

Abstract
Daptomycin (DAP) is bactericidal against methicillin-resistant Staphylococcus aureus (MRSA) in vitro, but it failed to eradicate MRSA in an experimental model of implant-associated infection. We therefore investigated various factors which could explain treatment failure by evaluating DAP activity, including the role of different cell wall components, adherence, biofilm, and calcium ions (Ca2+) in vitro and in vivo. In the tissue cage infection model, DAP was active only prophylactically and against low inocula. To identify the mechanisms of treatment failure, the in vitro activity of DAP against planktonic and adherent growing S. aureus and S. epidermidis mutants, differing in their capacity of biofilm formation and adherence, was determined. For planktonic staphylococci, the MIC was 0.625 μg/ml. For adherent staphylococci, DAP reduced biofilms at 30 μg/ml. However, it did not kill adherent bacteria up to 500 μg/ml, independent of biofilm biosynthesis (the ica mutant strain), nuclease (the nuc1/nuc2 mutant strain), LPXTG-anchored adhesin (the srtA mutant strain), autolysin (the atl mutant strain), or alanyl-LTA (the dltA mutant strain). Resistance of adherent staphylococci was not due to mutations of adherent bacteria, since staphylococci became DAP susceptible after detachment. Phenotypic tolerance was not explained by inactivation of DAP or inability of initial Ca2+-DAP complex formation. However, the addition of up to 100 mg/liter (2.5 mmol/liter) Ca2+ gradually improved bactericidal activity toward adherent staphylococci in vitro and increased the prevention rate in the cage model from 40% to 60%. In summary, adherent staphylococci are resistant to DAP killing unless Ca2+ is supplemented to physiologic concentrations.
INTRODUCTION
Prosthetic joints are increasingly used to maintain a good quality of life in patients with damaged joints. Hip and knee implants have good long-term results (19). However, they carry the risk of bacterial infection. Staphylococci (Staphylococcus epidermidis and Staphylococcus aureus) are the most frequent causes of periprosthetic hip and knee infections (S. epidermidis, 30 to 43% of infections; S. aureus, 12 to 23%) (43). Immediately after implantation, extracellular plasma proteins cover the implant surface. Staphylococci adhere to these proteins through their microbial surface components recognizing adhesive matrix molecules (MSCRAMMs). Subsequently, staphylococci aggregate to form a biofilm, which consists of polysaccharide intercellular adhesin (PIA) (10), extracellular DNA (32), or proteins (fibronectin binding proteins or Aap/SasG) (10, 26). The biofilm matures into a three-dimensional structure and undergoes quorum sensing-controlled dispersion at its surface (27). The biosynthetic enzymes of PIA are encoded by the ica operon. This is controlled by global regulatory networks, which suppress virulence factor gene expression and thereby maintain this special mode of growth (10, 15, 24, 27). The gene changes, which stabilize staphylococci in stationary phase in biofilm, may also explain the limited activity of antibiotics that target growing cells against bacteria in biofilms (5, 10, 27, 28). The biofilm further confers resistance against innate host defense by preventing bacterial complement binding and reducing phagocytosis (16, 40).
For successful treatment of device-related infections, drugs with bactericidal effects on surface-adhering, slow-growing, and biofilm-producing microorganisms are needed. These antimicrobial compounds should penetrate the biofilm, act independently of the bacterial physiological state, and prevent further adherence and biofilm formation. So far, antibiofilm drugs such as dispersin have been tested in vitro. However, none of the compounds significantly eradicated biofilms when applied alone (20, 27).
A promising candidate might be the cyclic lipopeptide daptomycin (DAP). Despite its high affinity for proteins, it exhibits concentration-dependent bactericidal activity against Gram-positive organisms, including methicillin-resistant S. aureus (MRSA) (35). It leads to rapid calcium-dependent cell death due to membrane depolarization (13, 34, 37). The bacterial membrane is the only target for DAP. It has been previously shown that DAP does not require cell division or active metabolism for bactericidal activity, although it is more active against growing staphylococci (12, 22). However, we and others could previously show that DAP was not able to eradicate adherent staphylococci in an implant-associated infection model at clinically relevant doses (12, 25). We therefore investigated the mechanism of phenotypic tolerance of adherent staphylococci to DAP in vitro and in vivo in the present study.
