

Abstract
The misfolding of the protein α-synuclein (αS) has been implicated in the molecular chain of events leading to Parkinson disease. Physiologically, αS undergoes a transition from a random coil to helical conformation upon encountering synaptic vesicle membranes. On analogous small unilamellar vesicles (SUVs), the conformation of αS is dominated by a single elongated αS helix. However, alternative broken helix states have been postulated, mandating experimental clarification. Here, the upper limit for the free energy difference between elongated and broken helix conformations on SUVs resembling synaptic vesicles was determined to be 1.2 ± 0.4 kcal/mol, which amounts to a population ratio of 7.6:1 between both states (12% broken helices). In response to helix breaks at different positions, αS rearranged in an opportunistic manner, thereby minimizing helix abrogations to as little as one to two turns. Enthalpy and entropy measurements of gel state SUV-αS interactions indicated that broken helix states retain the ability to relieve membrane-packing stress. Thus, broken helix states are a distinct physiological feature of the vesicle-bound αS state, making it a “checkered” protein of multiple parallel conformations. A continuous interconversion between structural states may contribute to pathological αS misfolding.
Keywords: Amyloid, Biophysics, Membrane Proteins, Protein Folding, Synuclein, Protein-Membrane Interaction
Introduction
The protein α-synuclein (αS)2 was discovered in amyloid plaques and as a gene whose expression is altered during the period of song learning in the zebra finch (1, 2), well representing subsequent discoveries regarding the pathological and physiological nature of this protein. In humans, each of the point mutations A30P, E46K, and A53T, as well as gene triplication, gives rise to familial parkinsonism and dementia (3–6). In conjunction with the propensity of αS to misfold into amyloid fibrils in vitro and in vivo (7, 8), this places αS at the center of molecular events leading to prevalent human neurodegenerative disorders such as Parkinson disease (9). Physiologically, αS is a presynaptic brain protein that co-localizes with synaptic vesicles (10, 11). The binding and concomitant stabilization of such vesicles may modulate the threshold of neurotransmitter release, i.e. neural plasticity (1, 12, 13). In addition, αS can promote SNARE (soluble NSF attachment protein receptor) complex assembly (14).
In aqueous solution, αS is highly soluble yet lacks secondary structure (15). It may therefore be considered to be an intrinsically unfolded protein. Based on the repetitive amphiphilic amino acid sequence of its N-terminal 89 residues (see Fig. 1A), it was early on proposed and verified that αS adopts predominantly helical conformation upon associating with small unilamellar vesicles (SUVs) (1, 16). Recently, the average structure of αS when bound to SUVs composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS) lipids was reported (17). This structure, which was based on EPR distance and membrane immersion measurements, assigned an uninterrupted elongated α-helix to the αS amphiphilic region, in agreement with additional independent long-range distance measurements (18–20). Nevertheless, studies using smaller diameter spheroidal micelles, but also certain vesicle preparations (e.g. containing exclusively 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) lipids), showed that the elongated helix can be broken, resulting in an antiparallel arrangement of two helices (21–27) or a mixture of elongated and broken helices (28). In addition, a distinct helix break can be avoided even on a micelle by rearranging the amphiphilic amino acid sequence (29). Moreover, on SUVs, multiple αS-binding modes (i.e. structural inhomogeneity) exist (30), in accordance with an inhomogeneous sequence distribution of surfactant affinities (29, 31). Thus, although an elongated helix will dominate vesicle-bound αS, it is unclear to what extent broken helix conformations are populated on SUVs resembling synaptic vesicles.
FIGURE 1.

Amino acid sequence and structural models of αS variants. A, the amino acid sequence of αS incorporates seven 11-residue imperfect repeats (shown in color) that, together with Asp2–Leu8 and Ala53–Ala56, form an amphiphilic sequence of helical periodicity (3.67 residues per turn). Asterisks indicate residues 11, 81, and 83, which were substituted with cysteine in the disulfide-restricted αS(Cys11–Cys81) and αS(Cys11–Cys83) constructs, respectively. The number sign highlights residues 4 and 39, which were used as fluorescence probes when substituted with tryptophan. B, models of αS(Cys11–Cys81) and αS(Cys11–Cys83) assuming helical conformation for residues 2–92. For clarity, the dynamically disordered tail region (residues 93–140) was omitted from the figure. C, models of αS(Cys11–Cys81) and αS(Cys11–Cys83) obtained when permitting a helix break at the beginning and center of the amphiphilic sequence, respectively. The disulfide bond and associated cysteines are shown in red. The hydrophobic face of the amphiphilic αS helices are depicted in green.
