FRET signal, the slight difference between the transfer efficiency suggested that protein disulfide isomerase may be the candidate redox enzyme to catalyze a selective formation of α/β disulfide linkage.
Furthermore, we observed that, without α/β interchain disulfide bonds, even though there was still a portion of GP Ibα in the GEMs, more than half of GP Ibα lost its capability in association with the GEMs. Our data thus suggested that (i) GP Ibα by itself is unable to associate with the GEMs, and (ii) another force may mediate the remaining amount of GP Ibα residing in the GEMs. One candidate force is the interchain interaction between GP Ibα and GP IX through their extracellular domains (40). Because the number of the GEM-associated mutant GP Ibα molecules (αSS) in CHOαSSβIX cells decreased whereas the total GP IX numbers in the GEMs remained unchanged, the ratio of the GEM-associated mutant GP Ibα (αSS) to GP IX would increase significantly (>2-fold). As a result, the interchain interaction between mutant GP Ibα(αSS) and IX in the GEMs could be strengthened dramatically, thereby becoming a major driving force to mediate part of mutant GP Ibα(αSS) localization to the GEMs. Thus, the function of GP IX may lie in not only stabilizing the structure of the GP Ib-IX complex but also facilitating GP Ibα association with the GEMs. Further characterization of the extracellular interaction between GP IX and GP Ibα is needed to clarify this possibility.
Through a number of techniques including the use of synthetic peptides (41), phage display (42), proteolytic or mutated fragments of GP Ibα (43, 44), canine-human GP Ibα chimeras (21), GP Ibα monoclonal antibody epitope mapping (17, 42), and most importantly, crystallography of the vWf A1 domain and GP Ibα N terminus complex (45–47), it has been demonstrated that the vWf binding domain of GP Ibα lies solely in the N terminus (Fig. 1), which has an estimated distance of 400 Å far from the cell membrane. Because our disulfide linkage mutations (C484S and C485S) are close to the cell membrane, it is unlikely that these mutations would alter the N-terminal structure of the GP Ibα and therein vWf binding. In consistent with this notion, we found that partial dissociation of GP Ibα with the GEMs did not affect GP Ibα interaction with vWf either in static condition (induced by ristocetin) or under low shear stress; instead, the greatest effect of inhibition was seen only when high shear stresses (10 or 20 dynes/cm2) were applied. In addition, it is worthy to note that because CHO cells are larger than platelets, the shear force acting on CHO cells was estimated to be 10-fold stronger than that on platelets under identical shear stress. Therefore, the shear stress of 10 or 20 dynes/cm2 we used on CHO cells will become 100 or 200 dynes/cm2 on platelets, high shear conditions that only appear in narrowed arteries (48). Because the mutant cells (CHOαSSβ9) were still able to roll continuously on the vWf surface at high shear, it suggested that the formation of the GP Ib-IX/vWf bonds is still normal. Furthermore, because immobilized vWf-mediated rolling of the GP Ib-IX complex-expressing CHO cells depends on the shear stress and the density of the receptors on cell surface available for interaction with the same amount of vWf on the coverslip, such dramatic difference in the rolling velocity can only be attributed to the difference of GP Ibα density on both cell surfaces. However, because the expressional level of GP Ibα in both is comparable, it should not be the number of GP Ibα molecules that triggered such a difference. Based on the fact that the spherical shape of CHO cell only allows a small contact region between cells and surface, it is likely that a bundle of GP Ib-IX molecules in one or several GEMs provides the forces to mediate the translocation of the cells. In agreement, it has been suggested that the GEM localization of GP Ibα may increase the local density of GP Ibα (clustering) (9, 49, 50), thereby providing short lived GP Ib-IX/vWf bonds at one time for slower rolling. Thus, dissociation of GP Ibα from these GEMs could decrease the local density of GP Ibα and reduce the multivalence of GP Ib-IX/vWf bonding and as a result, impair the resistance of the GP Ib-IX-expressing CHO cells to high shear force and cause faster movement of the cells under flow (Fig. 6). Should it be the case, elimination of GP Ibα association with the GEMs could cause a loss of function of vWf binding at high shear stress. Furthermore, because clustering of the GP Ib-IX complex plays an important role in the complex-induced αIIbβ3 activation (50), it is also possible that the non-GEM-associated GP Ibα cannot transmit a signal to activate αIIbβ3 upon vWf binding. Toward these ends, further characterization of GP Ibβ/GP IX-harboring GEM-targeting signals to permit a specific abolition of the GP Ibα-GEM association without compromising the functions of other GEM-associated receptors is needed to assure the specific roles of the GEMs in the GP Ib-IX complex function, where changes in the interaction with vWf and signal transmission to activate integrin due to specific disruption of the GEM association can be attributed to the dysfunction of the GP Ib-IX complex-associated GEMs.
Acknowledgments
We thank Dr. Jingfei Dong for insightful discussion and Dr. Paul Bray at Jefferson Medical College of Thomas Jefferson University for support in the early phase of this project.
