

Abstract
Expression of A20, a negative regulator of the NF-κB pathway, is frequently lost in several subtypes of Hodgkin and non-Hodgkin lymphoma. We report that A20 is expressed in Kaposi sarcoma-associated herpesvirus (KSHV)-infected primary effusion lymphoma cell lines, and its expression correlates closely with the expression of KSHV-encoded viral FLICE inhibitory protein K13. Ectopic expression of K13 induced A20 expression through NF-κB-mediated activation of A20 promoter. In turn, A20 blocked K13-induced NF-κB activity and up-regulation of proinflammatory cytokines CCL20 and IL-8 in a negative feedback fashion. Both the N-terminal deubiquitinating domain and the C-terminal zinc finger domain of A20 were involved in the inhibition of K13-induced NF-κB activity. Overexpression of A20 blocked K13-induced IκBα phosphorylation, NF-κB nuclear translocation, and cellular transformation. Consistent with the above, K13-induced IκBα phosphorylation and NF-κB transcriptional activation were enhanced in A20-deficient cells. Finally, A20 was found to interact physically with K13. Taken collectively, these results demonstrate that K13 is a key determinant of A20 expression in KSHV-infected cells, and A20 is a key negative regulator of K13-induced NF-κB activity. A20 might serve to control the inflammatory response to KSHV infection and protect KSHV-infected cells from apoptosis.
Keywords: Cytokine, Herpesvirus, NF-κB, Signal Transduction, Tumor Viruses, A20, KSHV, PEL, vFLIP
Introduction
The NF-κB2 pathway induces the expression of a number of genes involved in mediation of the inflammatory response, and attenuating NF-κB signaling is essential for modulating inflammatory reactions and maintaining homeostatic balance (1). As such, identification of genes that control NF-κB activation has been the focus of intense research. A20 gene (also known as tumor necrosis factor α-induced protein 3 or TNFAIP3) was first identified 20 years ago as a gene that is rapidly induced in human umbilical vein endothelial cells upon treatment with proinflammatory cytokines, such as TNFα and IL-1 (2). Subsequent studies reported that A20 is also induced by other stimuli, including phorbol esters, signaling via CD40 and TRAIL receptors, and activation of B cell surface receptor (3). In addition, Epstein-Barr virus (EBV)-encoded latent membrane protein 1 (LMP1) and human T cell lymphotropic virus 1 Tax protein are known to induce A20 expression (4, 5). Overexpression of A20 was shown to block NF-κB activation triggered by various stimuli, including TNF, IL-1, and lipopolysaccharide (LPS) (6, 7). Consistent with the above role, A20-deficient cells demonstrate prolonged NF-κB activation in response to TNFα, and mice deficient in A20 develop severe inflammation, show hypersensitivity to LPS and TNFα, and die early (8). Collectively, these studies have established A20 as a physiological regulator of NF-κB activation.
Kaposi sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8, is an oncogenic γ2 herpesvirus that is the etiological agent of Kaposi sarcoma (9) and has been associated with the pathogenesis of primary effusion lymphoma (PEL) and some aggressive forms of multicentric Castleman disease (10).
One of the KSHV proteins that is expressed in all cells latently infected with KSHV is K13 (11). Based on its structural homology to the prodomain of caspase 8/FLICE (12), K13 was originally classified as a viral FLICE inhibitory protein (vFLIP) (12). However, subsequent work showed that K13 is not an inhibitor of caspase 8/FLICE but is a potent activator of the NF-κB pathway and uses this pathway to promote cellular proliferation, survival, transformation, cytokine secretion, and KSHV latency (13–20). Ectopic expression of K13 in human vascular endothelial cells transforms them into spindle cells, reminiscent of the spindle cells found in the KS lesions, which is accompanied by increased expression of proinflammatory cytokines and chemokines known to be involved in the pathogenesis of KS lesions (18, 20). Because A20 is a key regulator of inflammatory response, in this study, we have examined the effect of K13 on A20 expression. Our results demonstrate that A20 expression is up-regulated by K13, and in turn A20 blocks K13-induced NF-κB activity.
MATERIALS AND METHODS
Cell Lines and Reagents
293T, BC1, BC3, BCBL1 cells were obtained from American Type Culture Collection (Manassas, VA), and APK1 cells were obtained from Dr. Jae Jung (University of Southern California). 293T and BCBL1 cells expressing an empty vector and K13 have been described previously (21, 22). A20+/+ and A20−/− mouse embryo fibroblast (MEF) cells were generously provided by Dr. Averil Ma (University of California, San Francisco, CA). NF-κB inhibitors Bay-11-7082 and PS-1145 were purchased from Calbiochem, and arsenic trioxide (As2O3) was from Sigma-Aldrich.
Plasmids
Plasmids encoding K13, E8, Tax, phosphorylation-resistant mutants of IκBα, kinase-defective mutants of IKK1/IKK2 and 4-hydroxytamoxifen (4OHT)-inducible K13-ERTAM have been described previously (14–15, 18, 23, 24). The A20 WT-Luc, A20 mNF-κB-Luc, and A20 m1-Luc luciferase reporter constructs were kind gifts from Dr. Dikstein (The Weizmann Institute of Science, Rehovot, Israel) (25). The wild-type and mutant A20 constructs were kindly provided by Dr. Lin Rongtuan (McGill University, Montreal, QC, Canada) (26). Recombinant retroviruses were generated and used to infect 293T, BCBL1, and MEF cells essentially as described previously (27).
Luciferase Reporter
The 293T cells were transfected in a 24-well plate with various test plasmids (250 ng/well) along with the WT or mutant A20 luciferase reporter constructs (75 ng/well) and a pRSV/LacZ (β-galactosidase) reporter construct (75 ng/well) using calcium phosphate as described previously (24). Cells were lysed 24–36 h later, and extracts were used for the measurement of firefly luciferase and β-galactosidase activities, respectively. Luciferase activity was normalized relative to the β-galactosidase activity to control for the difference in the transfection efficiency.
Western Blotting
Western blot analysis was performed essentially as described previously (28). Primary antibodies used in these experiments were: p65 (Santa Cruz Biotechnology), NEMO (Santa Cruz Biotechnology), IKKα/β (Santa Cruz Biotechnology), A20 (Cell Signaling), p-IκBα (Cell Signaling), total IκBα (Santa Cruz Biotechnology), tubulin (mouse monoclonal; Sigma), and mouse monoclonal M2 FLAG (Sigma). A mouse monoclonal antibody against K13 (8F6) was raised in our laboratory. FLAG, control mouse IgG, and goat anti-mouse IgG beads were obtained from Sigma and used in co-immunoprecipitation studies as described previously (28).