Using conformationally restricted αS variants, we determined the free energy difference between uninterrupted and broken helix states on SUVs incorporating synaptic vesicle lipids and calculated the relative population between both states. Furthermore, the structural arrangements of the broken helix states were examined, and their abilities to relieve membrane-packing stress were evaluated. Thus, the physiological incidence and the relevance of broken αS helix states are defined. Defining the SUV-bound αS population distribution bears relevance not only to elucidating αS function but also to understanding αS misfolding. At present, it is unclear what structure(s) of αS give rise to misfolding in vivo. Frequent structural interconversions of vesicle-bound αS segments may bear relevance to accessing its misfolding pathway (32, 33), which initially exhibits characteristics of a conformational search (34). At a given time, protein structures are generally characterized by a single well defined conformation, and the existence of multiple parallel αS conformations would be an interesting deviation from this paradigm.
EXPERIMENTAL PROCEDURES
Expression and Purification of αS Variants
Double αS(Cys11–Cys81) and αS(Cys11–Cys83) cysteine mutants were generated from the wild-type αS gene residing in the T7 promoter-controlled pRK172 vector by QuikChange mutagenesis (Stratagene) (24). Tryptophan substitutions in these constructs (see Fig. 1A) were generated analogously. Gene expression was induced in Escherichia coli BL21(DE3)pLysS cells growing in LB medium at 37 °C at an absorbance (600 nm) of 1.0 using 0.5 mm isopropyl β-d-thiogalactopyranoside. Cells were lysed by heat shock (80 °C, 10 min) under reducing conditions (10 mm β-mercaptoethanol and 1 mm EDTA, N2 atmosphere). Subsequent to acid precipitation (pH 3.5), random disulfide bond formation was allowed to take place by dialyzing overnight against 50 mm Tris-HCl (pH 7.5) and 50 mm NaCl. Following ion exchange chromatography using HiTrap Q-Sepharose (GE Healthcare), any residual DNA was digested with 500 Kunitz units of DNase I (bovine pancreas; EMD Chemicals, Inc.) for 1 h at 37 °C to exclude spectral contamination in concentration measurements. The desired intramolecularly disulfide-bonded proteins, αS(Cys11–Cys81) and αS(Cys11–Cys83), were then separated from covalently linked protein oligomers by gel filtration employing a Sephacryl S-100 HR 26/60 column (GE Healthcare) as described previously in detail (34). Sample purity was verified by SDS-PAGE (supplemental Fig. S1). Protein concentration measurements were carried out by UV spectroscopy at 280 nm using ϵ(Tyr) = 1490 m−1 cm−1, ϵ(Trp) = 5500 m−1 cm−1, and ϵ(cystine) = 125 m−1 cm−1 (35).
Preparation of SUVs
Liquid crystalline SUVs were prepared using POPC, POPS, and POPG lipids as well as a premixed 1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phospho-l-serine/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPC/DOPS/DOPE) formula (2:3:5, w/w/w) all purchased from Avanti Polar Lipids, Inc. Lipids were dissolved in 2 ml of 98% chloroform and 2% methanol to obtain the desired amounts of POPG, POPC/POPS (7:3 mol/mol), and DOPC/DOPS/DOPE, respectively. The solvent was subsequently evaporated under a stream of nitrogen, and the obtained lipid film was subjected to overnight desiccation. Lipids were hydrated in a defined volume of buffer (see below) to yield a total lipid concentration of 10 mm. This procedure resulted in a distinctly hazy solution characteristic of a suspension of large multilamellar vesicles (36). Following a standard protocol (37), large multilamellar vesicles were converted to SUVs by sonication in a water bath at 42 kHz in a borosilicate glass vial (12-mm diameter) for 30–40 min. Vesicle preparations were used subsequent to ultracentrifugation (80,000 × g, 15 min). As outlined in the literature (38–40), negative staining electron microscopy was utilized to verify the formation of SUVs (supplemental Fig. S2). Gel state SUVs were prepared analogously using 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) lipids. However, this SUV type was sonicated and stored at 45 °C, i.e. above the lipid phase transition temperature. The hydrodynamic radii of SUV preparations were evaluated by dynamic light scattering using a Wyatt DynaPro NanoStar instrument. Data were analyzed using Dynamics 7.0 software by performing a regularization fit using the Dynals algorithm on the resultant autocorrelation functions (see Table 1 and supplemental Fig. S3).
TABLE 1.
Hydrodynamic radii of SUVs
| SUV compositiona | Hydrodynamic radiusb | Percentage of total lipid massc |
|---|---|---|
| nm | ||
| POPC/POPS | 11.0 ± 0.2 | 100 |
| POPG | 10.5 ± 0.8 | 100 |
| DOPE/DOPC/DOPS | 13.4 ± 1.7 | 80 |
| DPPC | 17.9 ± 0.2 | 65 |
a Lipid ratios were 7:3 mol/mol POPC/POPS and 2:3:5 (w/w/w) DOPC/DOPS/DOPE. DPPC SUVs were in the gel state in contrast to the other liquid crystalline SUVs.
b SUVs were prepared in 10 mm KH2PO4/K2HPO4 (pH 7.4) and measured at 22 °C (POPC/POPS, POPG, and DOPE/DOPC/DOPS) and 30 °C (DPPC).
c Compare with supplemental Fig. S3.
