

Abstract
Transforming growth factor (TGF)-β family proteins are synthesized as precursors that are cleaved to generate an active ligand. Previous studies suggest that TGF-β activity can be controlled by lysosomal degradation of both precursor proteins and ligands, but how these soluble proteins are trafficked to the lysosome is incompletely understood. The current studies show that sortilin selectively co-immunoprecipitates with the cleaved prodomain and/or precursor form of TGF-β family members. Furthermore, sortilin co-localizes with, and enhances accumulation of a nodal family member in the Golgi. Co-expression of sortilin with TGF-β family members leads to decreased accumulation of precursor proteins and cleavage products and this is attenuated by lysosomal, but not proteosomal inhibitors. In Xenopus embryos, overexpression of sortilin leads to a decrease in phospho-Smad2 levels and phenocopies loss of nodal signaling. Conversely, down-regulation of sortilin expression in HeLa cells leads to an up-regulation of endogenous bone morphogenic protein pathway activation, as indicated by an increase in phospho-Smad1/5/8 levels. Our results suggest that sortilin negatively regulates TGF-β signaling by diverting trafficking of precursor proteins to the lysosome during transit through the biosynthetic pathway.
Keywords: Development, Intracellular Trafficking, Lysosomes, Transforming Growth Factor Beta (TGFβ), Xenopus, BMP, Sortilin, VPS10P
Introduction
Members of the TGF-β family, which include TGF-βs, activin, nodal, and bone morphogenic proteins (BMP)2 among others, play critical roles in lineage selection and differentiation of almost all cell types during embryonic development (1). Given their multifunctional nature, it is not surprising that TGF-β family activity is regulated at many levels, including the level of proteolytic activation (2). All family members are synthesized as inactive precursor proteins consisting of an amino (N)-terminal prodomain followed by a carboxyl-terminal mature ligand domain. Precursor proteins dimerize in the endoplasmic reticulum (ER) and are cleaved by members of the proprotein convertase (PC) family of serine proteases. Cleavage occurs during transit through the biosynthetic pathway or outside of cells, and is required to generate a mature ligand that can bind and activate its cognate receptor.
TGF-β ligands bind to a complex of transmembrane serine-threonine kinase receptors, which activate signaling by phosphorylating receptor-regulated Smad proteins (R-Smads) (1). Phosphorylated R-Smads bind to the common mediator Smad, Smad4, and this protein complex then moves into the nucleus to regulate transcription of target genes. Although TGF-β ligands bind their receptors at the cell surface, endocytosis of activated receptors is required in most cases for intracellular signal transduction (3).
Receptor-mediated endocytosis is not only required for TGF-β signal transduction, but it also plays an important role in signal termination by targeting the receptor and/or bound ligand to the lysosome for degradation (3). Several accessory proteins involved in targeting receptors for degradation have been identified. For example, Dapper 2, a late-endosomal protein, binds and facilitates lysosomal degradation of certain TGF-β type I receptors, leading to down-regulation of activin/nodal signaling in zebrafish, Xenopus, and mouse (4). The antagonistic Smad, Smad7, can also promote lysosomal targeting of some TGF-β type I receptors by recruiting the E3 ubiquitin ligases Smurf1 and Smurf2 and inducing ubiquitination (5, 6). Finally, the extracellular protein Bmper (BMP-binding endothelial cell precursor-derived regulator, also known as crossveinless2) binds BMP4 and stimulates receptor-dependent internalization and lysosomal degradation of BMP4 and its receptor (7).
TGF-β family precursor proteins and cleavage products can also be targeted for degradation independent of receptor binding. For example, the BMP4 precursor protein is sequentially cleaved at two sites within the prodomain and this regulates ligand stability (8). Cleavage at the first site generates a non-covalently associated ligand-prodomain complex that is targeted for lysosomal degradation (9), whereas subsequent cleavage at the second site, which occurs in a tissue-specific fashion (10), releases the ligand from the prodomain and thus stabilizes it. The prodomain of the zebrafish nodal-related protein, cyclops, contains a lysosomal targeting signal that destabilizes the precursor and thus restricts the signaling range of the mature ligand (11). By contrast, the prodomain of mouse nodal functions in cis to stabilize the precursor (12), whereas PC-mediated cleavage generates a mature ligand that is delivered to lysosomes via multivesicular bodies (13). The intrinsic instability of mature nodal is dependent on the prodomain because ligand generated from a chimeric precursor containing a dorsalin prodomain is substantially stabilized (14). The mechanism by which these soluble proteins are targeted for lysosomal degradation has not been investigated, but the observation that precursor proteins can be destabilized by elements in the prodomain prior to cleavage and receptor binding suggests the involvement of alternate trafficking receptors.
Sortilin is a good candidate for an intracellular sorting receptor involved in lysosomal targeting of TGF-β family members (15, 16). It is one of five members of a protein family that are structurally related to yeast vacuolar protein sorting 10 protein (Vps10p), which functions to traffic lysosomal enzymes from the Golgi to the vacuole (17). Sortilin is synthesized as a precursor molecule that is converted to its mature form by PC-catalyzed removal of its propeptide in a late Golgi compartment (18). The luminal portion of sortilin is composed almost entirely of the Vps10p domain, a 10-bladed β-propeller fold involved in ligand binding (19). This is followed by a transmembrane-spanning region and a short cytoplasmic tail that is homologous to that of the cation independent mannose 6-phosphate receptor, the major protein that routes soluble hydolases to the lysosome. The cytoplasmic tail of sortilin contains motifs that mediate rapid endocytosis and Golgi body to endosome trafficking (20). Newly synthesized sortilin moves to the cell surface, is internalized and shuttles between the trans-Golgi network (TGN), endosomal compartments, and the plasma membrane thereafter. Only a small fraction of sortilin (∼10%) is found at the cell surface, and yet sortilin has been shown to direct trafficking of substrates not only in the biosynthetic pathway but also during endocytic retrieval from the cell surface (20, 21).
In addition to sorting a subset of soluble hydrolases to the lysosome, sortilin modulates trafficking of a variety of non-lysosomal proteins, either restricting or facilitating their function (16). For example, within the biosynthetic pathway, sortilin facilitates secretion of apolipoprotein B100-containing lipoproteins, thereby regulating plasma cholesterol levels (22). It also controls sorting of brain-derived neurotrophic factor to the regulated secretory pathway in neurons (23) and enhances anterograde transport of Trk family receptors (24). At the cell surface, sortilin negatively regulates progranulin levels by facilitating their endocytosis and degradation (25). Furthermore, sortilin forms a tripartite complex with the nerve growth factor (NGF) precursor, pro-NGF, and its receptor p75NTR to induce rapid internalization and activate a signaling pathway that leads to neuronal apopotosis (26).
In the present study, we asked whether sortilin plays a role in intracellular trafficking of TGF-β family proteins. Our results demonstrate that sortilin is a novel inhibitor of TGF-β family signaling that can bind precursor proteins in the biosynthetic pathway and promote their trafficking to the lysosome for degradation.