We found that DAP treatment failed to eliminate adherent staphylococci independent of biofilm formation. However, by increasing the DAP or calcium concentration, the efficacy of DAP against adherent bacteria was improved in vitro and in the implant model in vivo.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The following staphylococcal strains were used: Staphylococcus aureus ATCC 43300, a PIA-negative (PIA−) clinical isolate resistant to methicillin (MRSA); PIA-positive (PIA+) (17) S. aureus SA113 wild type (wt) (ATCC 35556) and its isogenic Δnuc1/nuc2, ΔsrtA (PIA+), Δica, Δatl, and ΔdltA (PIA−) mutants (kindly provided by F. Götz and Andreas Peschel); S. epidermidis SE1457 wt, which forms a strong PIA-mediated biofilm in vitro and in vivo (11, 16), and the isogenic S. epidermidis SE1457 ΔluxS::ermB mutant (kindly provided by M. Otto). For the analysis of DAP concentrations in the tissue cage fluid, Kocuria rhizophila (ATCC 9341), formerly known as Micrococcus luteus, was used as an indicator organism in the bioassay.
The strains were stored at −70°C in a cryovial bead preservation system (Microbank; Pro-Lab Diagnostics, Richmond Hill, Ontario, Canada). For preparation of the inoculum, a bead was incubated in 1 ml of Trypticase soy broth (TSB) (Becton Dickinson and Company, Allschwil, Switzerland) for 7 h at 37°C, diluted 1:100 in fresh TSB, and incubated overnight at 37°C without shaking. The overnight culture was diluted 1:100 and further incubated 5 to 6 h at 37°C to reach the logarithmic growth phase. Afterwards, bacteria were washed twice with 0.9% saline (Bichsel, Interlaken, Switzerland) and diluted to the needed inoculum. Bacterial numbers were determined by plating aliquots from appropriate dilutions on agar, followed by colony counting after 24 h of incubation at 37°C.
Antimicrobial agent.
Daptomycin was supplied by Novartis Pharma Schweiz AG (Bern, Switzerland). Stock solutions were prepared in pyrogen-free 0.9% saline. It was supplemented with 50 mg/liter (1.25 mmol/liter) calcium ions (Ca2+) (CaCl2) in vitro, unless indicated otherwise.
Animal model.
We used a mouse model of foreign-body infection established by Kristian et al. (17). The method was approved by the review board of the Kantonale Veterinaeramt Basel-Stadt (permit no. 1710). Experiments were conducted according to the regulations of the Swiss veterinary law. Male C57BL/6 mice, 9 to 11 weeks old (Harlan Laboratories, Switzerland), kept in the Animal House of the Department of Biomedicine, University Hospital Basel, were anesthetized via intraperitoneal (i.p.) injection of 65 mg/kg ketamine (Ketalar; Warner-Lambert) and 13 mg/kg xylazium (Xylasol; Graeub). A sterile tissue cage (Angst + Pfister AG, Zurich, Switzerland) containing eight sintered-glass beads (Sikuf, Schott Schleifer, Muttenz, Switzerland) was implanted subcutaneously. After surgery, mice were treated with 0.05 mg/kg buprenorphine (Temgesic; Essex Chemie AG, Luzern) to treat postoperative pain. After complete wound healing (2 weeks), cages were tested for sterility by culturing the aspirated tissue cage fluid (TCF).
Pharmacokinetic study.
A single i.p. dose of 30 or 40 mg/kg DAP was injected (6 mice per group). The pharmacokinetic levels of DAP were investigated in TCF of uninfected mice at various time points (2, 4, 6, 8, and 24 h after drug administration). TCF was collected by cage puncture and centrifuged at 2,100 × g for 7 min. The supernatant was stored at −20°C until further analysis.
The concentration of DAP was evaluated by a previously described bioassay method (33). Briefly, 4 × 103 to 5 × 103 CFU/ml of a 6-h K. rhizophila (ATCC 9341) culture was added to antibiotic medium 11 (Difco, Becton Dickinson and Company, Allschwil, Switzerland) and used to fill bioassay dishes (Fisher Scientific, Wohlen, Switzerland). Samples in duplicate were applied in punched holes. A standard curve was established with a range from 1 to 128 μg/ml DAP in phosphate-buffered saline (PBS) (0.01 M, pH 7.4) supplemented with one volume of sterile TCF. To determine the DAP concentrations in TCF over time, the diameters of the inhibition zones of the standard probes were plotted against the logarithm of the concentrations.
Minimal infective dose (MID).
To evaluate the MID for MRSA, 102 to 105 CFU was injected into the tissue cage (3 mice per group). At different time points, TCF was collected in 1.5% EDTA (in 0.45% NaCl, pH 7.3) and bacterial numbers were determined by plating. The MID was defined as the count of CFU/tissue cage which was required to induce a persistent infection (15 days) in 100% of the tissue cages.