CD Spectroscopy and Analysis
CD measurements were carried out at 25 °C on a JASCO J-810 spectropolarimeter by acquiring spectra from 198 to 260 nm in a quartz cell of 1-mm path length. Eight spectra (recorded in 0.1-nm steps at a rate of 50 nm/min with a 0.1-nm bandwidth and a 0.5-s integration time) were accumulated. A starting protein concentration of 5 μm was prepared in 10 mm KH2PO4/K2HPO4 (pH 7.4), and SUVs were titrated (see Fig. 2 and supplemental Fig. S4). Spectra were corrected for solvent and lipid contributions as well as protein dilutions. The observed ellipticity at 222 nm in millidegrees (ϴ222) was converted to the mean residue ellipticity at 222 nm ([ϴ]MRW,222) using Equation 1,
where d is the path length in centimeters, c is the protein concentration in milligrams/ml, and MRW (mean residue weight) = MW/(n − 1), with MW denoting the molecular size of the polypeptide chain in daltons and n representing the number of amino acids in the chain. The total maximal percent helicity (% H) of the protein was estimated using Equation 2,
where [ϴ]MRW,222,sat refers to [ϴ]MRW,222 obtained at saturating lipid/protein ratios (41, 42).
FIGURE 2.

Association of αS variants with SUVs. Shown are isotherms of αS, αS(Cys11–Cys81), and αS(Cys11–Cys83) associations with SUVs composed of POPC/POPS (7:3 mol/mol), DOPC/DOPS/DOPE (2:3:5, w/w/w), and POPG, respectively. Fits to Equation 5 are shown. The starting protein concentration was 5 μm. Titrations with DOPC/DOPS/DOPE SUVs extended to a lipid/protein ratio of 600:1, but, for clarity, less than the full range is shown.
For a protein (P) exhibiting a single set of n identical independent binding sites, a two-state binding equilibrium with ligand (L) may be formulated as Pf* + Lf ↔ (P*L)b, where Pf* and Lf denote the molar concentrations of total unoccupied protein-binding sites and free ligand, respectively, and (P*L)b is the molar concentration of total occupied protein-binding sites or bound ligand (43, 44). The apparent dissociation and association constants (KD′ and KA′) for this interaction are given by Equation 3,
where Φ denotes the fraction of occupied lipid-binding sites on one protein, i.e. Equation 4,
where Lt and Pt represent the total molar concentrations of ligand and protein, respectively, and n is the (unknown) number of lipid-binding sites on protein. Combining Equations 3 and 4 and solving for Φ yields Equation 5.
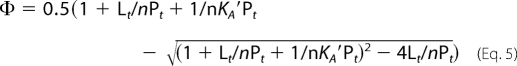 |
For the spectroscopic observation of the αS-lipid interaction, Φ corresponds to Equation 6,
 |
where [ϴ]MRW,222 is a function of Lt. KA′ and n were obtained by nonlinear curve fitting of experimental Δ[ϴ]MRW,222/Lt pairs (Fig. 2). With respect to the protein, the apparent association constant (KA) of the binding reaction equals n × KA′, which identifies n with the number of lipids associated with one protein.
Fluorescence Spectroscopy and Analysis
Fluorescence measurements were carried out at 25 °C using a Varian Cary Eclipse fluorometer. Emission spectra were recorded with an excitation wavelength of 290 nm and emission range of 300–400 nm with excitation and emission slit widths of 5 nm. The intrinsic fluorescence of αS variants incorporating a single tryptophan (see Table 3) was measured at a protein concentration of 0.5 μm in 25 mm KH2PO4/K2HPO4 (pH 7.4) and 100 mm KCl before and after adding saturating amount of SUVs. Corresponding solutions without protein were used to correct for background and SUV scattering contributions to the fluorescence signal. The accessibility of tryptophan was evaluated by monitoring its fluorescence quenching by I− in the absence and presence of SUVs, respectively. Quenching was assayed by adding increasing amounts of a solution of 4 m KI containing 1 mm Na2S2O3 (supplemental Fig. S5). The data were analyzed quantitatively by Stern-Volmer plots using Equation 7,
where F0 is the fluorescence intensity in the absence of quencher, F is the fluorescence intensity in the presence of quencher at molar concentration [Q], and KSV is the Stern-Volmer quenching constant (supplemental Table S1). A net accessibility factor was calculated from the ratios of KSV obtained from the quenching of tryptophan fluorescence in the presence and absence of SUVs, respectively.
TABLE 3.
Binding parameters of αS variant interactions with gel state DPPC SUVs
S.E. values are reported for experiments using independent SUV preparations in 10 mm KH2PO4/K2HPO4 (pH 7.4) and 100 mm KCl. Thermodynamic parameters were obtained at 30 °C.
| αS variant | ΔH | TΔS | ΔG | KDa | na |
|---|---|---|---|---|---|
| kcal/mol | kcal/mol | kcal/mol | ×10−6m | ||
| αS | −399.9 ± 25.7 | −389.7 ± 25.7 | −10.2 ± 0.1 | 0.039 ± 0.001 | 292 ± 14 |
| αS(Cys11–Cys83) | −24.6 ± 5.9 | −17.4 ± 5.6 | −7.2 ± 0.2 | 6.03 ± 2.1 | 76 ± 16 |
| αS(Cys11–Cys81) | −340.0 ± 3.6 | −330.0 ± 3.6 | −10.0 ± 0.1 | 0.055 ± 0.001 | 240 ± 3 |
a KD denotes the apparent dissociation constant, and n is the number of protein-associated lipids (Equations 3 and 4).