EXPERIMENTAL PROCEDURES
cDNA Constructs
cDNAs encoding HA- and myc-tagged Xenopus laevis BMP4 have been described previously (9). cDNAs encoding epitope-tagged activin, Xenopus nodal-related 2 (Xnr2), and wild type or mutant forms of sortilin were generated using the splicing by overlap extension method (27) or PCR-mediated introduction of appropriate restriction sites. All cDNAs were subcloned into vector pCS2+ for expression in Xenopus and mammalian cells. Regions of cDNAs generated by PCR were sequenced. Sequence encoding an HA-epitope tag was inserted in-frame within the prodomain of activin following the codon for the 98th amino acid (-HSLPP(HA)PGHS-), or within the prodomain of Xnr2 following the codon for the 56th amino acid (-SQGMR(HA)PSMMQL-). A full-length Xenopus sortilin cDNA (BI443658) was identified by searching the NCBI data base (ncbi.nlm.nih.gov) and obtained from Open BioSystems on behalf of the IMAGE consortium (28). A cDNA containing only the coding region of sortilin was generated by PCR and the sequence encoding a FLAG epitope tag was inserted following the codon for the 47th amino acid, which positions it immediately after the furin cleavage site (-RNRR(FLAG)LEQNR-). We have previously shown that the epitope tags in BMP4 have no effect on the folding or biological activity of the encoded ligand (9), and the same is true for the epitope-tagged versions of activin, Xnr2, and sortilin (data not shown). The FLAG-sortilin cDNA was used as a template to make plasmids encoding each of the mutant forms of sortilin that are illustrated in Fig. 5A. Expression constructs coding for transferrin, myc-tagged mNotch1, and His-tagged soluble ephrin were provided by Dr. Caroline Enns (OHSU), Dr. Wenbiao Chen (Vanderbilt), and Dr. Phillip Copenhaver (OHSU), respectively.
FIGURE 5.

Deletion mutant forms of sortilin interact with and cause a loss of TGF-β precursors. A, schematic illustration of wild type or deletion mutant forms of sortilin with predominant subcellular localization of each protein indicated to the right. Pro, prodomain; TMD, transmembrane domain; ND, not determined. Position of the FLAG tag is indicated by the yellow bar. Scissors indicate furin cleavage site. Blue bar represents intracellular trafficking signals in the cytoplasmic tail. Numbers indicate the position of the terminal amino acid for truncation mutants, or amino acids bordering deleted segments for internal deletions. B-D, HeLa cells were transfected with BMP4 or Xnr2 in the presence or absence of wild type or deletion mutant forms of sortilin. Western blots of cell lysates and/or medium (INPUT) or Western blots of anti-FLAG immunoprecipitates (FLAG IP) were probed with antibodies specific for the HA tag in BMP4 or Xnr2. In B and D, lanes from the same Western blot were aligned following removal of an intervening lane (denoted by black line) using Photoshop. In the experiment shown in D, levels of Xnr2 were less affected by sortN than by wild type or other deletion mutant forms of sortilin but this was not the case in two other experiments. Relative levels of BMP4 precursor proteins and cleavage products in the presence and absence of ectopic sortilin were quantified based on band intensity on Western blots from a minimum of three experiments for all constructs except for sortΔVPS and sortΔN in which case results from four experiments were analyzed. C, error bars represent S.D. Statistical analysis was carried out using one-way ANOVA and a Student's paired t test for significance at the p < 0.01 (**) or p < 0.05 (*) level. (E) HeLa cells were transfected with FLAG-tagged sortN and Western blots of cell lysates and media were probed with antibodies specific for FLAG.
Transient Transfection, Co-immunoprecipitation, and Western Blot Analysis
HeLa cells or HEK293 cells were grown in Dulbecco's modified Eagle's medium in the presence of 10% fetal bovine serum and penicillin/streptomycin. Cells were transiently transfected with GFP, sortilin, and/or TGF-β family expression plasmids using Lipofectamine 2000 (Invitrogen) and cells were cultured in Opti-MEM I (Invitrogen) for 20–24 h. Unless otherwise indicated, cells were transfected with 100 ng of DNA encoding TGF-β precursor proteins and 100 ng of cDNA encoding sortilin. The total amount of DNA transfected in each experiment was normalized to 2 μg by the addition of varying amounts of GFP DNA. Cells were lysed in modified RIPA buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1.0% Nonidet P-40, 1 mm PMSF, 1 mm NaF, 1 mm Na3VO4) with complete protease inhibitor mixture (Roche Applied Science), cleared by centrifugation, and protein concentration was estimated by BCA assay (Pierce). Proteins were precipitated from conditioned medium using 10% trichloroacetic acid. Proteins present in conditioned media and cell lysates (30 μg) were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membrane. Membranes were stained with Ponceau S to confirm equal loading and probed with antibodies specific for actin (Sigma), transferrin (a gift from C. Enns), phospho-Smad1/5/8 (Cell Signaling), Smad1 (Cell Signaling), neurotensin receptor 3 (BD Biosciences), or HA (3F10, Roche Applied Science), Myc (9E10), or FLAG (M2, Sigma) epitopes. For co-immunoprecipitation assays, sortilin was immunoprecipitated from clarified cell lysates by overnight incubation with anti-FLAG antibody (M2) and protein A-conjugated beads. Conjugates were washed 3 times with lysis buffer, separated by SDS-PAGE, and transferred onto PVDF membrane, which was probed with HA- or myc-specific antibodies. Immunoreactive proteins were detected using Enhanced Chemiluminescence reagent (Pierce), and band intensities were quantified using NIH Image J software and statistical analysis was performed using ANOVA Data Analysis function in Microsoft Office Excel.
Xnr Uptake
HeLa cells were transiently transfected with Xnr2-HA DNA (1 μg) or sortilin DNA (1 μg), and cultured in 1 ml of Opti-MEM I (Invitrogen) for 24 h. Mock transfected or sortilin-transfected cells were rinsed with PBS and then cultured in the presence of 0.5 ml of conditioned medium from Xnr-HA-transfected cells for 5, 10, 20, 30, or 60 min. Cells were washed twice on ice with acid wash buffer (10 mm glycine, 150 mm NaCl, pH 3.0) and intracellular pools of Xnr-HA were immunoprecipitated from clarified cell lysates by overnight incubation with anti-HA antibody (12Ca5) and protein A-conjugated beads, followed by Western blot analysis using anti-HA antibody (3F10) as described above.
Inhibition of Lysosomal or Proteosomal Function
HEK293 cells were transiently transfected with Xnr2 together with sortilin or sortN, cultured for 20 h, and then cultured for a further 6 h in fresh Opti-MEM I containing dimethyl sulfoxide, 10 mm epoxomycin, or 4 mm bafilomycin A1.
Immunofluorescent Staining of Transfected Cells
To visualize the subcellular localization of sortilin and/or Xnr2, cells were transfected on coverslips, cultured for 24 h, and then fixed in 4% paraformaldehyde for 15 min. Cells were permeabilized in 0.2% Triton X-100 and then incubated with rat anti-HA antibody (3F10, Roche), mouse anti-FLAG antibody (M1, Sigma), and sheep anti-TGN46 antibody (AbD Serotec) in 3% serum containing 1 mm CaCl2 overnight at 4 °C. Following three washes with PBS containing 1 mm CaCl2, cells were incubated with Alexa 488-conjugated anti-rat antibody, Cy5-conjugated anti-mouse antibody, and Alexa 546-conjugated anti-sheep antibody in 3% serum containing 1 mm CaCl2 for 1 h at room temperature, washed three times with PBS containing 1 mm CaCl2, and mounted in Elvanol on slides. Triple channel images were acquired with a ×100 oil immersion objective lens (NA = 1.45) using a Zeiss LSM5 Pascal confocal inverted microscope.
siRNA Transfection
Four siRNA duplexes designed against human SORTILIN1 (targets: 5′-GAAUUUGGCAUGGCUAUUG-3′, 5′-GAGCUAGGUCCAUGAAUAU-3′, 5′-GAAGGACUAUACCAUAUGG-3′, 5′-GAGACUAUGUUGUGACCAA-3′) and control siRNAs were obtained from Dharmacon. HeLa cells were transfected with siRNAs using Lipofectamine RNAiMAX (Invitrogen) when they reached 30–50% confluence (24 h after plating) and this was repeated 72 h later. Cell lysates were harvested 72 h after the second transfection.