NF-κB DNA Binding Assay
The DNA binding activity of the p65 subunit was measured in triplicate in the nuclear extracts using an enzyme-linked immunosorbent assay (ELISA)-based NF-κB DNA binding assay, as described previously (21).
ELISA
Human CCL20 and IL-8 were measured in the cell culture supernatant using ELISA kits from R&D Systems (Minneapolis, MN) and BD Pharmingen, respectively. The ELISA was performed following the recommendations of the manufacturer.
Soft Agar Assay
Soft agar assay was performed essentially as described previously (14). Briefly, Rat1 K13 cells expressing an empty vector or A20 were overlaid as a single cell suspension of 1000 cells in 1.0 ml of 0.4% Bacto-agar onto a 3.5-cm tissue culture dish containing a 0.6% agar base. All agar media were made with Dulbecco's modified Eagle's medium supplemented with 10% serum, penicillin/streptomycin. Triplicate plates were prepared for each tested cell line and inspected for colony formation after incubation at 37 °C for 14 days.
RNA Interference (RNAi)
siRNA oligonucleotides against A20 were purchased from Santa Cruz Biotechnology. p65 siRNA has been described previously (18). siRNA oligonucleotides were transfected using Neon transfection system (Invitrogen) and following the recommendations of the manufacturer. Following transfection, cells were immediately transferred to a 24-well plate containing prewarmed antibiotic-free medium and incubated at 37 °C. Cells were first examined for knock-down of A20 expression by Western blotting, and then the lysates were assayed for NF-κB luciferase reporter activity.
Statistical Analyses
A two-tailed paired Student's t test was used to test for differences between two groups. Differences with a p ≤ 0.05 were considered as statistically significant. All experiments were repeated a minimum of three times with triplicate samples.
RESULTS
A20 Expression in PEL Cell Lines Correlates with Their Level of Endogenous K13 Expression
A20 is a tumor suppressor, and recent studies have demonstrated a loss of its expression in several subtypes of Hodgkin and non-Hodgkin lymphoma (3). To determine whether A20 expression is also lost in PEL, we used Western blotting to examine its expression in four PEL cell lines, BC1, BC3, BCBL1, and APK1. The BC1 cells are dually infected with KSHV and EBV, whereas the BC3, BCBL1 and APK1 cells are infected with KSHV only. As shown in Fig. 1A, we observed robust expression of A20 in BC1 and BC3 and low level expression in BCBL1 and APK1 cell lines. Immunoblotting using a K13 monoclonal antibody revealed that expression of A20 in the four PEL cell lines correlated closely with their level of endogenous K13 expression, implying a causal association (Fig. 1A). To test this hypothesis further, we took advantage of BCBL1 cells that had been engineered to express FLAG-tagged K13 ectopically by retroviral-mediated gene transfer (21). Consistent with the notion that K13 up-regulates A20 expression, the BCBL1-K13 cells demonstrated a marked increase in A20 expression compared with the BCBL1-vector cells (Fig. 1B). Induction of A20 expression by ectopic K13 expression, however, was not limited to BCBL1 cells but was also observed in the 293T cell line (Fig. 1B). Taken collectively, the above results demonstrate that K13 is a key determinant of A20 expression in PEL.
FIGURE 1.
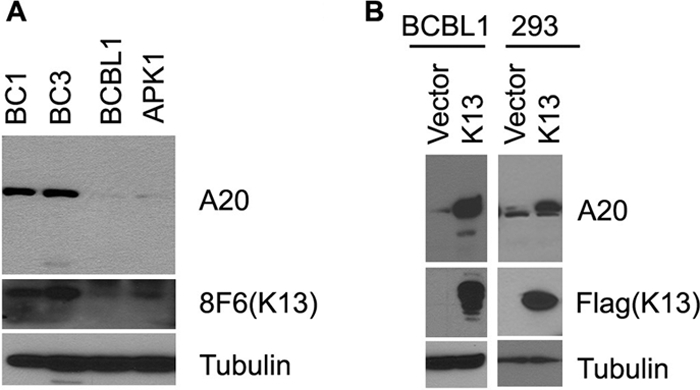
K13 up-regulates A20 expression in PEL cell lines. A, Western blots showing expression of A20 and K13 in PEL cell lines. B, Western blots showing up-regulation of A20 in BCBL1 and 293 cells by ectopic K13 expression. Stable pools of BCBL1 and 293 cells expressing an empty vector and FLAG-K13 were generated by retroviral gene transfer. Cell lysates from these cells were used for Western blot analysis. Tubulin blot shows equal protein loading (bottom panel).
K13 Activates A20 Promoter Activity
To determine the mechanism of A20 up-regulation by K13, we examined the effect of ectopic K13 expression on the transcriptional activity of a luciferase reporter construct (A20-Luc) driven by the A20 promoter region (25). We transfected the A20-Luc reporter plasmid into 293T cells along with an empty vector or an expression plasmid encoding K13 and measured the luciferase activity in the cell lysates 28–30 h after transfection. As shown in Fig. 2A, transient expression of K13 resulted in a >8-fold increase in A20 luciferase reporter activity compared with transfection with an empty vector, suggesting that K13 up-regulates A20 expression at the transcriptional level.
FIGURE 2.