Isothermal Titration Calorimetry and Population Ratio
Experiments were carried out at 30 °C using a MicroCal VP-ITC calorimeter. Following the methods of Nuscher et al. (13), aliquots of DPPC SUVs at a concentration of 45 mm were titrated into the 1.425-ml sample cell containing the protein solution at a concentration of 8.7 μm at 300-s intervals. Prior to data analysis, the measurements were corrected for the heat of dilution of the protein by performing titrations with buffer (25 mm KH2PO4/K2HPO4 (pH 7.4) and 100 mm KCl) alone. Assuming again a single set of n identical independent protein-binding sites for lipids (see above), at fractional saturation Φ of binding sites (Equation 4), the total heat content (Q) of the sample cell solution (V0) relative to unliganded protein is given by Equation 8,
where ΔH′ is the molar heat of lipid binding. Substituting Equation 5 into Equation 8 yields Equation 9.
 |
Upon titrating with a volume dVi, the heat detected at the ith injection, ΔQ(i), is given by Equation 10.
 |
The middle terms account for the volume displaced from the sample cell upon adding dVi. KA′, n, and ΔH′ were obtained by nonlinear curve fitting of ΔQ(i)/dVi pairs using Equations 9 and 10 (Fig. 4). The enthalpy change per mol of protein (ΔH) was obtained from the enthalpy changer per mol of lipid as ΔH = n × ΔH′ and the association constant with respect to the protein as KA = n × KA′. The free energy and entropy changes (ΔG and ΔS) with respect to the protein are given by Equations 11 and 12,
where R denotes the gas constant in calories/K/mol, and T is the absolute temperature in K. Assuming Boltzmann distributed states, the population ratio between the lower and higher energy states (N1 and N2) is given by Equation 13,
 |
where k denotes the Boltzmann constant.
FIGURE 4.

Association of αS variants with gel state SUVs. Shown is the isothermal titration of αS variants with gel state DPPC SUVs at 30 °C. A starting protein concentration of 8.7 μm was employed.
RESULTS AND DISCUSSION
To measure the energy of binding of elongated as well as broken αS helix states to SUVs, in addition to the wild type, two mutant proteins that covalently restrict the spatial arrangement of the αS amphiphilic sequence were examined. An engineered intramolecular Cys11–Cys81 disulfide bond forced any helical structure to alter its facial orientation at least once, whereas a Cys11–Cys83 disulfide bond could maintain the same helix face along the entire sequence (Fig. 1B). At the point of the disulfide bond, both helix ends would have to stack and thus cannot bind to a common surface, which, although a necessary design element, will invariably underestimate the incidence of a helix break. Based on the simple model depicted in Fig. 1B, which did not consider any deviations from helical structure, αS(Cys11–Cys83) appears better suited than αS(Cys11–Cys81) to interact with the vesicle membrane.
Observing a spectroscopic signal as a function of the lipid/protein ratio can monitor the strength of the association of αS variants with SUVs. The mean residue ellipticity at 222 nm, [ϴ]MRW,222, which responds to the peptide bond nπ* energy transition and which is sensitive to the overall helical content of the protein (41, 42, 45), was chosen for this purpose. To assess the dependence of the interaction on lipid composition, SUVs of three different lipid formulas were prepared (in the liquid crystalline state). Among POPG, DOPC/DOPS/DOPE (2:3:5, w/w/w), and POPC/POPS (7:3 mol/mol) SUVs, POPG bound αS most avidly, followed by DOPC/DOPS/DOPE, whereas POPC/POPS offered the weakest interaction surface as reflected by the differences in saturating lipid/protein ratios (Fig. 2). Because radii were similar between SUV types (Table 1), these differences indeed reflected differential αS-lipid interactions. Interestingly, the binding trends of αS(Cys11–Cys81) and αS(Cys11–Cys83) relative to each other and relative to αS also depended somewhat on lipid composition. For example, for DOPC/DOPS/DOPE SUVs and, to a lesser extent, for POPG SUVs, αS(Cys11–Cys81) was able to bind even more avidly than αS at lower lipid/protein ratios but, like αS(Cys11–Cys83) over all ratios, was unable to compete with αS at higher lipid/protein ratios (Fig. 2, B and C). A compact amphiphilic structure may allow αS(Cys11–Cys81) to initially require less lipid molecules to bind these SUV types efficiently. It is well established that αS binding strongly correlates with the content of net negatively charged lipids in SUVs (16). The low saturating lipid/protein ratio of ∼40:1 for all variants on POPG SUVs (Fig. 2C) is nevertheless noteworthy. It is even smaller than observed for SDS and sodium lauroyl sarcosinate micelles (23, 29) and, in accordance with literature reports (17, 46), suggests that αS can substantially remodel this SUV type in analogy to distorting native micelle shapes (24, 47).