Xenopus Embryological Methods
Xenopus embryos were obtained and injected with capped, synthetic RNA as described previously (29). Embryonic stages are according to Nieuwkoop and Faber (30). Whole mount in situ hybridization was performed as previously described (31) using BM purple substrate (Roche Applied Science). For anti-phospho-Smad2 immunostaining, embryos were fixed in MEMFA (0.1 m MOPS, pH 7.4, 2 mm EGTA, pH 8, 1 mm MgSO4, 3.7% formaldehyde) for 2 h, bisected along the dorsoventral axis using a surgical blade, refixed in MEMFA for 1 h, dehydrated in methanol, and then incubated in Dent's fixative (20% dimethyl sulfoxide in methanol) for 2 h. Embryos were washed four times in PBST (PBS, 0.1% Tween), incubated for 2 h at room temperature in blocking solution (PBST, 3% BSA, 20% goat serum), and then overnight at 4 °C in primary antibody (rabbit anti-phospho-Smad2, Cell Signaling, 1:25 dilution in blocking solution) followed by washes with PBST for 6 h. Secondary antibody incubation (HRP-conjugated goat anti-rabbit at 1:250 dilution) was carried out overnight at 4 °C, followed by washes as described above. Signal was detected by incubation in 0.3 mg/ml of 3,3′-diaminobenzidine and 0.064% NiCl. Pigment was bleached by incubation in 1% H2O2, 5% formamide, 0.5× SSC for 1–2 h on a fluorescent light box. Xenopus embryos were fixed in 4% paraformaldehyde in PBS, dehydrated, and embedded in paraffin wax. Sections (10 μm) were stained with hematoxylin and eosin.
RESULTS
Co-expression of Sortilin with TGF-β Family Members Leads to a Selective Loss of Precursor Proteins and Cleavage Products
To begin to address whether sortilin can influence intracellular trafficking of TGF-β family proteins, we transiently transfected cultured HeLa cells with DNA encoding precursor forms of HA-tagged BMP4, Xenopus nodal-related 2 (Xnr2), or activin, together with GFP or FLAG-tagged sortilin. Steady-state levels of precursor proteins and cleavage products (illustrated schematically above Fig. 1B) were analyzed 24 h later by probing Western blots of cell lysates and conditioned medium with antibodies specific for the HA tag in the prodomain and, in the case of BMP4, the myc tag in the mature ligand domain. Levels of BMP4, Xnr2, and activin cleavage products were reproducibly decreased by 70–80% relative to control levels in the presence of sortilin (Fig. 1, A–D) and the same was true for a fourth TGF-β family member, BMP7 (supplemental Fig. S1C). Identical results were obtained when sortilin was co-expressed with TGF-β family proteins in HEK 293 cells (supplemental Fig. S1), suggesting that these effects are not cell-type specific.
FIGURE 1.

Sortilin causes a selective loss of BMP4 and other TGF-β family precursor proteins. HeLa cells were transfected with DNA (100 ng) encoding HA- and myc-tagged BMP4 (A and C), HA-tagged Xnr2 (B and D), HA-tagged activin (B), myc-tagged Notch (E and F), or transferrin (G) in the absence (−) or presence (+) of DNA encoding FLAG-tagged sortilin (100 ng). Western blots of cell lysates or media (indicated at the left of each blot) were probed with antibodies specific for epitope tags (A, B, and E) or transferrin (G). Western blots of cell lysates or media were reprobed with antibodies specific for actin or were stained with Ponceau S, respectively, as a loading control. In all panels, two lanes from the same Western blot have been aligned following removal of an intervening lane using Photoshop. Relative levels of precursor proteins and cleavage products in the presence and absence of ectopic sortilin were quantified based on band intensity on Western blots from a minimum of three experiments (C, D, and F). Error bars represent S.D. Statistical analysis was carried out using one-way ANOVA and a Student's paired t test for significance at the p < 0.01 level.
Levels of Xnr2 and activin precursor proteins, both of which can escape intracellular cleavage and are thus readily detected in the culture media, were also decreased by 80% compared with control levels in the presence of sortilin (Fig. 1, B and D). By contrast, levels of BMP4 precursor were only modestly reduced in the presence of sortilin (Fig. 1, A and C), most likely because pro-BMP4 is primarily present in the ER at steady-state and this ER-resident pool is resistant to the effects of sortilin. As shown under supplemental Fig. S2A, intact BMP4 precursor protein was only detected in cell lysates and not in the medium, whereas cleavage products were only detected in the culture medium. Furthermore, only the endo-β-N-acetylglucosaminidase H-sensitive, ER-resident form of pro-BMP4 was detected inside of cells (supplemental Fig. S2, B and D). The observation that pro-BMP4 cannot be detected outside of the ER suggests that exit from the ER is rate-limiting for cleavage, and that cleavage is tightly coupled to secretion under steady-state conditions. Consistent with this idea, a cleavage mutant form of pro-BMP4 (cmBMP4) that it is resistant to PC-mediated proteolysis readily accumulated in culture medium (supplemental Fig. S2C). Whereas levels of BMP4 or cmBMP4 precursor protein found inside of cells showed little or no reduction in the presence of sortilin, the extracellular, post-ER pool of the cmBMP4 precursor was significantly decreased in the presence of sortilin (supplemental Fig. S2C). Collectively, these results suggest that sortilin-mediated loss of TGF-β family proteins occurs in post-ER compartments and is independent of cleavage.
To ask whether overexpressed sortilin specifically affects TGF-β family members, or whether it nonspecifically interferes with trafficking, cleavage, or secretion of all proteins that pass through the secretory pathway, we analyzed steady-state levels of the soluble protein transferrin and the membrane receptor Notch in the absence or presence of sortilin. The precursor form of the Notch receptor and the smaller, membrane-bound fragment generated by PC-mediated cleavage (illustrated schematically to the right of Fig. 1E) accumulated at equivalent levels in the presence or absence or sortilin (Fig. 1, E and F). Furthermore, co-expression of sortilin had no effect on secretion or accumulation of transferrin in cell lysates or culture medium (Fig. 1G). Co-expression of sortilin led to a dose-dependent reduction in steady-state levels of BMP4 or Xnr2 cleavage products when the two DNAs were transfected at stoichiometric or substoichiometric molar ratios, whereas sortilin had no effect on steady-state levels of transferrin or Notch when expressed at the same molar ratios (supplemental Fig. S3). Thus, sortilin specifically inhibits accumulation of TGF-β family proteins without impacting the ability of PCs to cleave other substrates, and without blocking trafficking through the secretory pathway in general. Collectively, our results are consistent with the possibility that sortilin functions as a sorting receptor to direct lysosomal targeting and degradation of TGF-β family precursor proteins, leading to a secondary loss of active ligand.