Prophylaxis study.
DAP (40 mg/kg, administered to 5 mice per group) was i.p. administered 6 h before the injection of 3 × 102 CFU MRSA cells. In a second approach, an additional i.p. dose of 40 mg/kg DAP was administered 6 h after the injection of MRSA. In a third approach, the Ca2+ concentration in the cage was increased by adding 50 mg/liter at the time of infection. In all three approaches, saline served as a control. TCF was collected in EDTA, and tissue cages were explanted 24 h after inoculation. TCF was used to determine the efficacy of DAP against planktonic bacteria by plating aliquots from appropriate dilutions on blood agar plates. The efficacy was expressed as the difference in bacterial counts (Δlog10 CFU/ml) between inoculation and 24 h later. The tissue cages were incubated in TSB for 48 h at 37°C before the prevention rate was determined by plating the supernatant. The prevention rate is defined as the number of cages without growth divided by the total number of inoculated cages.
Treatment study (1-day infection).
Mice were infected with 4 × 104 and 3 × 102 CFU MRSA cells, respectively. After 24 h, TCF was aspirated and bacterial numbers were determined by plating to prove an established infection. For therapy, infected mice were treated with saline (control group) and 40 mg/kg DAP i.p. every 24 h for 4 days before the animals were sacrificed 5 days later. On day 9, TCF was collected to quantify planktonic bacteria and tissue cages were explanted to determine the efficacy of DAP against adherent MRSA (cure rate). The efficacy of DAP against planktonic bacteria was quantified as the difference in bacterial counts (Δlog10 CFU/ml) before and 5 days after the end of treatment. The cure rate was defined as the number of cages without growth divided by the total number of cages in the individual treatment group.
Biofilm assay.
MRSA cells at 105 CFU/ml were seeded into flat-bottom 96-well plates (Becton Dickinson and Company, Allschwil, Switzerland) and treated with 30 μg/ml and 500 μg/ml DAP. After 24 h of incubation at 37°C, biofilm was stained using crystal violet as previously described (18). Briefly, supernatants were removed by using a dropper and plates were washed twice with PBS. The biofilm was fixed by incubating plates for 60 min at 60°C and stained with 100 μl of a 0.5% crystal violet solution for 20 min at room temperature (RT). After the plates were washed under running tap water, 100 μl of 33% acetic acid was added, and the optical density was measured at 590 nm using a Molecular Devices reader (Applied Biosystems, Rotkreuz, Switzerland).
In vitro susceptibility.
According to the CLSI guidelines, a standard inoculum of 1 × 105 to 5 × 105 CFU/ml was used. The MIC (lowest DAP concentration that inhibits visible bacterial growth) and the minimal bactericidal concentration in the logarithmic growth phase (MBClog) (lowest DAP concentration which kills ≥99.9% of the initial bacterial count in 24 h) (23) were determined by using 2-fold dilutions of DAP in Mueller-Hinton broth (4).
Daptomycin effects on adherent and detached staphylococci.
MRSA, SA113 wt and its isogenic mutants, and SE1457 wt and its isogenic mutant were incubated at 105 CFU/ml in TSB supplemented with additional 0.5% glucose for 18 h at 37°C in uncoated or 50% plasma-precoated (2 h, RT) flat-bottom 96-well plates. After nonadherent bacteria were washed away with PBS, adherent bacteria were treated with DAP at 0 μg/ml, 30 μg/ml, and 500 μg/ml for 24 h at 37°C. Then biofilm was stained using crystal violet as described above and adherent bacterial numbers were determined by plating after detachment. To avoid cell clusters, adherent bacteria were detached carefully by pipetting up and down.
To investigate the efficacy of DAP against detached bacteria, adherent MRSA cells were detached by pipetting up and down after 18 h of seeding. Detached MRSA cells were treated with DAP at 0 μg/ml and 30 μg/ml for 24 h, the optical density was measured at 590 nm, and viable counts were determined. DAP efficacy was also tested against dispersed MRSA ex vivo. To that aim, tissue cages without sintered glass beads from the 1-day infection study were incubated in TSB for 48 h at 37°C. Dispersed MRSA cells (105 CFU/ml) were incubated in a 96-well plate with 0 μg/ml, 0.625 μg/ml, or 30 μg/ml DAP. After incubation for 24 h at 37°C, the optical density at 590 nm was measured. TSB without MRSA and DAP served as a negative control.