Assuming a single set of identical independent lipid-binding sites on αS (Equations 3 and 4), adequate quantitative descriptions of the binding events were obtained (Fig. 2), as established previously for the wild type in the context of POPC/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate SUVs (44). In the presence of DOPC/DOPS/DOPE SUVs, the steeper ascent of αS(Cys11–Cys81) relative to αS was reflected by the halving of protein-associated lipids (Table 2), whereas the larger lipid/protein ratio required to reach binding saturation led to an apparent dissociation constant (KD) that was significantly larger compared with the wild type (Table 2). A similar trend was seen for POPG. Nevertheless, for both SUV types, the KD of αS(Cys11–Cys83) tended to be smaller than that of αS(Cys11–Cys81), as expected from the structural models (Fig. 1B). For POPC/POPS SUVs, both variants were difficult to differentiate (Fig. 2A). The low number of protein-associated lipids for POPG SUVs was again indicative of the extensive reorganization of the vesicle by αS. When considering the structural rearrangements that must ensue from the disulfide bond restrictions (e.g. Fig. 1B), the observed reductions in SUV affinities relative to wild-type αS appear modest. Furthermore, the oftentimes similar KD values between αS(Cys11–Cys83) and αS(Cys11–Cys81) showed that the amphiphilic region possesses the ability to reorganize in a highly opportunistic manner. What is the structural basis of these observations?
TABLE 2.
Binding parameters of αS variant interactions with liquid crystalline SUVs
S.E. values are reported for experiments using independent SUV preparations in 10 mm KH2PO4/K2HPO4 (pH 7.4).
| SUV composition/αS varianta | KDb | nb | ΔGc | Hd | Protein bounde | [Θ]208/[Θ]222 |
|---|---|---|---|---|---|---|
| ×10−6m | kcal/mol | % | % | |||
| DOPC/DOPS/DOPE | ||||||
| αS | 0.17 ± 0.07 | 67 ± 1 | −9.2 ± 0.3 | 67 ± 1 | 100 | 1.12 ± 0.02 |
| αS(Cys11–Cys83) | 1.3 ± 0.7 | 60 ± 6 | −8.0 ± 0.3 | 62 ± 1 | 99.9 | 1.21 ± 0.03 |
| αS(Cys11–Cys81) | 2.4 ± 1.4 | 34 ± 1 | −7.7 ± 0.3 | 64 ± 4 | 99.9 | 1.17 ± 0.04 |
| POPC/POPS | ||||||
| αS | 0.31 ± 0.01 | 133 ± 2 | −8.9 ± 0.1 | 59 ± 2 | 100 | 1.13 ± 0.14 |
| αS(Cys11–Cys83) | 2.4 ± 1.9 | 117 ± 5 | −7.7 ± 0.5 | 47 ± 7 | 100 | 1.25 ± 0.14 |
| αS(Cys11–Cys81) | 1.0 ± 0.7 | 221 ± 25 | −8.2 ± 0.4 | 40 ± 2 | 100 | 1.17 ± 0.16 |
| POPG | ||||||
| αS | 0.43 ± 0.55 | 9 ± 3 | −8.7 ± 0.7 | 77 ± 2 | 99.8 | 1.13 ± 0.01 |
| αS(Cys11–Cys83) | 0.66 ± 0.12 | 11 ± 2 | −8.4 ± 0.1 | 56 ± 1 | 99.7 | 1.24 ± 0.04 |
| αS(Cys11–Cys81) | 2.3 ± 2.6 | 7 ± 3 | −7.7 ± 0.7 | 55 ± 6 | 98.8 | 1.19 ± 0.03 |
a Wild-type αS and variant proteins incorporated Y39W substitutions.
b KD denotes the apparent dissociation constant, and n is the number of protein-associated lipids (Equations 3 and 4).
c ΔG denotes the free energy change upon SUV binding at 25 °C (Equation 11).
d Percent H denotes the protein helical content at the titration end point (Equation 2; lipid/protein ratios of 600:1, 600:1, and 40:1 for DOPC/DOPS/DOPE, POPC/POPS, and POPG, respectively) (see Fig. 2).
e Values are the percentage of SUV-bound protein at the titration end point for the respective KD.