K13 activates A20 promote through NF-κB pathway. A and B, the 293T cells were transfected with a control vector and a vector encoding FLAG-K13 (250 ng/ml) along with either A20-Luc or A20 mNF-κB-Luc or A20 m1-Luc reporter construct (75 ng/well) and a pRSV/LacZ (β-galactosidase) reporter construct (75 ng/well), and the reporter assay was performed as described under “Materials and Methods.” The values shown are averages (mean ± S.E.) of one representative experiment of three in which each transfection was performed in duplicate. Expression of the K13 was confirmed by Western blot analysis with an antibody against the FLAG epitope tag. C, K13-ERTAM induces A20 promoter activity upon treatment with 4OHT. The experiment was performed essentially as described in A. The values shown are averages (mean ± S.E.) of one representative experiment ofthree in which each transfection was performed in duplicate. D, wild-type K13 but not vFLIP E8- or NF-κB-defective mutants of K13 induces A20 promoter activity. 293T cells were transfected with the indicated FLAG-tagged expression constructs along with a A20 promoter luciferase construct. The experiment was performed as described for A. The values shown are averages (mean ± S.E.) of one representative experiment of three in which each transfection was performed in duplicate. Lower panel shows the expression of the transfected proteins as dermined by Western blotting with a FLAG antibody. E, dominant negative mutants of IκBα (IκBαΔN and IκBαS32A/S36A), IKK1 and IKK2 block K13-induced A20 promoter activity. The 293T cells were transfected either with an empty vector or K13, along with an A20 luciferase reporter construct and a reporter construct, as described in A. The amount of inhibitor plasmids (500 ng/well) was five times the amount of vector or K13 (100 ng/well) plasmid, and the total amount of transfected DNA was kept constant by adding empty vector. The values shown are averages (mean ± S.E.) of one representative experiment of three in which each transfection was performed in duplicate. Expression of the K13 was confirmed by Western blot analysis with the FLAG antibody. Asterisks indicate significance at levels of p ≤ 0.05. F, pharmacologic inhibitors of NF-κB block K13-induced A20 promoter activation. The 293T cells were transfected with an empty vector or a vector encoding K13 along with A20-Luc and pRSV/LacZ reporter constructs. Approximately 3 h after transfection, cells were treated with dimethyl sulfoxide (vehicle) or the indicated compounds for 18 h before cell lysis and measurement of reporter activities. Asterisks indicate significance at levels of p ≤ 0.05 compared with vehicle-treated cells.
The NF-κB Sites at −54 and −66 in the A20 Promoter Are Essential for K13-mediated Activation
The A20 promoter region contains two NF-κB sites residing at −54 and −66 nucleotides from the putative transcription start site which have been shown to play a key role in response to TNFα stimulation (25, 29). In addition, the A20 promoter has a negative upstream promoter element called ELIE (Elongation Inhibitory Element) (30). To examine the involvement of these NF-κB and ELIE sites in K13-induced A20 promoter activation, we transfected 293T cells with either a luciferase reporter construct containing the wild-type A20 promoter (A20-WT-Luc) or reporter constructs bearing mutations in the NF-κB (A20-mNF-κB-Luc) and ELIE sites (A20-m1-Luc), respectively. As shown in Fig. 2B, K13-induced A20 promoter activity was markedly attenuated in the A20-mNF-κB-Luc construct. On the other hand, mutation of the ELIE site had no effect on K13-induced A20 promoter activation. The role of the NF-κB sites in K13-induced A20 promoter activation was confirmed by an independent experiment involving expression of a K13-ERTAM fusion protein in which K13 activity is controlled at the post-translational level by fusion with a mutated estrogen receptor (18). The mutated estrogen receptor (ERTAM) in this construct does not bind to the physiological ligand estrogen but binds with very high affinity to the synthetic ligand 4OHT and regulates the activity of K13 in a 4OHT-dependent fashion (18). Consistent with the previous results, 4OHT treatment strongly activated the luciferase activity in cells transfected with K13-ERTAM and A20-WT-Luc construct but not in those transfected with K13-ERTAM and A20-mNF-κB-Luc (Fig. 2C). Collectively, these results demonstrate the importance of the NF-κB sites in K13-mediated A20 gene expression.
K13 Stimulates A20 Promoter Activity via NF-κB Activation
To confirm the role of the NF-κB pathway in K13-induced A20 promoter activation, we used three previously described point mutants of K13, K13-58AAA (E58A/C59A/L60A), K13-65AAA (R65A/R66A/D67A), and K13-67AAA (D67A/L68A/L69A), which are defective in NF-κB activation (14) Consistent with their loss of NF-κB activating ability, these mutants failed to activate the A20-Luc construct (Fig. 2D). Furthermore, no A20 promoter activation was observed upon co-expression of the vFLIP E8 from the equine herpesvirus 2, which resembles K13 in structure but lacks the ability to activate the NF-κB pathway (24, 31) (Fig. 2D). On the other hand, co-expression of human T cell leukemia-encoded Tax protein, which resembles K13 in activating the NF-κB pathway, led to A20 promoter activation (Fig. 2D). Collectively, these results indicate that K13-induced NF-κB activity is likely involved in A20 promoter activation.
Abrogation of K13-induced A20 Promoter Activation by Genetic and Pharmacological Inhibitors of the NF-κB Pathway
K13 activates the NF-κB pathway by activating a ∼700-kDa IKK complex consisting of IKK1/IKKα, IKK2/IKKβ, and NEMO/IKKγ which phosphorylates IκBα, targeting it for ubiquitination and subsequent degradation via the proteasome (28). We examined the effect of dominant negative mutants of IκBα, IKK1/IKKα, and IKK2/IKKβ on K13-induced A20 promoter activity. As shown in Fig. 2E, K13-induced A20 promoter activity was completely blocked by a phosphorylation-resistant mutant of IκBα in which the two critical serine residues have been mutated to alanine (IκBα S32A/S36A), and a deletion mutant of IκBα lacking the N-terminal 36 amino acids (IκBαΔN). Similarly, kinase-dead mutants of IKK1 and IKK2 effectively blocked K13-induced A20 promoter activity (Fig. 2E).
We next examined whether K13-induced A20 promoter activity could be antagonized by pharmacologic inhibitors of the NF-κB pathway. We have previously reported that Bay-11-7082, a specific inhibitor of the NF-κB, can block K13-induced NF-κB activity and spindle cell transformation of human vascular endothelial cells (18). Consistent with these results, treatment with Bay-11-7082 also blocked K13-induced A20 promoter activity in 293T cells (Fig. 2F). Because K13 activates the NF-κB pathway by activating the IKK complex (24, 28), we examined the ability of a specific inhibitor of this complex, PS1145 (32), to block K13-induced A20 promoter activity. As shown in Fig. 2F, this inhibitor effectively blocked K13-transactivated A20 promoter activity. Arsenic trioxide, another known inhibitor of K13-induced NF-κB activation (33), also blocked K13-induced A20 promoter activity (Fig. 2F). Taken collectively, these results confirm the importance of the IKK complex and the NF-κB pathway in K13-induced A20 promoter activation.