TGF-β Family Precursors Interact with Sortilin in Vivo
Co-immunoprecipitation assays were used to determine whether sortilin can interact with TGF-β family members in vivo. HeLa cells were transfected with HA-tagged BMP4, HA-tagged Xnr2, or myc-tagged Notch in the presence or absence of FLAG-tagged sortilin. Protein complexes containing sortilin were immunoprecipitated from cell lysates using an antibody specific for the FLAG epitope and Western blots of cell lysates or immunoprecipitates were probed with antibodies specific for HA or myc. Precursor forms of BMP4 and Xnr2 co-immunopurified with sortilin in this assay, as did BMP7, whereas Notch, transferrin, and a soluble form of ephrin did not (Fig. 2A and supplemental Fig. S4). Notably, in cells transfected with pro-BMP4 and sortilin, both the BMP4 precursor protein and lesser amounts of cleaved prodomain consistently co-immunoprecipitated with sortilin (Fig. 2B), suggesting that interactions are mediated, at least in part, by elements in the prodomain. Consistent with this possibility, when cells were transfected with DNA encoding only the prodomain of BMP4, the isolated prodomain co-immunopurified with sortilin (Fig. 2C).
FIGURE 2.

Sortilin selectively co-immunoprecipitates with TGF-β family prodomain and/or precursors. A–C, HeLa cells were transfected with DNA encoding HA-tagged BMP4, HA-tagged Xnr2, Myc-tagged Notch, or HA-tagged BMP4 prodomain in the absence (−) or presence (+) of FLAG-tagged sortilin. Immunoblots of cell lysates (input) or anti-FLAG immunoprecipitates (FLAG IP) were probed with antibodies specific for HA, myc, or sortilin (sort) as indicated to the right of each blot. The asterisk in A indicates a more slowly migrating immunoreactive protein that was not observed in other experiments. The arrowhead in B indicates cleaved BMP4 prodomain. D, HeLa cells were transfected with DNA encoding HA-tagged BMP4 or Xnr2 and immunoblots of cell lysates or anti-HA immunoprecipitates (IP) were probed with antibodies specific for HA or sortilin. The arrow indicates endogenous sortilin, whereas the asterisk denotes a nonspecific background band observed in all HA IPs but not in the input samples. In B and D, lanes from the same Western blot were aligned following removal of an intervening lane using Photoshop.
Co-immunoprecipitation assays were also used to test whether TGF-β family members can associate with endogenous sortilin. HeLa cells were transfected with DNA encoding HA-tagged BMP4 or Xnr2, precursor proteins were immunoprecipitated from cell lysates using an antibody specific for the HA epitope, and Western blots of cell lysates or immunoprecipitates were probed with antibodies specific for sortilin. As shown in Fig. 2D, endogenous sortilin co-immunoprecipitated with pro-BMP4 and pro-Xnr2. Collectively, these results suggest that sortilin selectively interacts in vivo with TGF-β family precursor proteins.
Sortilin Does Not Affect Endocytosis of Xnr2
Sortilin functions as a trafficking receptor in the biosynthetic pathway but can also mediate endocytosis of bound proteins from the cell surface (15). To ask whether sortilin present at the cell surface can facilitate endocytosis and/or lysosomal targeting of TGF-β family proteins, we compared levels of HA-tagged Xnr2 taken up into HeLa cells in the presence or absence of ectopic sortilin DNA. HeLa cells were mock transfected, or transfected with DNA encoding sortilin, and the next morning equivalent amounts of Xnr2 precursor protein and cleavage products were added to the media. Western blot analysis revealed that increasing amounts of pro-Xnr2 and cleaved prodomain were taken up into cells over time but no difference was observed in steady-state levels of internalized Xnr2 precursor protein or prodomain in the presence or absence of sortilin (Fig. 3). These results suggest that sortilin does not enhance endocytic retrieval of TGF-β family proteins from the cell surface.
FIGURE 3.

Sortilin does not enhance endocytic uptake of Xnr2. A, culture media containing equivalent amounts of HA-tagged Xnr precursor and prodomain fragments was added to HeLa cells that had been mock transfected (−) or transfected with DNA encoding sortilin (+). Cells were harvested at increasing times after addition of Xnr2 and immunoblots of cell lysates were probed with antibodies specific for the HA tag. Results were reproduced in a minimum of three independent experiments. B, relative levels of precursor protein and prodomain in the presence and absence of ectopic sortilin at the 60-min time point were quantified based on band intensity on Western blots from four independent experiments. Error bars represent S.D. Statistical analysis was carried out using one-way ANOVA and a Student's paired t test for significance at the p < 0.05 level.
Sortilin Co-localizes with and Promotes Accumulation of Xnr2 in Golgi Compartments
To begin to test the possibility that sortilin interacts with TGF-β precursors in the biosynthetic pathway and diverts their intracellular trafficking to the lysosome, we asked whether these proteins co-localize to the same subcellular compartment(s). HeLa cells were transfected with DNA encoding HA-tagged Xnr2 in the absence or presence of FLAG-tagged sortilin and triple label immunostaining was performed using antibodies specific for the HA and FLAG tags together with ER- or TGN-specific markers. In the absence of ectopic sortilin, immunoreactive Xnr2 was detected in puncta that were evenly distributed throughout the cytoplasm (Fig. 4, A and B, arrowheads), consistent with previous studies suggesting that Xnr2 bypasses the TGN on the way to the cell surface (32). Sortilin staining overlapped with TGN markers and was also detected in vesicles dispersed throughout the cell body, consistent with previously published studies (33) (Fig. 4, B and C). No differences were detected in the pattern of sortilin staining in the presence or absence of Xnr2. By contrast, Xnr2 staining was markedly enriched in TGN compartments in cells that co-expressed sortilin (arrows) relative to those that did not (arrowheads) (Fig. 4B). Significant overlap in Xnr2 and sortilin staining was detected in TGN compartments (Fig. 4, B and C, arrows), but there was little or no overlap in vesicular staining in post-TGN compartments when cells were examined at higher magnification (Fig. 4C, bottom right panel). Xnr2 staining outside of the TGN may represent the fraction of the precursor protein that escapes interaction with sortilin and thus traffics to the cell surface independent of sortilin and/or this staining may be a precursor or cleaved prodomain that is internalized after secretion. We also attempted to examine the subcellular localization of BMP4 in the presence and absence of sortilin but the intense ER staining in cells transfected with BMP4 (supplemental Fig. S2D) obscured signals in other subcellular compartments and precluded this analysis. Collectively, these data are consistent with the possibility that sortilin interacts with Xnr2 in the biosynthetic pathway and enhances trafficking through the TGN to the lysosome.
FIGURE 4.

Sortilin co-localizes with and promotes accumulation of Xnr2 in the TGN. HeLa cells were transfected with HA-tagged Xnr2 in the absence (A) or presence (B and C) of FLAG-tagged sortilin. Double (A and C) or triple (B) label immunostaining was performed using antibodies specific for HA, FLAG, and TGN46. The region of the TGN is indicated by arrowheads in cells that express Xnr alone and by arrows in cells that co-express Xnr2 and sortilin. The boxed area in the lower left panel of C is shown at higher magnification in the lower right panel. Scale bars represent 10 μm.