To evaluate the activity of DAP after 24 h of treatment of adherent MRSA, the supernatant was collected, filtered under sterile conditions, and added to 105 CFU/ml fresh planktonic MRSA cells. After 24 h at 37°C, the optical density at 590 nm was measured.
Calcium competition assay.
Adherent MRSA cells were treated with nonpreincubated or Ca2+-preincubated DAP (30 μg/ml with 50 mg/liter Ca2+ for 2 h at RT). After 24 h of incubation at 37°C, adherent bacterial numbers were determined after detachment.
To confirm the effect of additional Ca2+, adherent MRSA cells were treated with 30 μg/ml DAP supplemented with either 75 mg/liter (1.8 mmol/liter) or 100 mg/liter (2.5 mmol/l) Ca2+. The difference in adherent bacterial counts before and after 24 h of incubation was plotted.
Statistical analysis.
Treatment effects were analyzed with the Mann-Whitney U test. A two-way analysis of variance (ANOVA) test was used for statistical analysis of the in vitro data. A P value of <0.05 was considered statistically significant. Statistical analysis was done with 5.0a Prism (GraphPad Software).
RESULTS
Daptomycin efficacy is influenced by infection time and inoculum in vivo.
We previously reported that DAP monotherapy was not successful against implant-associated MRSA infection in guinea pigs (12). The aim of the present study was to elucidate this treatment failure by evaluating DAP in the tissue cage model in mice. This model is suitable to study phenotypic staphylococcal resistance against antimicrobial treatment (9, 18). We first performed pharmacokinetic studies in sterile tissue cage fluid (TCF) of mice after intraperitoneal (i.p.) administration of 30 and 40 mg/kg DAP. The peak concentrations (Cmax) for the 30-and 40-mg/kg doses were 25 and 35 μg/ml, respectively. For both doses, the Cmax was reached after 6 h and was above the MBC in the stationary growth phase (MBCstat) of 20 μg/ml (12) during 4 to 8 h. The DAP concentration after 24 h (Cmin) remained above the MIC and MBClog of 0.625 μg/ml (12) (Fig. 1). For the following treatment studies, DAP at 40 mg/kg was used.
Fig. 1.

Pharmacokinetics of daptomycin (DAP) in sterile cage fluid of mice after a single intraperitoneal administration of a 30-mg/kg (squares) and 40-mg/kg (circles) dose. Values are means ± SD. The horizontal dotted lines indicate the MIC and MBClog (below) and MBCstat (above) of MRSA (S. aureus ATCC 43300) for DAP.
We asked whether DAP efficacy was better when a lower inoculum was used or when given prophylactically, i.e., before establishment of a biofilm. The minimal infective dose (MID) of MRSA cells for induction of a persistent infection in tissue cages of C57BL/6 mice was 3 × 102 CFU/cage. Therefore, all experiments were performed with at least this inoculum.
First, the efficacy of a 4-day treatment of DAP against planktonic and adherent MRSA was assessed in a 1-day infection study with an inoculum of either ∼104 or ∼102 CFU/cage (Fig. 2). One day after infection, planktonic bacterial numbers had increased more than 2-fold over numbers in the initial inoculum (data not shown). Five days after the end of treatment, the numbers of planktonic bacteria were not significantly reduced (0.6 log10 CFU/ml) and DAP failed to cure any tissue cages infected with the high inoculum (Fig. 2A and B). In contrast, DAP killed 3 log10 CFU/ml planktonic bacteria and cured 33% of tissue cage-associated infections in mice with the low inoculum (Fig. 2A and B). As expected, untreated mice showed an increase of 2.7 log10 planktonic CFU/ml and no spontaneous cure (Fig. 2A and B).
Fig. 2.

Influence of infection time and inoculum on the efficacy of daptomycin at 40 mg/kg (open bars) in a 1-day infection and a prophylaxis study against planktonic (A) and adherent (B) MRSA. Inocula of the 1-day infection and prophylaxis study were 4 × 104 CFU/cage and 3 × 102 to 4 × 102 CFU/cage, respectively. Saline (gray bars) served as control. In panel A, positive values denote growth and negative values killing. In panel B, efficacy against adherent MRSA is expressed as cure or prevention rate.