Inhibition of K13-induced NF-κB Activation by Overexpression of A20
A20 is a key player in the negative feedback regulation of NF-κB signaling in response to multiple stimuli, including TNFα and IL-1β (34). In contrast, NF-κB activation by human T cell lymphotropic virus 1 Tax protein, which resembles K13 in activating NF-κB by directly binding to NEMO, has been reported to be resistant to A20 (7). Therefore, we next examined the effect of A20 on K13- and Tax-induced NF-κB activity. For this purpose, we transfected 293T cells with an NF-κB driven luciferase-based reporter construct along with K13 and Tax in the presence or absence of A20 and ∼24 h after transfection examined the induction NF-κB promoter activity by a luciferase assay. A20 effectively blocked K13-induced NF-κB activity but had only a minor effect of Tax induced NF-κB activity (Fig. 3, A and B). Furthermore, A20 co-expression led to near complete inhibition of NF-κB reporter activity in 293T cells that had been transfected with the K13-ERTAM construct and treated with 4OHT (Fig. 3C). A20 contains two ubiquitin-editing domains, an N-terminal deubiquitinating domain of the OTU family, and a C-terminal ubiquitin ligase domain that contains seven zinc fingers (3). Both these domains have been shown to play a role in inhibiting NF-κB activity. To determine the role of these domains in K13-induced NF-κB activity, we tested the ability of A20 deletion mutants containing its N-terminal (1–380) and C-terminal (373–790) domains to block K13-induced NF-κB activity. We observed that both domains of A20 contributed to inhibition of K13-induced NF-κB activity (Fig. 3D). Essentially similar results were obtained when the experiment was repeated using the K13-ERTAM construct (Fig. 3E).
FIGURE 3.

A20 inhibits K13-induced NF-κB activity. A and B, 293T cells were transfected with control vector, K13, or Tax along with an NF-κB-Luc construct (75 ng/well) and a pRSV/LacZ (β-galactosidase) reporter construct (75 ng/well) in the presence or absence of A20. Cells were lysed 20 h after transfection for reporter assays. The values shown are the averages (mean ± S.E.) of one representative experiment of three in which each transfection was performed in duplicate. The expression of the transfected A20 and FLAG-tagged K13 and Tax proteins was demonstrated by immunoblotting (lower panel). C, A20 inhibits K13-ERTAM-induced NF-κB activity. The experiment was performed essentially as described in 2A. The values shown are averages (mean ± S.E.) of one representative experiment of three in which each transfection was performed in duplicate. D and E, 293T cells were transfected with K13 or K13-ERTAM along with NF-κB-Luc and pRSV/LacZ reporter constructs in the presence or absence of the indicated A20 constructs. The cells transfected with K13-ERTAM were treated with 4OHT (20 nm) for 24 h prior to cell lysis and measurement of reporter activities. The values shown are the averages (mean ± S.E.) of one representative experiment of three in which each transfection was performed in duplicate. Lower panel shows the expression of the transfected proteins as determined by Western blotting with a FLAG antibody. Asterisks indicate statistical significance at levels of p ≤ 0.05.
A20 Blocks K13-induced CCL20 and IL-8 Production
We and others have shown that K13 induces the expression of a large number of proinflammatory chemokines and cytokines that have been implicated in the pathogenesis of Kaposi sarcoma (15, 18–19, 35–37). Because A20 is known to block the inflammatory response, we examined the effect of A20 on K13-induced promoter activation of two inflammatory chemokines, CCL20 and IL-8. As shown in Fig. 4, A–D, luciferase-based reporter assays showed that A20 blocks the induction of CCL20 and IL-8 by K13 by blocking the transcriptional activation of their promoters. Furthermore, co-expression of A20 blocked the induction of both these chemokines by K13 as measured by ELISA (Fig. 4, E and F).
FIGURE 4.

A20 blocks K13-induced up-regulation of CCL20 and IL-8 and cellular transformation. A–D, A20 blocks K13-induced up-regulation of CCL20 and IL-8. 293T cells were transfected with an empty vector, K13, or K13-ERTAM along with luciferase reporter constructs driven by CCL20 (CCL20-Luc) or IL-8 promoters (IL-8-Luc) (75 ng/well) and a pRSV/LacZ (β-galactosidase) reporter construct (75 ng/well) in the presence or absence of A20. The cells transfected with K13-ERTAM were subsequently treated with 4OHT (20 nm) for 24 h and the luciferase reporter assay performed essentially as described in Fig. 2A. Lower panels demonstrate the expression of the transfected proteins as measured by immunoblotting with the indicated antibodies. The values shown are the averages (mean ± S.E.) of one representative experiment of three in which each transfection was performed in duplicate. E and F, cellular supernatants from 293T cells transfected, as explained in A–D, were collected before doing luciferase assay and used to measure the secretion of CCL20 and IL-8 by ELISA. The values shown are averages (mean ± S.E.) of one representative experiment of three in which the level of CCL20 or IL-8 secretion was measured in duplicate. Asterisks (*) indicate significance at levels of p ≤ 0.05. G, immunoblot shows increased A20 expression in Rat1-K13 cells transfected with an A20 expression construct. H, soft agar colony formation by Rat1-K13 cells transfected with an A20 expression vector was reduced compared with those transfected with an empty vector. The values shown are mean ± S.D. of two independent experiments performed in triplicate. *, p ≤ 0.05 versus vector cells.
A20 Blocks K13-induced Cellular Transformation
Anchorage-independent growth is a characteristic feature of transformed cells. We have demonstrated previously that ectopic expression of K13 in Rat1 fibroblast cells confers on them the ability to grow in an anchorage-independent manner and form colonies in soft agar. To examine the effect of A20 on K13-induced cellular transformation, we generated stable populations of Rat1-K13 cells expressing an empty vector and A20 (Fig. 4G). We next used a soft agar colony formation assay to test the effect of A20 on K13-induced cellular transformation. As shown in Fig. 4H, we observed greater than 4-fold reduction in soft agar colony formation in the Rat1-K13 cells transfected with the A20 expression vector compared with those transfected with the control vector.