With virtually all protein bound to DOPC/DOPS/DOPE SUVs, the helical contents of αS(Cys11–Cys81) and αS(Cys11–Cys83) were reduced by 3 and 5%, respectively, compared with αS (Table 2). This indicated that, relative to the wild type, an average of one to two helix turns “broke” in both variants. For the other studied lipid compositions, more extensive helix breaks were observed (Table 2). In addition, for these SUV types, wild-type αS exhibited different helical contents compared with DOPC/DOPS/DOPE SUVs. The precise nature of the vesicle-αS interactions is therefore conspicuously sensitive to the membrane lipid composition. To localize regions that experienced helix breaks, tryptophan accessibility measurements were carried out using fluorescence spectroscopy. Single tryptophan fluorescence probes were incorporated at the strategic Phe4 and Tyr39 positions, respectively (Fig. 1A), to compare their net accessibility to the collisional quencher iodide. For DOPC/DOPS/DOPE SUVs, significant scattering contributions hampered measurements (supplemental Fig. S3), whereas for POPC/POPS SUVs, relatively weak KI concentration-dependent binding rendered measurements unreliable. However, data of adequate quality were obtained for POPG SUVs (supplemental Fig. S5). This SUV type will bias αS toward antiparallel conformations (26), but because both αS(Cys11–Cys81) and αS(Cys11–Cys83) invariably have to adopt such states (Fig. 1B), any differences detected between both forms reflect their lipid association and structural preferences. Protection of Trp39 in αS(Cys11–Cys81) was comparable with that in wild-type αS, whereas in αS(Cys11–Cys83), Trp39 was significantly more exposed (Fig. 3). In contrast, Trp4 was more protected in αS(Cys11–Cys83) than in αS(Cys11–Cys81). Thus, in αS(Cys11–Cys81), helical structure must have been disrupted near Phe4, whereas in αS(Cys11–Cys83), this occurred predominantly near Tyr39. Based on these results, our initial structural models of αS(Cys11–Cys81) and αS(Cys11–Cys83) were updated (Fig. 1C). When tolerating disorder near residue 11, αS(Cys11–Cys81) can reduce helix curvature and maintain a continuous helix that faces the membrane. When tolerating disorder near the middle of the amphiphilic region, αS(Cys11–Cys83) can maintain two shorter essentially linear helices that face a common membrane surface. Especially the differences in helix lengths between these models may be reflected in differences in the ellipticity ratio between the peptide bond ππ*‖ and nπ* energy transitions, [ϴ]208/[ϴ]222 (45). This is indeed the case for all SUV types: αS(Cys11–Cys83) exhibited the highest [ϴ]208/[ϴ]222 ratio, αS(Cys11–Cys81) was intermediate, and αS (which exhibited the most elongated helix) showed the lowest ratio (Table 2). In conclusion, by tolerating disorder (i.e. a helix break) at variable positions, αS has the capacity to bind SUVs in several different helical conformations.
FIGURE 3.

Tryptophan accessibility in αS variants. For single tryptophan-substituted αS variants, the net accessibility factors, defined as the ratio of Stern-Volmer quenching constants (KSV), obtained in the presence and absence of POPG lipid vesicles, respectively (supplemental Table S1), are compared.
To better judge the physiological relevance of a broken helix state, it is pertinent to address to what degree the disulfide restrictions impair the ability of αS to alleviate packing defects in vesicle membranes. The interaction of wild-type αS with electroneutral gel state SUVs is fundamentally different from such interactions in the liquid crystalline state (13). The binding event experiences large enthalpy and entropy contributions arising from lipid ordering in the vesicle membrane (13). We exploited this phenomenon to compare this functional ability of wild-type αS with the disulfide-restricted variants. Changes in enthalpy and entropy upon complex formation with gel state SUVs composed of DPPC lipids were obtained from isothermal titration calorimetry (Fig. 4). If both αS(Cys11–Cys81) and αS(Cys11–Cys83) would show significantly decreased DPPC SUV associations, it may be argued that broken helix states have no functional relevance and are mere byproducts of the flexibility of the elongated helix. This was not the case; enthalpy and entropy changes of the DPPC SUV-αS(Cys11–Cys81) interaction were reduced by only 15.0 and 15.3%, respectively, relative to the wild-type interaction (Table 3). A highly curved and broken αS helix is therefore still capable of effecting lipid ordering, aiding the relief of membrane-packing stress (13). In contrast, αS(Cys11–Cys83), with a putative proximate antiparallel helix arrangement (Fig. 1C), has limited functional significance as judged from its weak association with DPPC SUVs (Fig. 4 and Table 3). However, αS(Cys11–Cys83) may suffer from the putative extreme proximity of both helices, which we regard as unfavorable for reducing lipid-packing stress. Upon breaking the elongated αS helix near its midpoint, it is unlikely that a proximate distinctly antiparallel helix arrangement would ensue in the absence of a disulfide bond restriction. In conclusion, the ability to compensate membrane-packing stress does not rest solely with an elongated αS helix; broken and curved helices are still efficient, albeit proximate arrangements of broken helices appear unfavorable.