Mutant Forms of Sortilin Lacking Lysosomal Trafficking Signals and Other Functional Domains Interact with and Cause a Loss of TGF-β Precursors When Co-expressed
To ask whether sortilin directly mediates trafficking of TGF-β precursors to the lysosome, steady-state levels of BMP4 were compared in the presence or absence of a mutant form of sortilin (sortΔCT, illustrated in Fig. 5A) that lacks the cytoplasmic tail. If sortilin binds pro-BMP4 and directs it to the lysosome via trafficking signals present in its cytoplasmic tail, then sortΔCT should have no effect on steady-state levels of BMP4. Contrary to these predictions, sortΔCT caused a loss of pro-BMP4 and cleaved prodomain equivalent to that observed in cells expressing wild type sortilin (Fig. 5, B, input, results are quantified in C). This result suggests that the luminal portion of sortilin contains (or binds to a distinct protein containing) intracellular trafficking signals that function independent of the cytoplasmic tail.
We then tested additional deletion mutant forms of sortilin for their ability to interact with and cause a decrease in steady-state levels of BMP4 and Xnr2 when co-expressed in HeLa cells, and to be secreted into the culture medium as analyzed by Western blotting. As illustrated in Fig. 5A, these mutants included forms of sortilin lacking all but the N-terminal most fragment of the VPS10 cargo binding domain (sortΔVPS), containing only the N-terminal portion of the luminal domain (sortN), lacking only the N-terminal portion of the luminal domain (sortΔN), or containing only the signal peptide and prodomain (sortPro). Each of these deletion mutants, with the exception of sortPro, caused a decrease in steady-state levels of the BMP4 precursor and its cleaved prodomain that was comparable with that mediated by wild type sortilin (Fig. 5B, input, results quantified in Fig. 5C), and identical results were obtained for Xnr2 (Fig. 5D). Furthermore, even sortN, which lacks a transmembrane domain, was primarily retained inside of cells rather than being secreted into the medium (Fig. 5E). Protein complexes containing native or mutant forms of sortilin were then immunoprecipitated from cell lysates using an antibody specific for the FLAG epitope and Western blots of immunoprecipitates were probed with antibodies specific for the HA tag present in BMP4 or Xnr2. Pro-BMP4 and Xnr2 co-immunopurified with each of the deletion mutants, with the exception of sortPro (Fig. 5, B and D, FLAG IP). Collectively, these data demonstrate that a single TGF-β-interacting domain cannot be identified in sortilin. The most logical explanation for these findings is that sortilin interacts with, and mediates trafficking of TGF-β precursors as part of a multiprotein complex that bridges different domains of sortilin and TGF-β family proteins.
The subcellular distribution of each active deletion mutant of sortilin was analyzed by immunostaining transfected cells with anti-FLAG monoclonal antibody M1, which only recognizes FLAG epitopes at the free N terminus of proteins (supplemental Fig. S5, results summarized in Fig. 5A). The FLAG tag in sortilin (yellow bar in illustration in supplemental Fig. S5) is positioned immediately following the prodomain such that the M1 antibody will only detect the mature form of sortilin that has been proteolytically activated in post-ER compartments. When cells expressing sortΔCT were stained with the M1 antibody immunoreactive protein was enriched at the cell membrane, and was also detected in puncta distributed throughout the cell body (supplemental Fig. S5). By contrast, sortΔVPS and sortN were concentrated in TGN compartments but staining was also visible in puncta distributed throughout the cytoplasm, similar to the pattern of staining for wild type sortilin. SortΔN was detected in puncta throughout the cell body. Collectively, these results suggest that an N-terminal fragment of sortilin that lacks most of the VPS domain contains (or binds to a distinct protein containing) trafficking and/or retention signals that can direct TGN localization independent of signals in the cytoplasmic tail.
Sortilin-mediated Degradation of Xnr2 Is Sensitive to Lysosomal, but Not Proteosomal Inhibitors
To address whether sortilin-mediated loss of TGF-β family proteins requires lysosomal or proteosomal function, we asked whether treatment with the lysosomal inhibitor bafilomycin A1, or with the proteosomal inhibitor epoxomicin, could rescue steady-state levels of Xnr2 in cells made to co-express either sortilin or the deletion mutant, sortN. Bafilomycin A1 treatment led to a significant increase in steady-state levels of pro-Xnr2 in the absence of sortilin, consistent with previous reports that nodal precursor proteins are targeted for lysosomal degradation (11), whereas treatment with epoxomicin had no effect (Fig. 6). Co-expression of sortilin or sortN led to a decrease in steady-state levels of pro-Xnr2 relative to untreated controls and expression was substantially rescued by lysosomal, but not proteosomal inhibition (Fig. 6). These results are consistent with the hypothesis that sortilin and sortN can target pro-Xnr2 to the lysosome for degradation.
FIGURE 6.
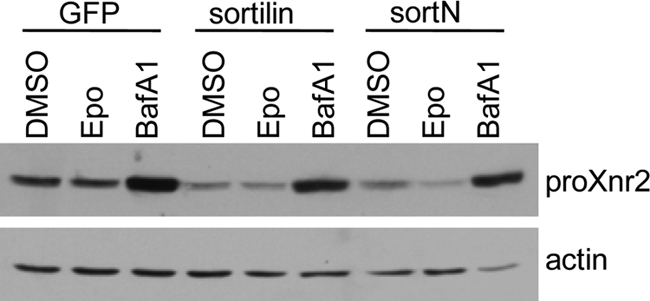
Sortilin-mediated degradation of Xnr2 is sensitive to lysosomal, but not proteosomal inhibitors. HEK 293 cells expressing Xnr2 together with GFP, sortilin, or sortN were cultured for 6 h in medium containing vehicle alone (dimethyl sulfoxide, DMSO) or vehicle containing epoxomycin (Epo, 10 μm) or bafilomycin A1 (BafA1, 4 μm). Cell lysates were harvested and expression of Xnr2 was analyzed by Western blot. Results were reproduced in three independent experiments.
Ectopic Expression of Sortilin Inhibits Endogenous TGF-β Family Signaling in Xenopus Embryos
The above results suggest that sortilin can direct lysosomal trafficking and degradation of TGF-β family precursor proteins when both are overexpressed in cultured cells. To determine whether sortilin can negatively regulate endogenous TGF-β family signals, we overexpressed wild type or a subset of deletion mutant forms of sortilin in Xenopus embryos and looked for phenotypic defects characteristic of loss of early TGF-β-mediated patterning events. Signaling downstream of Xnrs is essential for induction and patterning of endodermal and mesodermal germ layers (reviewed in Ref. 34). RNA encoding sortN or wild type sortilin (500 pg to 2 ng) was injected near the dorsal midline of four-cell Xenopus embryos and expression of the mesodermal marker, xbra, was analyzed at the early gastrula stage by in situ hybridization. In control embryos, xbra was expressed in a continuous ring throughout the mesoderm (Fig. 7A, n = 68/75 embryos in three experiments). Ectopic expression of wild type sortilin (Fig. 7B) or sortN (Fig. 7C) completely repressed expression of xbra on the dorsal side of the embryo (arrows), and substantially repressed expression throughout the remainder of the mesoderm (n = 45/60 sortilin-injected and 76/92 sortN-injected embryos in three experiments). Identical results were obtained following injection of RNA encoding sortΔVPS (data not shown). Furthermore, expression of phospho-Smad2, which is directly activated downstream of Xnr2, was repressed in mesendodermal cells of embryos made to ectopically express sortilin or sortN, with the strongest repression being observed in dorsal mesoderm (Fig. 7, D–F, circled regions). At the tailbud stage, embryos injected with RNA encoding sortilin or sortN showed a loss of anterior structures, a shortened, curved body axis, and/or spina bifida (Fig. 7, G–I; n = 39/60 sortilin-injected and 65/70 sortN-injected embryos in two experiments). Embryos injected with RNA encoding sortΔVPS showed an identical phenotype (data not shown). Histological analysis revealed that differentiated muscle, notochord, and neural tissue was reduced or disorganized, and in some cases absent in tailbud stage embryos made to overexpress sortilin or sortN (Fig. 7, J–L). These defects phenocopy those observed in embryos in which all TGF-β family signaling is inhibited in dorsal cells by removal of Smad4, or by overexpression of the inhibitory Smad, Smad7 (35, 36). Collectively, these results demonstrate that overexpression of wild type or deletion mutant forms of sortilin can repress endogenous TGF-β signaling in vivo.