To study whether DAP prevents colonization of implants, we administered 40 mg/kg DAP prophylactically 6 h before infection with the MID inoculum of 3 × 102 CFU. One day after infection, untreated mice showed an increase of 1.3 log10 CFU/ml (Fig. 2A). DAP reduced the planktonic bacteria by 2.3 log10 CFU/ml to minimal CFU numbers, and it was able to prevent 40% tissue cage-associated infections (Fig. 2B). In a second series of experiments, a second 40-mg/kg DAP dose was applied 6 h after infection in order to achieve prolonged bactericidal drug levels in the cage. Under these conditions, CFU numbers were reduced by 3 log10 CFU/ml to a few counts within 24 h and DAP prevented infection in all tissue cages (Fig. 2B).
Taken together, DAP was partially efficacious in the 4-day therapy only when a low inoculum was used. Complete prevention of cage-associated infection was reached only with 2 consecutive doses of DAP before and after inoculation.
Daptomycin is not bactericidal on adherent staphylococci in vitro.
Since DAP did not eradicate tissue cage-associated infection when treatment was started 1 day after inoculation, we asked whether the biofilm prevented killing. In addition, the effect of cell wall modifications on DAP susceptibility was assessed in order to link phenotypic resistance of adherent staphylococci to defined molecules participating in adherence.
First, we compared the susceptibility of planktonic wild-type (wt) and mutant staphylococci to DAP in vitro (Table 1). All tested SA113 and SE1457 strains had a MIC of 0.625 μg/ml and MBClog of 1.25 μg/ml except for the ΔdltA mutant, which was more sensitive. The ΔdltA mutant lacks alanylation of lipoteichoic acid (LTA) and expresses less biofilm (10). DAP was similarly efficient against planktonic staphylococci independent of PIA biofilm (the Δica mutant strain) or biofilm regulation by luxS. The ΔluxS mutant shows a higher ica gene transcription and more biofilm (41). Further, the MIC for DAP was unaltered in the Δnuc1/nuc2 mutant, which accumulates undegraded extracellular DNA and has more PIA-independent biofilm (21). Finally, in the ΔsrtA mutant, lacking LPXTG-anchored cell wall molecules, and in the Δatl mutant, missing autolysins and biofilm (3), the MIC for DAP was also unaffected. These results suggest that only the positive charge conferred by alanine to LTA affected DAP efficacy on planktonic staphylococci.
Table 1.
In vitro susceptibility to daptomycin
| Strain and genotype | MICa (μg/ml) | MBClogb (μg/ml) |
|---|---|---|
| S. aureus SA113 | ||
| wt | 0.625 | 1.25 |
| Δica | 0.625 | 1.25 |
| Δnuc1/nuc2 | 1.25 | 1.25 |
| ΔsrtA | 0.625 | 1.25 |
| Δatl | 0.625 | 1.25 |
| ΔdltA | 0.157 | 0.313 |
| S. epidermidis SE1457 | ||
| wt | 0.625 | 1.25 |
| ΔluxS | 0.625 | 1.25 |
MIC, minimal inhibition concentration.
MBClog, minimal bactericidal concentration under logarithmic growth phase conditions.
In addition, the effect of DAP on adherent staphylococci was assessed. The DAP effects both on PIA-positive or PIA-independent biofilm and on adherent staphylococci were quantified 24 h after incubation with 30 and 500 μg/ml DAP. Biofilm was measured with crystal violet staining. This stain does not discriminate between live and dead bacteria; it binds to negatively charged surface molecules and polysaccharides (29). The crystal violet staining was moderate in untreated SA113 wt and the ΔsrtA mutant, lower in the Δica, Δatl, and ΔdltA mutants, and 3-fold higher in the Δnuc1/nuc2 mutant (Fig. 3 A). DAP eliminated biofilm in all S. aureus strains except the Δnuc1/nuc2 mutant, where it remained unchanged with 30 μg/ml DAP (Fig. 3A) and was decreased only after 500 μg/ml DAP (data not shown). Untreated SE1457 wt and its ΔluxS mutant produced very strong biofilm, which was reduced 2- to 3-fold by DAP in both strains (Fig. 3A).
Fig. 3.

Efficacy without (gray bars) and with (black bars) 30 μg/ml daptomycin on adherent staphylococci in vitro. After daptomycin exposure, biofilm formation (A) using crystal violet staining and adherent bacterial numbers (B) using plating after detachment were determined. Values are means ± SD of results of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.01.
Adherent staphylococci numbered 107 to 108 CFU per well except for the Δnuc1/nuc2 mutant, which showed higher numbers of 1011 CFU/well. DAP did not significantly lower CFU counts independent of biofilm, nucleases, adhesins, autolysins, and alanyl-LTAs (Fig. 3B). Precoating of the plates with plasma proteins did also not allow a significant eradication of adherent staphylococci (data not shown).