For liquid crystalline DOPC/DOPS/DOPE vesicles, the differences in KD values between αS and the examined variants translated into a minimal free energy difference (ΔΔG) of 1.2 ± 0.4 kcal/mol for αS(Cys11–Cys83) (Table 2). For POPG and POPC/POPS SUVs, the smallest obtained ΔΔG values were 0.3 ± 0.7 and 0.7 ± 0.4 kcal/mol, respectively (Table 2). Synaptic vesicles exhibit an average PC/PS/PE lipid ratio of 2:0.7:2.3 (w/w/w) and contain a substantial fraction of cholesterol (48). Based on the three examined quite diverse membrane systems and the limitations of the disulfide-enforced structural constraint (Fig. 1, B and C), we regard the largest ΔΔG value of 1.2 ± 0.4 kcal/mol to represent the upper limit for a helix break on synaptic vesicles. For Boltzmann distributed states, an energy difference of 1.2 kcal/mol corresponds to a population ratio of 7.6:1 between the elongated and broken helix states (∼12% of broken helices). It is therefore evident that vesicle-bound αS contains a permanent subpopulation exhibiting a broken helix. This also shows that studies of micelle-bound αS carry physiological relevance.
Our result successfully integrates the findings of the many diverse biophysical αS studies (see the Introduction), including the reports of αS conformational inhomogeneity (28–31). It is also in agreement with SUV-bound αS distance measurements where an elongated helix was detected (17–20). Variable helix break positions and variable relative helix-helix orientations give rise to non-uniform broad interhelical distance distributions that appear to be difficult to detect in the presence of the predominant elongated helix state. In support of this view, we note that, on sodium lauroyl sarcosinate micelles, little preference exists to arrange the broken helices in either NC or CN surface-bound arrangements (24). In sum, based on free energy considerations, a helix break of vesicle-bound αS is a common event, substantially clarifying the structural biology of vesicle-bound αS. The relevance of these finding may also apply to the misfolding pathway of αS where deviations from helical conformation are required to transition to misfolded species that are rich in β-sheet conformations (32, 33).
Supplementary Material
Acknowledgments
We thank Keith Bromley and Janet Oldak for assistance with dynamic light scattering and Jobin Varkey for advice regarding electron microscopy.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and Table S1.
- αS
- α-synuclein
- SUV
- small unilamellar vesicle
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- POPG
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- DOPC/DOPS/DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phospho-l-serine/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- DPPC
- 1,2-dipalmitoyl-sn-glycero-3-phosphocholine.
REFERENCES
- 1. George J. M., Jin H., Woods W. S., Clayton D. F. (1995) Neuron 15, 361–372 [DOI] [PubMed] [Google Scholar]
- 2. Uéda K., Fukushima H., Masliah E., Xia Y., Iwai A., Yoshimoto M., Otero D. A., Kondo J., Ihara Y., Saitoh T. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 11282–11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., Riess O. (1998) Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 4. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Di Iorio G., Golbe L. I., Nussbaum R. L. (1997) Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 5. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., de Yebenes J. G. (2004) Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 6. Singleton A. B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R., Lincoln S., Crawley A., Hanson M., Maraganore D., Adler C., Cookson M. R., Muenter M., Baptista M., Miller D., Blancato J., Hardy J., Gwinn-Hardy K. (2003) Science 302, 841 [DOI] [PubMed] [Google Scholar]
- 7. Han H., Weinreb P. H., Lansbury P. T., Jr. (1995) Chem. Biol. 2, 163–169 [DOI] [PubMed] [Google Scholar]
- 8. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., Goedert M. (1997) Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 9. Nussbaum R. L., Ellis C. E. (2003) N. Engl. J. Med. 348, 1356–1364 [DOI] [PubMed] [Google Scholar]
- 10. Iwai A., Masliah E., Yoshimoto M., Ge N., Flanagan L., de Silva H. A., Kittel A., Saitoh T. (1995) Neuron 14, 467–475 [DOI] [PubMed] [Google Scholar]
- 11. Jensen P. H., Nielsen M. S., Jakes R., Dotti C. G., Goedert M. (1998) J. Biol. Chem. 273, 26292–26294 [DOI] [PubMed] [Google Scholar]
- 12. Abeliovich A., Schmitz Y., Fariñas I., Choi-Lundberg D., Ho W. H., Castillo P. E., Shinsky N., Verdugo J. M., Armanini M., Ryan A., Hynes M., Phillips H., Sulzer D., Rosenthal A. (2000) Neuron 25, 239–252 [DOI] [PubMed] [Google Scholar]