A20 Blocks K13-induced NF-κB Activity by Blocking p65 Nuclear Translocation and IκBα Phosphorylation
The mechanism by which A20 blocks NF-κB is not entirely clear, and contradictory results have been reported on the effects of A20 on NF-κB nuclear translocation and DNA binding. For example, it has been reported that A20 blocks the TNF-induced NF-κB luciferase activity in HEK293 cells without blocking NF-κB nuclear localization or DNA binding (38). To understand the mechanism by which A20 blocks K13-induced NF-κB, we examined its effect on K13-induced nuclear translocation of p65/RelA subunit of NF-κB. For this purpose, we transfected 293T cells with an EGFP-p65 fusion construct in the presence and absence of co-transfected K13 and A20 and examined the cellular localization of EGFP-p65 by fluorescence microscopy. Consistent with the fact that under basal conditions p65 is retained in the cytosol by its interaction with IκBα, the cells transfected with the EGFP-p65 construct alone primarily showed cytoplasmic localization of EGFP-p65 (Fig. 5A). On the other hand, EGFP-p65 was localized in the nucleus in a significant proportion of cells cotransfected with K13 (Fig. 5A). However, K13-induced nuclear localization of EGFP-p65 was markedly reduced upon coexpression of A20 (Fig. 5A).
FIGURE 5.

A20 blocks K13-induced NF-κB activation by blocking IκBα phosphorylation and p65 nuclear translocation. A, 293T cells were transfected with an EGFP-p65 fusion construct along with the indicated expression constructs, and cellular localization of EGFP-p65 was examined by fluorescence microscopy. K13-induced nuclear accumulation of EGFP-p65 is blocked by co-expression of A20. B, 293T cells were transfected with K13 in the presence or absence of A20, and 24 h after transfection, total cell lysates were prepared and used to examine the effect of A20 on K13-induced phosphorylation of IκBα. The expression of total IκBα is shown in the lower panel. C, immunoblot shows increase in phosphorylation of IκBα in K13-ERTAM-expressing A20−/− MEFs compared with the corresponding A20+/+ MEFs. A20+/+ and A20−/− MEFs expressing FLAG-K13-ERTAM were treated with 4OHT for the indicated time points, and the phosphorylation of IκBα and A20 expression was measured using the indicated antibodies. Equivalent expression of the FLAG-tagged K13-ERTAM protein and equal protein loading was confirmed by blotting with FLAG and tubulin antibodies, respectively. D, A20+/+ and A20−/− MEFs expressing FLAG-K13-ERTAM were transfected with NF-κB promoter luciferase construct (75 ng/well) and a Renilla reporter construct (75 ng/well, normalization control) using Lipofectamine-mediated transfection as described previously (33). The transfected cells were subsequently treated with 4OHT (20 nm) for 48 h, and the luciferase reporter assay was performed essentially as described in Fig. 2A. The values shown are the averages (mean ± S.E.) of one representative experiment of three in which each transfection was performed in duplicate. Asterisks (*) indicate significance at levels of p ≤ 0.05. E, upper, BC1-NFκB-Luc cells were transfected with a control siRNA (lamin A/C) or siRNAs directed against A20 or p65, respectively. Approximately 72 h after transfection, cell lysates were prepared and used for the measurement of luciferase activity. The data represent mean ± S.E. of a representative of two independent experiments with similar results. *, p ≤ 0.05 versus control cells. E, lower, immunoblot confirms specific knock-down of A20 and p65 proteins following siRNA-mediated gene silencing.
K13 activates NF-κB by inducing phosphorylation of IκB which results in its degradation by the ubiquitin-proteasome pathway, allowing the NF-κB subunits to enter the nucleus (28). To examine the mechanism by which A20 blocks K13-induced nuclear translocation of p65/RelA, we examined the phosphorylation status of IκBα in 293T cells that have been transfected with K13 in the presence or absence of A20. As shown in Fig. 5B, transfection of K13-induced significant phosphorylation of IκBα compared with empty vector-transfected cells. However, phosphorylation of IκBα was markedly reduced in cells in which K13 was co-transfected with A20 (Fig. 5B).
K13-induced NF-κB Activity Is Enhanced in A20−/− MEFs
To confirm the physiological relevance of the above studies, we generated stable pools of A20+/+ and A20−/− MEFs expressing FLAG-K13-ERTAM and confirmed equivalent expression of the transduced protein by Western blotting (Fig. 5C). Consistent with our previous results, treatment of A20+/+K13-ERTAM cells with 4OHT resulted in up-regulation of A20 expression, which was evident as early as 6 h after treatment and continued to increase up to 12 h after treatment (Fig. 5C). More importantly, 4OHT-induced phosphorylation of IκBα was substantially increased in the A20−/−K13-ERTAM MEFs compared with the A20+/+ K13-ERTAM MEFs (Fig. 5C). Furthermore, 4OHT-induced a greater increase in NF-κB luciferase activity in A20−/− K13-ERTAM cells compared with A20+/+ K13-ERTAM cells, as measured by the luciferase-based reporter assay (Fig. 5D). Collectively, these results demonstrate that A20 negatively regulates K13-induced NF-κB activity by blocking phosphorylation of IκBα.
A20 Silencing Enhances NF-κB Activity in BC1 PEL Cells
To demonstrate that A20 also controls NF-κB activity in PEL cells, we took advantage of BC1 cells that are naturally infected with KSHV and express high levels of K13 endogenously (Fig. 1A). We generated stable populations of these cells expressing an NF-κB-driven luciferase reporter construct using lentiviral-mediated gene transfer. We next used siRNA-mediated gene silencing to study the effect of A20 depletion on the NF-κB-Luc activity in the BC1-NF-κB-Luc cells. Lamin A/C and p65 siRNAs were used as negative and positive controls, respectively. As shown in Fig. 5E, we observed increased NF-κB-Luc activity in the A20-depleted cells compared with those depleted of lamin A/C, whereas reduced NF-κB-Luc activity was observed in the p65-depleted cells. These results demonstrate that A20 negatively regulates NF-κB activity in the KSHV-infected PEL cells.
A20 Interacts with K13
To examine the mechanism by which A20 blocks K13-induced IκBα phosphorylation, we used a co-immunoprecipitation assay to examine the interaction between the two proteins. We immunoprecipitated FLAG-K13 from K13-expressing BC3 cells using FLAG or control antibody beads and examined the presence of endogenously expressed A20 in the immunoprecipitated samples by Western blotting. As shown in Fig. 6A, A20 was readily detected in the samples immunoprecipitated with the FLAG antibody. A strong interaction between K13 and NEMO was also observed (Fig. 6A). To confirm the results, we repeated the experiment in the K13-expressing A20+/+ and A20−/− MEFs. A band corresponding to A20 was readily detected in the samples immunoprecipitated with the FLAG antibody from K13-expressing A20+/+ cells but not the A20−/− cells (Fig. 6B).
FIGURE 6.