Tolerance to daptomycin is linked to physiological status of bacteria.
These surprising results suggest that, despite an effect on the biomass, DAP was not bactericidal on adherent cells. To investigate whether DAP resistance of adherent staphylococci was reversible, the efficacy of 30 μg/ml DAP against detached MRSA was determined. DAP was found to be bactericidal against detached MRSA cells 24 h after incubation (Fig. 4 A and B). To confirm these in vitro data, dispersed MRSA cells from untreated (saline) or treated (40 mg/kg DAP) tissue cages of the 1-day infection study were treated with 30 μg/ml DAP ex vivo (Fig. 4C). DAP was bactericidal, and the effect was independent of a previous in vivo DAP treatment. These data indicate that adherent bacteria were not genotypically resistant to DAP but tolerant. This tolerance was linked to the physiological status of the bacteria and therefore fully reversible after detachment.
Fig. 4.

Efficacy of daptomycin (DAP) against detached MRSA in vitro (A, OD measurement; B, viable counts) and against dispersed MRSA derived from untreated and treated tissue cages of the 1-day infection study (C, OD measurement). Values are means ± SD of results of three experiments. **, P < 0.01; ***, P < 0.01.
To determine whether adherent bacteria inactivated DAP, the bactericidal activity of a supernatant of adherent, treated MRSA was tested with fresh planktonic MRSA. DAP in the supernatant was able to inhibit growth of planktonic MRSA, indicating that adherent bacteria did not inactivate DAP (Fig. 5).
Fig. 5.
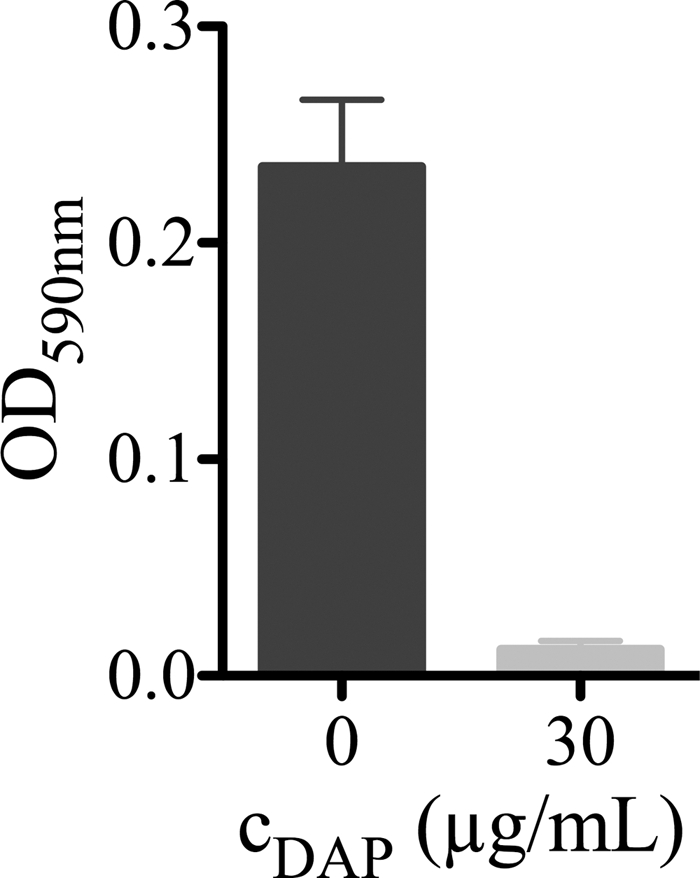
Efficacy of daptomycin (DAP) in sterile-filtered supernatant, derived from adherent DAP-treated MRSA against fresh planktonic 105 CFU/ml MRSA cells. Values are means ± SD of results of three experiments.
Daptomycin efficacy is partially overcome by increasing Ca2+ concentrations.
Adherent staphylococci did not inactivate DAP but might restrict accessible calcium ions (Ca2+) required for killing by DAP. Therefore, we asked whether there was a calcium competition between DAP and staphylococci. To answer this question, 30 μg/ml DAP was preincubated with 50 mg/liter Ca2+ for 2 h to lock DAP in its active conformation. This time period was considered sufficient for the formation of the active DAP conformation because time-kill studies showed bactericidal activity against stationary-phase-grown staphylococci above 20 μg/ml of DAP within 1 to 2 h (12). The efficacy of Ca2+-preincubated DAP against adherent MRSA was similar to that of nonpreincubated DAP, since numbers of adherent MRSA cells were similar (9.8 × 106 and 1.7 × 107 CFU/ml, respectively).