- 13. Nuscher B., Kamp F., Mehnert T., Odoy S., Haass C., Kahle P. J., Beyer K. (2004) J. Biol. Chem. 279, 21966–21975 [DOI] [PubMed] [Google Scholar]
- 14. Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M. R., Südhof T. C. (2010) Science 329, 1663–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weinreb P. H., Zhen W., Poon A. W., Conway K. A., Lansbury P. T., Jr. (1996) Biochemistry 35, 13709–13715 [DOI] [PubMed] [Google Scholar]
- 16. Davidson W. S., Jonas A., Clayton D. F., George J. M. (1998) J. Biol. Chem. 273, 9443–9449 [DOI] [PubMed] [Google Scholar]
- 17. Jao C. C., Hegde B. G., Chen J., Haworth I. S., Langen R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Georgieva E. R., Ramlall T. F., Borbat P. P., Freed J. H., Eliezer D. (2008) J. Am. Chem. Soc. 130, 12856–12857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trexler A. J., Rhoades E. (2009) Biochemistry 48, 2304–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferreon A. C., Gambin Y., Lemke E. A., Deniz A. A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5645–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eliezer D., Kutluay E., Bussell R., Jr., Browne G. (2001) J. Mol. Biol. 307, 1061–1073 [DOI] [PubMed] [Google Scholar]
- 22. Chandra S., Chen X., Rizo J., Jahn R., Südhof T. C. (2003) J. Biol. Chem. 278, 15313–15318 [DOI] [PubMed] [Google Scholar]
- 23. Ulmer T. S., Bax A., Cole N. B., Nussbaum R. L. (2005) J. Biol. Chem. 280, 9595–9603 [DOI] [PubMed] [Google Scholar]
- 24. Rao J. N., Jao C. C., Hegde B. G., Langen R., Ulmer T. S. (2010) J. Am. Chem. Soc. 132, 8657–8668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bisaglia M., Tessari I., Pinato L., Bellanda M., Giraudo S., Fasano M., Bergantino E., Bubacco L., Mammi S. (2005) Biochemistry 44, 329–339 [DOI] [PubMed] [Google Scholar]
- 26. Drescher M., Veldhuis G., van Rooijen B. D., Milikisyants S., Subramaniam V., Huber M. (2008) J. Am. Chem. Soc. 130, 7796–7797 [DOI] [PubMed] [Google Scholar]
- 27. Bortolus M., Tombolato F., Tessari I., Bisaglia M., Mammi S., Bubacco L., Ferrarini A., Maniero A. L. (2008) J. Am. Chem. Soc. 130, 6690–6691 [DOI] [PubMed] [Google Scholar]
- 28. Robotta M., Braun P., van Rooijen B., Subramaniam V., Huber M., Drescher M. (2011) ChemPhysChem 12, 267–269 [DOI] [PubMed] [Google Scholar]
- 29. Rao J. N., Kim Y. E., Park L. S., Ulmer T. S. (2009) J. Mol. Biol. 390, 516–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bodner C. R., Dobson C. M., Bax A. (2009) J. Mol. Biol. 390, 775–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bartels T., Ahlstrom L. S., Leftin A., Kamp F., Haass C., Brown M. F., Beyer K. (2010) Biophys. J. 99, 2116–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chiti F., Dobson C. M. (2006) Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 33. Glabe C. G. (2006) Neurobiol. Aging 27, 570–575 [DOI] [PubMed] [Google Scholar]
- 34. Suk J. E., Lokappa S. B., Ulmer T. S. (2010) Biochemistry 49, 1533–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gill S. C., von Hippel P. H. (1989) Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 36. Bangham A. D., Standish M. M., Watkins J. C. (1965) J. Mol. Biol. 13, 238–252 [DOI] [PubMed] [Google Scholar]
- 37. Huang C. H. (1969) Biochemistry 8, 344–352 [DOI] [PubMed] [Google Scholar]
- 38. Tenchov B. G., Yanev T. K., Tihova M. G., Koynova R. D. (1985) Biochim. Biophys. Acta 816, 122–130 [DOI] [PubMed] [Google Scholar]
- 39. Egerdie B., Singer M. (1982) Chem. Phys. Lipids 31, 75–85 [DOI] [PubMed] [Google Scholar]
- 40. Parente R. A., Lentz B. R. (1984) Biochemistry 23, 2353–2362 [DOI] [PubMed] [Google Scholar]
- 41. Morrisett J. D., David J. S., Pownall H. J., Gotto A. M., Jr. (1973) Biochemistry 12, 1290–1299 [DOI] [PubMed] [Google Scholar]
- 42. Greenfield N., Fasman G. D. (1969) Biochemistry 8, 4108–4116 [DOI] [PubMed] [Google Scholar]
- 43. Tamm L. K. (1991) Biochim. Biophys. Acta 1071, 123–148 [DOI] [PubMed] [Google Scholar]
- 44. Pfefferkorn C. M., Lee J. C. (2010) J. Phys. Chem. B 114, 4615–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Manning M. C., Woody R. W. (1991) Biopolymers 31, 569–586 [DOI] [PubMed] [Google Scholar]
- 46. Drescher M., van Rooijen B. D., Veldhuis G., Subramaniam V., Huber M. (2010) J. Am. Chem. Soc. 132, 4080–4082 [DOI] [PubMed] [Google Scholar]
- 47. Ulmer T. S., Bax A. (2005) J. Biol. Chem. 280, 43179–43187 [DOI] [PubMed] [Google Scholar]
- 48. Takamori S., Holt M., Stenius K., Lemke E. A., Grønborg M., Riedel D., Urlaub H., Schenck S., Brügger B., Ringler P., Müller S. A., Rammner B., Gräter F., Hub J. S., De Groot B. L., Mieskes G., Moriyama Y., Klingauf J., Grubmüller H., Heuser J., Wieland F., Jahn R. (2006) Cell 127, 831–846 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.