K13 interacts with A20. A, cell lysates (C.L.) prepared from BC3 cells expressing an empty vector or FLAG-tagged K13 were immunoprecipitated (I.P.) using control (C) or FLAG (F) beads, and the presence of interacting A20 and NEMO proteins was detected by Western blotting with the indicated antibodies. B, co-immunoprecipitation assay shows interaction of K13 with A20 in A20+/+ MEFs. The experiment was performed essentially as described for A. A tubulin blot shows equal protein loading. C, cell lysates (C.L.) prepared from BC1 and BC3 cells were immunoprecipitated using control antibody or 8F6 antibody raised against K13, and the presence of interacting A20 and IKK α/β proteins was detected by Western blotting with the indicated antibodies. D, A20 does not block the interaction between K13 and IKK complex subunits. 293T cells were transfected with an empty vector or a vector expressing FLAG-tagged K13, either alone or with A20, and a co-immunoprecipitation assay was performed essentially as described for A. A tubulin blot shows equal protein loading.
To confirm that KSHV-derived endogenous K13 also interacts with endogenously expressed A20, we immunoprecipitated K13 from BC1 and BC3 cells using 8F6, a monoclonal antibody against K13, and examined its interaction with A20 and IKKα/β (positive control) by Western blotting. As shown in Fig. 6C, A20 and IKKα/β were readily detected in the samples immunoprecipitated with the 8F6 mAb but not in those immunoprecipitated with a control mAb.
Finally, we examined whether the K13-A20 interaction will interfere with the known interaction of K13 with the IKK complex. For this purpose, we transfected 293T cells with FLAG-tagged K13, either alone or in combination with A20, and studied the effect of A20 on the interaction between K13 and endogenously expressed IKK complex subunits by co-immunoprecipitation assay. As shown in Fig. 6D, co-expression of A20 did not impair the interaction between K13 and IKK complex subunits. These results demonstrate that A20 blocks K13-induced NF-κB without physically disrupting the K13-IKK complex.
DISCUSSION
A20 is known to act as a tumor suppressor in several subtypes of non-Hodgkin and Hodgkin lymphomas, including marginal zone lymphoma, diffuse large B cell lymphoma, mantle cell lymphoma, mucosa-associated lymphoid tissue lymphoma, primary mediastinal lymphoma, Burkitt lymphoma, natural killer cell lymphoma, adult T cell leukemia/lymphoma, and classical Hodgkin lymphoma (3, 39–41). A20 expression is lost in a substantial number of these lymphomas by gene deletion, promoter methylation, frameshift mutations, and/or point mutations (3). Because A20 is a negative regulator of NF-κB activity, its inactivation is believed to contribute to development of these lymphomas by promoting unchecked NF-κB activity and enhancing cell survival (3). However, KSHV-infected PEL cells are known to possess an alternative mechanism for activating the NF-κB pathway, namely the expression of KSHV-encoded K13 protein which is a potent activator of the IKK complex (28, 42). Consistent with their lack of requirement for A20 inactivation to activate NF-κB, we observed A20 expression, albeit of varying magnitude, in all four PEL-derived cell lines tested in the current study, which correlated closely with their level of K13 expression. Interestingly, similar to K13, EBV-encoded LMP1 results in constitutive NF-κB activation (43), and a recent study revealed that A20 mutation frequency and mutation patterns in classical Hodgkin lymphoma differed significantly according to their EBV status (40). Thus, whereas A20 genetic alterations were relatively rare among 16 EBV-positive classical Hodgkin lymphomas, they were quite frequent among the EBV-negative classical Hodgkin lymphomas and frequently involved both copies of the TNFAIP3 gene (40). Taken collectively with this study, our results suggest that A20 inactivation is not a common feature of KSHV and EBV-associated lymphomas due, in part, to the constitutive NF-κB activation induced by the K13 and LMP1 proteins.
In this report we demonstrate that K13 up-regulates A20 expression via NF-κB activation. The involvement of K13-induced NF-κB pathway in up-regulating A20 expression is supported by the following lines of evidence. First, the expression of A20 correlated closely with their level of K13 expression and their previously reported level of NF-κB activity (21). Thus, the BC1 cell line, which possesses high level K13 expression and constitutive NF-κB activity (21, 28, 42), showed high A20 expression, whereas the BCBL1 cell line, which has low K13 expression and NF-κB activity (21, 28, 42), showed weak A20 expression. Furthermore, A20 expression was up-regulated in the BCBL1-K13 cells, which possess increased NF-κB activity (21). Second, we observed a strong correlation between the ability of vFLIPs to activate NF-κB and their ability to activate A20 promoter. Thus, whereas the wild-type K13 strongly activated A20 promoter, its NF-κB-defective mutants failed to do so. Similarly, no A20 promoter activation was observed upon expression of the vFLIP E8 that lacks NF-κB activity (24). Third, K13 failed to activate an A20 promoter construct containing mutations in the two NF-κB sites. Finally, K13-induced A20 promoter activity was blocked by genetic (i.e. IκBα S32A/S36A, ΔNIκBα, IKK1-KM, and IKK2-KM) and pharmacological (i.e. Bay-11-7082, PS1145, and arsenic trioxide) inhibitors of the NF-κB pathway.
We observed that A20 overexpression effectively blocked K13-induced NF-κB activity, induction of NF-κB-responsive genes, such as CCL20 and IL-8, and K13-induced cellular transformation. The physiological relevance of these studies was confirmed by results showing that K13-induced IκBα phosphorylation and NF-κB transcriptional activity are increased in A20−/− cells. The N-terminal region of A20 contains a deubiquitinating domain of the OTU family that deubiquitinates Lys-63-polyubiquitinated proteins, whereas the C-terminal region contains 7 Zn fingers that catalyze the addition of Lys-48-linked polyubiquitin chains on the target proteins, targeting them for proteasomal degradation (3, 44). A recent study showed that A20, together with the regulatory molecule TAX1BP1, interacted with E2 ubiquitin-conjugating enzymes Ubc13 and UbcH5c, triggering their ubiquitination and proteasome-dependent degradation, which in turn blocked IL-1β-induced NF-κB by blocking Lys-63-linked polyubiquitination (45). Both the OTU and zinc finger 4 domains of A20 were found to be important for binding to Ubc13 and UbcH5c and for inhibition of IL-1β-induced NF-κB by A20 (45). In the present study, we discovered that both the N-terminal and C-terminal domains of A20 are involved in inhibition of K13-induced NF-κB activity. Therefore, it is conceivable that similar to its inhibition of IL-1β signaling, A20 blocks K13-induced NF-κB by inducing degradation of Ubc13 and UbcH5c. We also observed that although A20 physically interacted with K13, it did not block K13-IKK interaction, thereby arguing against the possibility that A20 blocks K13-induced NF-κB by interfering with the assembly of the K13-IKK complex. Finally, we have observed that co-expression of A20 and K13 sometimes results in the appearance of their high molecular weight products upon immunoblotting (e.g. Fig. 6, A and B). Studies are currently in progress to examine whether these slow migrating products of A20 and K13 represent their polyubiquitinated forms and their role in the regulation of K13-induced NF-κB activity.