FIGURE 7.

Ectopic expression of sortilin in Xenopus embryo phenocopies loss of Xnr function. Xenopus embryos were injected near the dorsal midline at the four-cell stage with a control RNA or RNA encoding sortilin or sortN. A–C, expression of the Xnr target gene, xbra, was analyzed by in situ hybridization at the early gastrula stage. Arrows indicate the dorsal side of embryos. D–F, embryos were immunostained with antibodies specific for phospho-Smad2 at the early gastrula stage. Dorsal mesoderm, where phospho-Smad2 levels are normally highest, is circled. G–I, gross morphology and J–L, histological sections of tailbud stage embryos. Nt, neural tissue; No, notochord; Mu, muscle.
Endogenous Sortilin Negatively Modulates BMP Signaling in Vivo
To directly address whether endogenous sortilin negatively modulates TGF-β signaling in vivo, we asked whether signaling downstream of BMP or other TGF-β family members is up-regulated when expression of sortilin is suppressed. We initially assayed TGF-β family activity in Xenopus embryos injected with morpholino antisense oligonucleotides specific for sortilin but results were inconclusive due to high levels of maternally stored sortilin protein, which could not be depleted in early embryos (data not shown). As an alternate approach, HeLa cells were transfected with increasing amounts of siRNA specific for SORTILIN, a nonspecific control siRNA, or a cDNA encoding BMP4 (as a positive control for pathway activation) and levels of phospho-Smad1/5/8 were assayed as a sensitive and direct readout for BMP activity. We observed a reproducible, and dose-dependent decrease in endogenous sortilin protein levels in response to target-specific siRNAs (Fig. 8A, top panel). The reduction in endogenous sortilin was accompanied by an increase in levels of phosphorylated Smad1/5/8 (Fig. 8A, second panel from the top). When cells were exposed to 1-nm siRNA, sortilin levels were repressed to 30% of controls and levels of pSmad1/5/8 showed a 2.6-fold increase (Fig. 8B). The level of total Smad1 protein was not affected by sortilin siRNAs, indicating that the increase in phospho-Smad1/5/8 immunoreactivity was due to pathway activation. These results demonstrate that endogenous sortilin dampens endogenous BMP signaling in HeLa cells.
FIGURE 8.

BMP signaling is up-regulated when expression of endogenous sortilin is suppressed. A, HeLa cells were transfected with increasing amounts of siRNA designed against human sortilin, a nonspecific control siRNA (NC), or a cDNA encoding BMP4 (as a positive control for pathway activation). Western blots of cell lysates were probed with antibodies specific for endogenous sortilin and then stripped and reprobed with antibodies specific for pSmad1/5/8, total Smad1, or actin (as a loading control). B, relative levels of sortilin and pSmad1/5/8 in cells transfected with 1 nm control or sortilin siRNA were quantified based on band intensity on Western blots from a minimum of three experiments. Error bars represent S.D. Statistical analysis was carried out using one-way ANOVA and a Student's paired t test; significant differences at the level of p < 0.01 (**) or p < 0.05 (*) are indicated.
DISCUSSION
The current studies suggest that sortilin functions as a novel negative regulator of TGF-β family activity. Specifically, our data demonstrate that overexpressed sortilin facilitates trafficking of TGF-β precursor proteins to the lysosome for degradation. The physiological relevance of this negative regulatory mechanism is supported by our data showing that depletion of endogenous sortilin leads to enhanced BMP pathway activation, whereas enhancement of sortilin levels leads to a decrease in endogenous TGF-β signaling.
Although our studies support a role for endogenous sortilin as a negative regulator of TGF-β family activity, mice mutant for Sortilin are grossly normal (37), whereas mice carrying mutations in critical negative regulators of TGF-β activity show a variety of defects, in some cases leading to lethality (38). These phenotypic differences raise the possibility that sortilin functions primarily as a buffer to fine-tune the activity of its many substrates rather than providing an on-off switch for activity. Alternatively, other members of the VPS10 protein family may function redundantly to complement sortilin function in vivo. Consistent with either possibility, mice lacking Sortilin do not phenocopy mice mutant for other known sortilin substrates, although defects can often be uncovered on a sensitized background. For example, Sortilin nullizygous mice do not exhibit signs of lysosomal pathologies despite evidence that sortilin functions as a trafficking receptor for several soluble lysosomal proteins (39). Furthermore, although sortilin is recognized as a crucial component of the proneurotrophin-p75NTR signaling complex that controls apoptotic cell death during neurogenesis (26), Sortilin-deficient mice show reduced apoptosis in only a small subset of neurons known to undergo developmentally regulated, p75NTR-dependent apoptosis (37). As a final example, sortilin facilitates hepatic export of lipoproteins in vivo, which is required to prevent hypercholesterolemia. This function is only apparent in Sortilin mutants, however, when plasma low-density lipoprotein levels are elevated above physiological levels by either diet or genetic background (22). Further in-depth analysis in mice mutant for multiple members of the VPS10 protein family is needed to document whether redundancy accounts for the relatively minor defects present in Sortilin mutant mice.
Mechanisms that regulate intracellular trafficking, stability, and proteolytic maturation of TGF-β family precursor proteins are poorly understood. Cleavage of pro-BMP4 is proposed to occur within the TGN and/or endosomes as the precursor traverses the biosynthetic pathway (9). By contrast, the nodal precursor arrives at the cell surface intact via a pathway that bypasses the TGN, but then undergoes rapid endocytic uptake, prodomain removal, and delivery to lysosomes (13, 32). The receptor involved in cell surface retrieval and lysosomal targeting of nodal is unknown. Interestingly, the precursor form of NGF is also secreted intact and must undergo endocytic uptake and intracellular cleavage to generate the mature ligand (40). Sortilin binds to the NGF precursor at the cell surface, which results in its rapid internalization and cleavage (26, 41), raising the possibility that sortilin might function in a similar manner to direct endocytic retrieval and lysosomal targeting of nodal. Contrary to this prediction, however, we found that ectopic sortilin did not enhance uptake of nodal from the cell surface, but instead appeared to divert and/or retain nodal within the TGN as it trafficked through the biosynthetic pathway. This function is analogous to the role of the sortilin family member, SorLA, in intracellular trafficking of the amyloid precursor protein (APP) (15). Specifically, SorLA does not affect retrograde trafficking of APP from the cell surface, but instead acts as a retention factor to prolong residence time for nascent APP molecules in the TGN, and may also shuttle APP back to the TGN from endosomes (42, 43). In the case of APP, this prevents release of the precursor into pathways that promote proteolytic processing into disease causing, amyloidogenic peptides (reviewed in Ref. 15). Collectively, our results are consistent with a role for endogenous sortilin in enhancing trafficking of nodal and other TGF-β precursors through the TGN to the lysosome. It is possible that assays of endogenous nodal trafficking, which are not currently feasible, and/or analysis of trafficking in cells that express nodal receptors and co-receptors will show that sortilin has a role in endocytic uptake of precursors as well.