This work was supported, in whole or in part, by National Institutes of Health Grant HL095676 (to Y. P.). This work was also supported by the American Heart Association Scientist Development Grant 0635155N (to Y. P.), the Alkek Endowment Fund (to Y. P.), and the Fondren Foundation.
- GP
- glycoprotein
- GEM
- glycosphingolipid-enriched membrane
- MβCD
- methyl-β-cyclodextrin
- TMD
- transmembrane domain
- vWf
- von Willebrand factor.
REFERENCES
- 1. Savage B., Almus-Jacobs F., Ruggeri Z. M. (1998) Cell 94, 657–666 [DOI] [PubMed] [Google Scholar]
- 2. López J. A., Andrews R. K., Afshar-Kharghan V., Berndt M. C. (1998) Blood 91, 4397–4418 [PubMed] [Google Scholar]
- 3. Luo S. Z., Mo X., Afshar-Kharghan V., Srinivasan S., López J. A., Li R. (2007) Blood 109, 603–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanaji T., Russell S., Ware J. (2002) Blood 100, 2102–2107 [DOI] [PubMed] [Google Scholar]
- 5. Bergmeier W., Piffath C. L., Goerge T., Cifuni S. M., Ruggeri Z. M., Ware J., Wagner D. D. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 16900–16905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andrews R. K., Gardiner E. E., Shen Y., Whisstock J. C., Berndt M. C. (2003) Int. J. Biochem. Cell Biol. 35, 1170–1174 [DOI] [PubMed] [Google Scholar]
- 7. Bodnar R. J., Xi X., Li Z., Berndt M. C., Du X. (2002) J. Biol. Chem. 277, 47080–47087 [DOI] [PubMed] [Google Scholar]
- 8. Bergmeier W., Piffath C. L., Cheng G., Dole V. S., Zhang Y., von Andrian U. H., Wagner D. D. (2004) Circ. Res. 95, 677–683 [DOI] [PubMed] [Google Scholar]
- 9. Shrimpton C. N., Borthakur G., Larrucea S., Cruz M. A., Dong J. F., López J. A. (2002) J. Exp. Med. 196, 1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin W., Inoue O., Tamura N., Suzuki-Inoue K., Satoh K., Berndt M. C., Handa M., Goto S., Ozaki Y. (2007) J. Thromb. Haemost. 5, 1034–1040 [DOI] [PubMed] [Google Scholar]
- 11. Melkonian K. A., Ostermeyer A. G., Chen J. Z., Roth M. G., Brown D. A. (1999) J. Biol. Chem. 274, 3910–3917 [DOI] [PubMed] [Google Scholar]
- 12. Koziak K., Kaczmarek E., Kittel A., Sévigny J., Blusztajn J. K., Schulte Am Esch J., 2nd, Imai M., Guckelberger O., Goepfert C., Qawi I., Robson S. C. (2000) J. Biol. Chem. 275, 2057–2062 [DOI] [PubMed] [Google Scholar]
- 13. Scheiffele P., Roth M. G., Simons K. (1997) EMBO J. 16, 5501–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown D. A. (2006) Physiology 21, 430–439 [DOI] [PubMed] [Google Scholar]
- 15. Muszbek L., Laposata M. (1989) J. Biol. Chem. 264, 9716–9719 [PubMed] [Google Scholar]
- 16. López J. A., del Conde I., Shrimpton C. N. (2005) J. Thromb. Haemost. 3, 1737–1744 [DOI] [PubMed] [Google Scholar]
- 17. Peng Y., Berndt M. C., Cruz M. A., López J. A. (2004) Blood 104, 3971–3978 [DOI] [PubMed] [Google Scholar]
- 18. López J. A., Weisman S., Sanan D. A., Sih T., Chambers M., Li C. Q. (1994) J. Biol. Chem. 269, 23716–23721 [PubMed] [Google Scholar]
- 19. Gu M., Xi X., Englund G. D., Berndt M. C., Du X. (1999) J. Cell Biol. 147, 1085–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin P. J., Williams W. P., Kobiljski J., Numata M. (2007) Cell. Signal. 19, 978–988 [DOI] [PubMed] [Google Scholar]
- 21. Shen Y., Romo G. M., Dong J. F., Schade A., McIntire L. V., Kenny D., Whisstock J. C., Berndt M. C., López J. A., Andrews R. K. (2000) Blood 95, 903–910 [PubMed] [Google Scholar]
- 22. Michal P., Rudajev V., El-Fakahany E. E., Dolezal V. (2009) Eur. J. Pharmacol. 606, 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pike L. J. (2004) Biochem. J. 378, 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Du X., Beutler L., Ruan C., Castaldi P. A., Berndt M. C. (1987) Blood 69, 1524–1527 [PubMed] [Google Scholar]