Similar to K13, human T cell lymphotropic virus 1 Tax activates NF-κB by directly binding to NEMO (45). However, Tax-induced NF-κB has been reported to be relatively resistant to A20 (7, 45), a finding confirmed in the current study. Tax is believed to circumvent A20 inhibition by binding to Tax1BP1 and disrupting the interaction among A20, Tax1BP1, and Ubc13, thereby preventing Ubc13 degradation (45). Our results showing that K13-induced NF-κB can be blocked by A20 suggest that although both K13 and Tax require binding to NEMO for NF-κB activation, they employ distinct underlying mechanisms.
What is the physiological significance of these results? K13-induced NF-κB activation is known to play a key role in the natural history of KSHV infection and in the pathogenesis of KSHV-associated malignancies (46). K13 has been shown to promote cellular proliferation, transformation, cytokine secretion, and viral latency (13–20). However, unchecked activation of the NF-κB pathway has the potential of resulting in uncontrolled inflammatory response. By blocking K13-induced NF-κB activation, A20 may serve to attenuate the inflammatory response and cellular transformation and thereby help maintain a balance between the virus and the host. Finally, in addition to its role in blocking the inflammatory response, A20 is known to serve a cytoprotective role and has been shown to protect vascular endothelial cells against TNFα, Fas, and natural killer cell-induced cell death (47). Although based on its structural homology to the prodomain of caspase 8/FLICE, K13 was originally believed to protect against apoptosis induced by the death receptors of the TNF family by directly binding to FADD and/or caspase 8/FLICE (12), subsequent studies have questioned this interaction as the basis of its cytoprotective ability (16). In contrast, it is now generally believed that K13 exerts its antiapoptotic and cytoprotective effect primarily via NF-κB activation (46). However, the NF-κB-responsive genes responsible for this effect have not been identified. Based on our results, it is conceivable that K13-induced A20 up-regulation may protect the KSHV-infected vascular endothelial cells against death receptor-induced apoptosis and thereby contribute to the pathogenesis of Kaposi sarcoma.
Acknowledgments
We thank Dr. Averil Ma for A20−/− MEFs, Dr. Dikstein for A20 luciferase reporter constructs, Dr. Lin Rongtuan for wild-type and mutant A20 expression constructs, and Dr. Ciaren Graham for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants CA85177, CA124621, and DE019811. This work was also supported by the Leukemia and Lymphoma Society.
- NF-κB
- nuclear factor κB
- EBV
- Epstein-Barr virus
- ELIE
- elongation inhibitory element
- ER
- estrogen receptor
- HTLV
- human T cell lymphotropic virus
- IKK
- IκB kinase
- KSHV
- Kaposi sarcoma-associated herpesvirus
- LMP1
- latent membrane protein 1
- MEF
- mouse embryonic fibroblast
- 4OHT
- 4-hydroxytamoxifen
- PEL
- primary effusion lymphoma
- vFLIP
- viral FLICE inhibitory protein
- OTU
- ovarian tumor.
REFERENCES
- 1. Aggarwal B. B. (2004) Cancer Cell 6, 203–208 [DOI] [PubMed] [Google Scholar]
- 2. Dixit V. M., Green S., Sarma V., Holzman L. B., Wolf F. W., O'Rourke K., Ward P. A., Prochownik E. V., Marks R. M. (1990) J. Biol. Chem. 265, 2973–2978 [PubMed] [Google Scholar]
- 3. Hymowitz S. G., Wertz I. E. (2010) Nat. Rev. Cancer 10, 332–341 [DOI] [PubMed] [Google Scholar]
- 4. Laherty C. D., Hu H. M., Opipari A. W., Wang F., Dixit V. M. (1992) J. Biol. Chem. 267, 24157–24160 [PubMed] [Google Scholar]
- 5. Laherty C. D., Perkins N. D., Dixit V. M. (1993) J. Biol. Chem. 268, 5032–5039 [PubMed] [Google Scholar]
- 6. Jäättelä M., Mouritzen H., Elling F., Bastholm L. (1996) J. Immunol. 156, 1166–1173 [PubMed] [Google Scholar]
- 7. Heyninck K., De Valck D., Vanden Berghe W., Van Criekinge W., Contreras R., Fiers W., Haegeman G., Beyaert R. (1999) J. Cell Biol. 145, 1471–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee E. G., Boone D. L., Chai S., Libby S. L., Chien M., Lodolce J. P., Ma A. (2000) Science 289, 2350–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dourmishev L. A., Dourmishev A. L., Palmeri D., Schwartz R. A., Lukac D. M. (2003) Microbiol. Mol. Biol. Rev. 67, 175–212, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moore P. S., Chang Y. (2001) Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 499–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stürzl M., Hohenadl C., Zietz C., Castanos-Velez E., Wunderlich A., Ascherl G., Biberfeld P., Monini P., Browning P. J., Ensoli B. (1999) J. Natl. Cancer Inst. 91, 1725–1733 [DOI] [PubMed] [Google Scholar]
- 12. Thome M., Schneider P., Hofmann K., Fickenscher H., Meinl E., Neipel F., Mattmann C., Burns K., Bodmer J. L., Schröter M., Scaffidi C., Krammer P. H., Peter M. E., Tschopp J. (1997) Nature 386, 517–521 [DOI] [PubMed] [Google Scholar]
- 13. Sun Q., Matta H., Chaudhary P. M. (2003) Blood 101, 1956–1961 [DOI] [PubMed] [Google Scholar]
- 14. Sun Q., Zachariah S., Chaudhary P. M. (2003) J. Biol. Chem. 278, 52437–52445 [DOI] [PubMed] [Google Scholar]