Our data showing that deletion mutant forms of sortilin lacking most of the VPS10 ligand-binding domain are able to co-immunopurify with TGF-β precursor proteins are unexpected but not unprecedented. For example, following the initial demonstration that SorLA and APP interact directly via the luminal domain of each protein (44), subsequent studies demonstrated a secondary binding site between the cytoplasmic domains of each protein (42). Similarly, although it has been show that sortilin interacts directly with glucose transporter isoform 4 (GLUT4) and plays a major role in the formation of GLUT4-containing insulin responsive vesicles (45), more recent structure-function studies have revealed that this is not a direct binary interaction but instead requires formation of a protein complex consisting of multiple additional partners (46, 47). Our data, which show that deletion mutant forms of sortilin lacking complimentary regions of the luminal domain are able to co-immunopurify with TGF-β family precursors, suggest a similar model in which additional binding partners function as a scaffold to mediate interactions between different domains of sortilin and TGF-β precursor proteins.
In overexpression assays, deletion mutant forms of sortilin that lack the cytoplasmic tail and its attendant trafficking signals retain the ability to accelerate degradation of TGF-β precursor proteins. There are several potential explanations for this seemingly anomalous result. First although chimeric analysis shows that signals in the sortilin tail domain are sufficient to direct Golgi to endosome transport and retrieval, additional intrinsic targeting signals appear to be present within the luminal domain because truncated forms of sortilin that lack either specific sorting signals or the entire cytoplasmic domain localize to the late Golgi rather than reaching the plasma membrane (20, 48). Our observation that SortN is enriched in the Golgi suggests that the extreme N terminus of sortilin contains information sufficient to direct Golgi localization independent of signals in the cytoplasmic tail. Using similar functional assays, previously published studies have shown that a wide range of deletion mutant forms of sortilin, or SorLA, retain function in mediating assembly of GLUT4-containing vesicles and in trafficking of APP (42, 46). These findings have been interpreted to suggest the involvement of additional accessory proteins in directing intracellular trafficking and other functions of sortilin containing protein complexes. As one example of this, the neurotrophin receptor p75NTR has been shown to function as a trafficking switch that impairs lysosomal sorting of sortilin and its cargo, pro-NGF, and instead diverts trafficking to the cell surface (49). A second possible explanation for the ability of the deletion mutants to induce degradation of TGF-β precursors is that these mutants are misfolded and aggregate in the ER, leading to ER stress and protein degradation. Sortilin-dependent degradation is selective for TGF-β proteins, however, and is not dependent on proteosomal function for the subset of the proteins that we have tested, demonstrating that the ER-associated degradation pathway has not been activated. Alternatively, it is possible that sortilin mutants aggregate in post-ER compartments, leading to lysosmal targeting similar to that observed for the PC family member, furin. Signals within the cytoplasmic tail of furin direct localization predominantly to the TGN, but in the absence of these signals, determinants in the luminal domain induce aggregation in the TGN followed by lysosomal targeting (50). It is possible that similar signals function to direct aggregation and lysosomal trafficking of TGF-β family proteins bound to sortilin mutants. If so, ectopically expressed deletion mutant forms of sortilin may mediate degradation of TGF-β family members through a distinct mechanism (aggregation induced lysosomal targeting) than that used by wild type sortilin (targeting mediated by signals in the cytoplasmic tail). Further studies will be required to determine whether ER-associated degradation, ER stress-induced autophagy (51), or aggregation contribute to degradation induced by individual sortilin mutants.
Taken together with previously published studies, our results are most consistent with a model in which a trafficking complex, consisting of TGF-β family precursor proteins and multiple other binding partners, assembles and escorts precursor proteins to the cell surface. We propose that sortilin is one component of this trafficking complex that can dominantly route a subset of precursor proteins to the lysosome for degradation. Our results suggest that the level of lysosomal degradation depends on the relative level of sortilin expressed by a given cell type. An alternate model is that TGF-β proteins normally associate with an endogenous trafficking complex that escorts precursors to the cell surface via a TGN-independent route. Sortilin may compete with components of this complex for binding to TGF-β precursors, thereby diverting trafficking through the TGN to the lysosome. This scenario is consistent with our observation that ectopic sortilin can divert trafficking of TGF-β precursor proteins even when expressed at substoichiometric ratios. Future studies will be required to definitively test these models and identify additional components of TGF-β trafficking complexes.
Supplementary Material
Acknowledgments
We thank Caroline Enns and Alex Nechiporuk for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R03HD058841 and RO1 HD37976 (to J. L. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- BMP
- bone morphogenic protein
- Xnr
- Xenopus nodal-related protein
- VPS10p
- vacuolar protein sorting 10 protein
- ER
- endoplasmic reticulum
- PC
- proprotein convertase
- GFP
- green fluorescent protein
- APP
- amyloid precursor protein
- GLUT4
- glucose transporter isoform 4
- ANOVA
- analysis of variance.
REFERENCES
- 1. Moustakas A., Heldin C. H. (2009) Development 136, 3699–3714 [DOI] [PubMed] [Google Scholar]
- 2. Sopory S., Christian J. L. (2006) in Methods in Signal Transduction (Whitman M., Sater A. ed) pp. 37–60, CRC Press, Boca Raton, FL [Google Scholar]
- 3. Chen Y. G. (2009) Cell Res. 19, 58–70 [DOI] [PubMed] [Google Scholar]
- 4. Zhang L., Zhou H., Su Y., Sun Z., Zhang H., Zhang L., Zhang Y., Ning Y., Chen Y. G., Meng A. (2004) Science 306, 114–117 [DOI] [PubMed] [Google Scholar]
- 5. Ebisawa T., Fukuchi M., Murakami G., Chiba T., Tanaka K., Imamura T., Miyazono K. (2001) J. Biol. Chem. 276, 12477–12480 [DOI] [PubMed] [Google Scholar]
- 6. Kavsak P., Rasmussen R. K., Causing C. G., Bonni S., Zhu H., Thomsen G. H., Wrana J. L. (2000) Mol. Cell 6, 1365–1375 [DOI] [PubMed] [Google Scholar]