Increasing Ca2+ may enhance bactericidal activity of DAP on adherent bacteria. Therefore, the influence of a Ca2+ concentration above 50 mg/liter on DAP efficiency against adherent MRSA was determined. Increasing physiological Ca2+ concentrations added to 30 μg/ml DAP progressively reduced adherent bacterial numbers; 100 mg/liter Ca2+ led to a significant reduction of adherent MRSA by 2.5 log10 CFU/ml (Fig. 6 A). Ca2+ alone had no effect on bacterial survival (data not shown). To confirm these in vitro data, we investigated the effect of DAP treatment in a tissue cage MRSA infection with increased Ca2+ concentrations. One day after infection, DAP with an additional 50 mg/liter Ca2+ reduced the planktonic MRSA cell count by 2.7 log10 CFU/ml (Fig. 6B). Furthermore, DAP prophylaxis with additional Ca2+ was able to prevent 60% of the cage-associated infections (Fig. 6C). These data indicate that with increasing physiologic Ca2+ concentrations the phenotypic resistance of adherent bacteria can be partially overcome.
Fig. 6.

(A) Influence of increasing calcium concentrations on efficacy of 30 μg/ml daptomycin. The difference in adherent MRSA cell numbers before and after daptomycin exposure is shown. Negative values denote killing. Values are means ± SD of results of six experiments. **, P < 0.01. (B and C) Influence of additional calcium ions (50 mg/liter) on daptomycin (DAP) efficiency (light gray bars) against planktonic (B) and adherent MRSA (C) in vivo. Inocula were 6 × 102 CFU/cage. Saline (open bars) and DAP without additional calcium (dark gray bars) served as controls. In panel B, positive values denote growth and negative values killing. Values are means ± SD. In panel C, efficacy against adherent MRSA was expressed as the prevention rate.
DISCUSSION
Daptomycin (DAP) has potent activity against a wide range of Gram-positive bacteria, including beta-lactam- and vancomycin-resistant strains (35). It is approved for skin infections, bacteremia, and endocarditis (7). However, in experimental implant-associated infection, DAP monotherapy failed for unknown reasons (12).
Earlier results showed that stationary-phase bacteria had a 16-fold-higher MBC for DAP than bacteria in logarithmic growth phase (22). As the fraction of stationary-phase bacteria increases with time after inoculation, a time-dependent failure rate of DAP treatment (12) probably resulted. Also in the present study in mice, DAP was ineffective after 1 day of infection; only the prophylactic application of DAP was highly efficacious. This suggests that treatment failure was due to the high quantity and/or the physiological state of the bacteria in the tissue cage. In this model, staphylococci change from the planktonic to the adherent phenotype and are increasingly embedded in a biofilm. Therefore, we investigated the effect of adherence and of biofilms on DAP responsiveness.
We observed the expected strong biofilm in S. epidermidis SE1457 wt and even more so in the ΔluxS mutant, which lacks the quorum system with autoinducer 2-mediated ica repression (41). We confirmed low biofilm formation by all S. aureus strains except for the Δnuc1/nuc2 mutant, which lacks extracellular DNase and therefore accumulates more DNA-containing biofilm (2). Independent of the amount of biofilm in untreated cells, DAP strongly reduced the biofilm at concentrations above the MBClog except in the Δnuc1/nuc2 mutant, which was more resistant against DAP. It was shown earlier that DAP is able to enter a biofilm (36); it may lead to biofilm dispersion via modulation of the cell membrane.
Our data show that the adherent growth mode and not the extracellular polysaccharide matrix formation was responsible for the DAP resistance to killing. A similar phenomenon was shown before for other antibiotics (31, 38). These authors described a high resistance against oxacillin, vancomycin, teicoplanin, ciprofloxacin, and rifampin of adherent biofilm-positive and biofilm-negative S. epidermidis and of S. aureus strains, which were sensitive to all antibiotics in their planktonic state. Based on these results, the resistance to DAP in our study was likely independent of the antibiotic structure and mechanism of action. Accordingly, we found no evidence for inactivation of DAP by staphylococci. Furthermore, the reversibility of the resistant phenotype upon detachment excluded the genetic perturbations, which had been previously associated with DAP resistance, including point mutations of mprF, rpoB, and yyCG (1, 6, 8). However, it appeared that adherent staphylococci adapted to DAP and became tolerant, resulting in treatment failure. This may be due to their metabolic status, with a higher net positive charge, which accompanies all adherence (39), and with an enhanced membrane stability. Support for this hypothesis is provided by three observations. First, the ΔdltA mutant, which has a lower positive charge and thus adheres less to polystyrene (30), expressed an enhanced DAP susceptibility. Second, a DAP-resistant clinical isolate showed enhanced ΔdltA expression (42). Finally, DAP resistance was found associated with an mprF mutation, which led to higher lysyl-phosphatidylglycerol in the membrane and thus better membrane integrity (14).