- 15. Sun Q., Matta H., Lu G., Chaudhary P. M. (2006) Oncogene 25, 2717–2726 [DOI] [PubMed] [Google Scholar]
- 16. Chugh P., Matta H., Schamus S., Zachariah S., Kumar A., Richardson J. A., Smith A. L., Chaudhary P. M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 12885–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guasparri I., Keller S. A., Cesarman E. (2004) J. Exp. Med. 199, 993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18. Matta H., Surabhi R. M., Zhao J., Punj V., Sun Q., Schamus S., Mazzacurati L., Chaudhary P. M. (2007) Oncogene 26, 1656–1660 [DOI] [PubMed] [Google Scholar]
- 19. Xu Y., Ganem D. (2007) J. Gen. Virol. 88, 46–50 [DOI] [PubMed] [Google Scholar]
- 20. Grossmann C., Podgrabinska S., Skobe M., Ganem D. (2006) J. Virol. 80, 7179–7185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao J., Punj V., Matta H., Mazzacurati L., Schamus S., Yang Y., Yang T., Hong Y., Chaudhary P. M. (2007) PLoS ONE 2, e1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matta H., Mazzacurati L., Schamus S., Yang T., Sun Q., Chaudhary P. M. (2007) J. Biol. Chem. 282, 24858–24865 [DOI] [PubMed] [Google Scholar]
- 23. Chaudhary P. M., Eby M. T., Jasmin A., Hood L. (1999) J. Biol. Chem. 274, 19211–19219 [DOI] [PubMed] [Google Scholar]
- 24. Chaudhary P. M., Jasmin A., Eby M. T., Hood L. (1999) Oncogene 18, 5738–5746 [DOI] [PubMed] [Google Scholar]
- 25. Ainbinder E., Amir-Zilberstein L., Yamaguchi Y., Handa H., Dikstein R. (2004) Mol. Cell. Biol. 24, 2444–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin R., Yang L., Nakhaei P., Sun Q., Sharif-Askari E., Julkunen I., Hiscott J. (2006) J. Biol. Chem. 281, 2095–2103 [DOI] [PubMed] [Google Scholar]
- 27. Matta H., Chaudhary P. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 9399–9404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu L., Eby M. T., Rathore N., Sinha S. K., Kumar A., Chaudhary P. M. (2002) J. Biol. Chem. 277, 13745–13751 [DOI] [PubMed] [Google Scholar]
- 29. Krikos A., Laherty C. D., Dixit V. M. (1992) J. Biol. Chem. 267, 17971–17976 [PubMed] [Google Scholar]
- 30. Amir-Zilberstein L., Dikstein R. (2008) J. Biol. Chem. 283, 1317–1323 [DOI] [PubMed] [Google Scholar]
- 31. Matta H., Punj V., Schamus S., Mazzacurati L., Chen A. M., Song R., Yang T., Chaudhary P. M. (2008) Oncogene 27, 5243–5253 [DOI] [PubMed] [Google Scholar]
- 32. Hideshima T., Chauhan D., Richardson P., Mitsiades C., Mitsiades N., Hayashi T., Munshi N., Dang L., Castro A., Palombella V., Adams J., Anderson K. C. (2002) J. Biol. Chem. 277, 16639–16647 [DOI] [PubMed] [Google Scholar]
- 33. Matta H., Sun Q., Moses G., Chaudhary P. M. (2003) J. Biol. Chem. 278, 52406–52411 [DOI] [PubMed] [Google Scholar]
- 34. Vereecke L., Beyaert R., van Loo G. (2009) Trends Immunol. 30, 383–391 [DOI] [PubMed] [Google Scholar]
- 35. Punj V., Matta H., Schamus S., Chaudhary P. M. (2009) BMC Med. Genomics 2, 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sakakibara S., Pise-Masison C. A., Brady J. N., Tosato G. (2009) J. Virol. 83, 2140–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Punj V., Matta H., Schamus S., Yang T., Chang Y., Chaudhary P. M. (2009) Blood 113, 5660–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Natoli G., Costanzo A., Guido F., Moretti F., Bernardo A., Burgio V. L., Agresti C., Levrero M. (1998) J. Biol. Chem. 273, 31262–31272 [DOI] [PubMed] [Google Scholar]
- 39. Kato M., Sanada M., Kato I., Sato Y., Takita J., Takeuchi K., Niwa A., Chen Y., Nakazaki K., Nomoto J., Asakura Y., Muto S., Tamura A., Iio M., Akatsuka Y., Hayashi Y., Mori H., Igarashi T., Kurokawa M., Chiba S., Mori S., Ishikawa Y., Okamoto K., Tobinai K., Nakagama H., Nakahata T., Yoshino T., Kobayashi Y., Ogawa S. (2009) Nature 459, 712–716 [DOI] [PubMed] [Google Scholar]
- 40. Schmitz R., Hansmann M. L., Bohle V., Martin-Subero J. I., Hartmann S., Mechtersheimer G., Klapper W., Vater I., Giefing M., Gesk S., Stanelle J., Siebert R., Küppers R. (2009) J. Exp. Med. 206, 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Honma K., Tsuzuki S., Nakagawa M., Tagawa H., Nakamura S., Morishima Y., Seto M. (2009) Blood 114, 2467–2475 [DOI] [PubMed] [Google Scholar]
- 42. Keller S. A., Schattner E. J., Cesarman E. (2000) Blood 96, 2537–2542 [PubMed] [Google Scholar]
- 43. Paine E., Scheinman R. I., Baldwin A. S., Jr., Raab-Traub N. (1995) J. Virol. 69, 4572–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wertz I. E., O'Rourke K. M., Zhou H., Eby M., Aravind L., Seshagiri S., Wu P., Wiesmann C., Baker R., Boone D. L., Ma A., Koonin E. V., Dixit V. M. (2004) Nature 430, 694–699 [DOI] [PubMed] [Google Scholar]
- 45. Shembade N., Ma A., Harhaj E. W. (2010) Science 327, 1135–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chaudhary P. M., Nicholas J. (2008) in Human Cancer Viruses: Principles of Transformation and Pathogenesis (Nicholas J., Jeang K.-T., Wu T.-.C. eds) pp. 186–210, Karger, Basel [Google Scholar]
- 47. Daniel S., Arvelo M. B., Patel V. I., Longo C. R., Shrikhande G., Shukri T., Mahiou J., Sun D. W., Mottley C., Grey S. T., Ferran C. (2004) Blood 104, 2376–2384 [DOI] [PubMed] [Google Scholar]