- 7. Kelley R., Ren R., Pi X., Wu Y., Moreno I., Willis M., Moser M., Ross M., Podkowa M., Attisano L., Patterson C. (2009) J. Cell Biol. 184, 597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cui Y., Hackenmiller R., Berg L., Jean F., Nakayama T., Thomas G., Christian J. L. (2001) Genes Dev. 15, 2797–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Degnin C., Jean F., Thomas G., Christian J. L. (2004) Mol. Biol. Cell 15, 5012–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goldman D. C., Hackenmiller R., Nakayama T., Sopory S., Wong C., Kulessa H., Christian J. L. (2006) Development 133, 1933–1942 [DOI] [PubMed] [Google Scholar]
- 11. Tian J., Andrée B., Jones C. M., Sampath K. (2008) Development 135, 2649–2658 [DOI] [PubMed] [Google Scholar]
- 12. Le Good J. A., Joubin K., Giraldez A. J., Ben-Haim N., Beck S., Chen Y., Schier A. F., Constam D. B. (2005) Curr. Biol. 15, 31–36 [DOI] [PubMed] [Google Scholar]
- 13. Blanchet M. H., Le Good J. A., Oorschot V., Baflast S., Minchiotti G., Klumperman J., Constam D. B. (2008) Sci. Signal. 1, ra13 [DOI] [PubMed] [Google Scholar]
- 14. Constam D. B., Robertson E. J. (1999) J. Cell Biol. 144, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Willnow T. E., Petersen C. M., Nykjaer A. (2008) Nat. Rev. Neurosci. 9, 899–909 [DOI] [PubMed] [Google Scholar]
- 16. Hermey G. (2009) Cell Mol. Life Sci. 66, 2677–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marcusson E. G., Horazdovsky B. F., Cereghino J. L., Gharakhanian E., Emr S. D. (1994) Cell 77, 579–586 [DOI] [PubMed] [Google Scholar]
- 18. Munck Petersen C., Nielsen M. S., Jacobsen C., Tauris J., Jacobsen L., Tauris J., Jacobsen L., Gliemann J., Moestrup S. K., Madsen P. (1999) EMBO J. 18, 595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quistgaard E. M., Madsen P., Groftehauge M. K., Nissen P., Petersen C. M., Thirup S. S. (2009) Nat. Struct. Mol. Biol. 16, 96–98 [DOI] [PubMed] [Google Scholar]
- 20. Nielsen M. S., Madsen P., Christensen E. I., Nykjaer A., Gliemann J., Kasper D., Pohlmann R., Petersen C. M. (2001) EMBO J. 20, 2180–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petersen C. M., Nielsen M. S., Nykjaer A., Jacobsen L., Tommerup N., Rasmussen H. H., Roigaard H., Gliemann J., Madsen P., Moestrup S. K. (1997) J. Biol. Chem. 272, 3599–3605 [DOI] [PubMed] [Google Scholar]
- 22. Kjolby M., Andersen O. M., Breiderhoff T., Fjorback A. W., Pedersen K. M., Madsen P., Jansen P., Heeren J., Willnow T. E., Nykjaer A. (2010) Cell Metab. 12, 213–223 [DOI] [PubMed] [Google Scholar]
- 23. Chen Z. Y., Ieraci A., Teng H., Dall H., Meng C. X., Herrera D. G., Nykjaer A., Hempstead B. L., Lee F. S. (2005) J. Neurosci. 25, 6156–6166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vaegter C. B., Jansen P., Fjorback A. W., Glerup S., Skeldal S., Kjolby M., Richner M., Erdmann B., Nyengaard J. R., Tessarollo L., Lewin G. R., Willnow T. E., Chao M. V., Nykjaer A. (2011) Nat. Neurosci. 14, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu F., Padukkavidana T., Vaegter C. B., Brady O. A., Zheng Y., Mackenzie I. R., Feldman H. H., Nykjaer A., Strittmatter S. M. (2010) Neuron 68, 654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nykjaer A., Lee R., Teng K. K., Jansen P., Madsen P., Nielsen M. S., Jacobsen C., Kliemannel M., Schwarz E., Willnow T. E., Hempstead B. L., Petersen C. M. (2004) Nature 427, 843–848 [DOI] [PubMed] [Google Scholar]
- 27. Horton R. M., Cai Z. L., Ho S. N., Pease L. R. (1990) BioTechniques 8, 528–535 2357375 [Google Scholar]
- 28. Lennon G., Auffray C., Polymeropoulos M., Soares M. B. (1996) Genomics 33, 151–152 [DOI] [PubMed] [Google Scholar]
- 29. Moon R. T., Christian J. L. (1989) Technique 1, 76–89 [Google Scholar]
- 30. Nieuwkoop P. D., Faber J. (1967) Normal Table of Xenopus laevis, North Holland Publishing Co., Amsterdam [Google Scholar]
- 31. Harland R. M. (1991) Methods Cell Biol. 36, 685–695 [DOI] [PubMed] [Google Scholar]
- 32. Blanchet M. H., Le Good J. A., Mesnard D., Oorschot V., Baflast S., Minchiotti G., Klumperman J., Constam D. B. (2008) EMBO J. 27, 2580–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ni X., Morales C. R. (2006) Traffic 7, 889–902 [DOI] [PubMed] [Google Scholar]
- 34. Heasman J. (2006) Development 133, 1205–1217 [DOI] [PubMed] [Google Scholar]
- 35. Chang C., Brivanlou A. H., Harland R. M. (2006) J. Biol. Chem. 281, 30794–30803 [DOI] [PubMed] [Google Scholar]
- 36. Nakayama T., Snyder M. A., Grewal S. S., Tsuneizumi K., Tabata T., Christian J. L. (1998) Development 125, 857–867 [DOI] [PubMed] [Google Scholar]
- 37. Jansen P., Giehl K., Nyengaard J. R., Teng K., Lioubinski O., Sjoegaard S. S., Breiderhoff T., Gotthardt M., Lin F., Eilers A., Petersen C. M., Lewin G. R., Hempstead B. L., Willnow T. E., Nykjaer A. (2007) Nat. Neurosci. 10, 1449–1457 [DOI] [PubMed] [Google Scholar]
- 38. Walsh D. W., Godson C., Brazil D. P., Martin F. (2010) Trends Cell Biol. 20, 244–256 [DOI] [PubMed] [Google Scholar]
- 39. Zeng J., Racicott J., Morales C. R. (2009) Exp. Cell Res. 315, 3112–3124 [DOI] [PubMed] [Google Scholar]
- 40. Boutilier J., Ceni C., Pagdala P. C., Forgie A., Neet K. E., Barker P. A. (2008) J. Biol. Chem. 283, 12709–12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bronfman F. C., Tcherpakov M., Jovin T. M., Fainzilber M. (2003) J. Neurosci. 23, 3209–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spoelgen R., von Arnim C. A., Thomas A. V., Peltan I. D., Koker M., Deng A., Irizarry M. C., Andersen O. M., Willnow T. E., Hyman B. T. (2006) J. Neurosci. 26, 418–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt V., Sporbert A., Rohe M., Reimer T., Rehm A., Andersen O. M., Willnow T. E. (2007) J. Biol. Chem. 282, 37906–37912 [DOI] [PubMed] [Google Scholar]
- 44. Andersen O. M., Reiche J., Schmidt V., Gotthardt M., Spoelgen R., Behlke J., von Arnim C. A., Breiderhoff T., Jansen P., Wu X., Bales K. R., Cappai R., Masters C. L., Gliemann J., Mufson E. J., Hyman B. T., Paul S. M., Nykjaer A., Willnow T. E. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13461–13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi J., Kandror K. V. (2005) Dev. Cell 9, 99–108 [DOI] [PubMed] [Google Scholar]
- 46. Shi J., Kandror K. V. (2007) J. Biol. Chem. 282, 9008–9016 [DOI] [PubMed] [Google Scholar]
- 47. Bogan J. S., Kandror K. V. (2010) Curr. Opin. Cell Biol. 22, 506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lefrancois S., Zeng J., Hassan A. J., Canuel M., Morales C. R. (2003) EMBO J. 22, 6430–6437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim T., Hempstead B. L. (2009) EMBO J. 28, 1612–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wolins N., Bosshart H., Küster H., Bonifacino J. S. (1997) J. Cell Biol. 139, 1735–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lamark T., Johansen T. (2010) Curr. Opin. Cell Biol. 22, 192–198 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.