- 25. Cranmer S. L., Ulsemer P., Cooke B. M., Salem H. H., de la Salle C., Lanza F., Jackson S. P. (1999) J. Biol. Chem. 274, 6097–6106 [DOI] [PubMed] [Google Scholar]
- 26. Schuck S., Honsho M., Ekroos K., Shevchenko A., Simons K. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 5795–5800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berndt M. C., Gregory C., Kabral A., Zola H., Fournier D., Castaldi P. A. (1985) Eur. J. Biochem. 151, 637–649 [DOI] [PubMed] [Google Scholar]
- 28. Mo X., Luo S. Z., Munday A. D., Sun W., Berndt M. C., López J. A., Dong J. F., Li R. (2008) J. Thromb. Haemost. 6, 1789–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gosse J. A., Wagenknecht-Wiesner A., Holowka D., Baird B. (2005) J. Immunol. 175, 2123–2131 [DOI] [PubMed] [Google Scholar]
- 30. Perschl A., Lesley J., English N., Hyman R., Trowbridge I. S. (1995) J. Cell Sci. 108, 1033–1041 [DOI] [PubMed] [Google Scholar]
- 31. Bock J., Gulbins E. (2003) FEBS Lett. 534, 169–174 [DOI] [PubMed] [Google Scholar]
- 32. Munro S. (1995) EMBO J. 14, 4695–4704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nezil F. A., Bloom M. (1992) Biophys. J. 61, 1176–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perrault C., Moog S., Rubinstein E., Santer M., Baas M. J., de la Salle C., Ravanat C., Dambach J., Freund M., Santoso S., Cazenave J. P., Lanza F. (2001) Thromb. Haemost. 86, 1238–1248 [PubMed] [Google Scholar]
- 35. Essex D. W., Li M., Miller A., Feinman R. D. (2001) Biochemistry 40, 6070–6075 [DOI] [PubMed] [Google Scholar]
- 36. Jordan P. A., Stevens J. M., Hubbard G. P., Barrett N. E., Sage T., Authi K. S., Gibbins J. M. (2005) Blood 105, 1500–1507 [DOI] [PubMed] [Google Scholar]
- 37. Essex D. W. (2004) Antioxid. Redox Signal. 6, 736–746 [DOI] [PubMed] [Google Scholar]
- 38. Burgess J. K., Hotchkiss K. A., Suter C., Dudman N. P., Szöllösi J., Chesterman C. N., Chong B. H., Hogg P. J. (2000) J. Biol. Chem. 275, 9758–9766 [DOI] [PubMed] [Google Scholar]
- 39. Sullam P. M., Hyun W. C., Szöllösi J., Dong J., Foss W. M., López J. A. (1998) J. Biol. Chem. 273, 5331–5336 [DOI] [PubMed] [Google Scholar]
- 40. Wu G., Meloni F. J., Shapiro S. S. (1996) Blood 87, 2782–2787 [PubMed] [Google Scholar]
- 41. Katagiri Y., Hayashi Y., Yamamoto K., Tanoue K., Kosaki G., Yamazaki H. (1990) Thromb. Haemost. 63, 122–126 [PubMed] [Google Scholar]
- 42. Cauwenberghs N., Vanhoorelbeke K., Vauterin S., Westra D. F., Romo G., Huizinga E. G., Lopez J. A., Berndt M. C., Harsfalvi J., Deckmyn H. (2001) Blood 98, 652–660 [DOI] [PubMed] [Google Scholar]
- 43. Ward C. M., Andrews R. K., Smith A. I., Berndt M. C. (1996) Biochemistry 35, 4929–4938 [DOI] [PubMed] [Google Scholar]
- 44. Marchese P., Saldívar E., Ware J., Ruggeri Z. M. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 7837–7842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huizinga E. G., Tsuji S., Romijn R. A., Schiphorst M. E., de Groot P. G., Sixma J. J., Gros P. (2002) Science 297, 1176–1179 [DOI] [PubMed] [Google Scholar]
- 46. Uff S., Clemetson J. M., Harrison T., Clemetson K. J., Emsley J. (2002) J. Biol. Chem. 277, 35657–35663 [DOI] [PubMed] [Google Scholar]
- 47. Dumas J. J., Kumar R., McDonagh T., Sullivan F., Stahl M. L., Somers W. S., Mosyak L. (2004) J. Biol. Chem. 279, 23327–23334 [DOI] [PubMed] [Google Scholar]
- 48. Fredrickson B. J., Dong J. F., McIntire L. V., López J. A. (1998) Blood 92, 3684–3693 [PubMed] [Google Scholar]
- 49. Savage B., Saldívar E., Ruggeri Z. M. (1996) Cell 84, 289–297 [DOI] [PubMed] [Google Scholar]
- 50. Kasirer-Friede A., Ware J., Leng L., Marchese P., Ruggeri Z. M., Shattil S. J. (2002) J. Biol. Chem. 277, 11949–11956 [DOI] [PubMed] [Google Scholar]