Adherence in a polystyrene plate does not mirror biofilm formation in a medical device, because in the first case hydrophobicity and atl (3) play a role, while in vivo the interaction between MSCRAMMs and host extracellular matrix molecules initiates biofilm (27). We used both adherence settings, with and without plasma coating, and the in vitro assays fairly predicted the in vivo effects.
Interestingly, we could overcome DAP resistance of adherent bacteria in vitro and in vivo by increasing Ca2+ concentrations. This effect was not due to early DAP-Ca2+ complex formation, but it may have altered the ionic forces involved in adhesion.
In conclusion, our data revealed that DAP is inefficient in experimental implant-associated infections and showed that this effect is independent of biofilm but influenced by physiological modulations of extracellular Ca2+.
ACKNOWLEDGMENTS
We thank Werner Zimmerli for critical review of the manuscript, Manuel Battegay for helpful discussion, and Zarko Rajacic for technical assistance.
This study was supported by Thommen Medical AG (Waldenburg, Switzerland).
Footnotes
Published ahead of print on 16 May 2011.
REFERENCES
- 1. Baltz R. H. 2009. Daptomycin: mechanisms of action and resistance, and biosynthetic engineering. Curr. Opin. Chem. Biol. 13:144–151 [DOI] [PubMed] [Google Scholar]
- 2. Bayles K. W. 2007. The biological role of death and lysis in biofilm development. Nat. Rev. Microbiol. 5:721–726 [DOI] [PubMed] [Google Scholar]
- 3. Biswas R., et al. 2006. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol. Lett. 259:260–268 [DOI] [PubMed] [Google Scholar]
- 4. Clinical and Laboratory Standards Institute 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 8th ed. NCCLS documents M07–A8. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 5. Costerton J. W., Stewart P. S., Greenberg E. P. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322 [DOI] [PubMed] [Google Scholar]
- 6. Cui L., et al. 2010. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 54:5222–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eisenstein B. I. 2004. Lipopeptides, focusing on daptomycin, for the treatment of Gram-positive infections. Expert Opin. Invest. Drugs 13:1159–1169 [DOI] [PubMed] [Google Scholar]
- 8. Friedman L., Alder J. D., Silverman J. A. 2006. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 50:2137–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gordon O., et al. 2010. Silver coordination polymers for prevention of implant infection: thiol interaction, impact on respiratory chain enzymes, and hydroxyl radical induction. Antimicrob. Agents Chemother. 54:4208–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gotz F. 2002. Staphylococcus and biofilms. Mol. Microbiol. 43:1367–1378 [DOI] [PubMed] [Google Scholar]
- 11. Hudetz D., et al. 2008. Weak effect of metal type and ica genes on staphylococcal infection of titanium and stainless steel implants. Clin. Microbiol. Infect. 14:1135–1145 [DOI] [PubMed] [Google Scholar]
- 12. John A. K., et al. 2009. Efficacy of daptomycin in implant-associated infection due to methicillin-resistant Staphylococcus aureus: importance of combination with rifampin. Antimicrob. Agents Chemother. 53:2719–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jung D., Rozek A., Okon M., Hancock R. E. 2004. Structural transitions as determinants of the action of the calcium-dependent antibiotic daptomycin. Chem. Biol. 11:949–957 [DOI] [PubMed] [Google Scholar]
- 14. Kilelee E., Pokorny A., Yeaman M. R., Bayer A. S. 2010. Lysyl-phosphatidylglycerol attenuates membrane perturbation rather than surface association of the cationic antimicrobial peptide 6W-RP-1 in a model membrane system: implications for daptomycin resistance. Antimicrob. Agents Chemother. 54:4476–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kong K. F., Vuong C., Otto M. 2006. Staphylococcus quorum sensing in biofilm formation and infection. Int. J. Med. Microbiol. 296:133–139 [DOI] [PubMed] [Google Scholar]
- 16. Kristian S. A., et al. 2008. Biofilm formation induces C3a release and protects Staphylococcus epidermidis from IgG and complement deposition and from neutrophil-dependent killing. J. Infect. Dis. 197:1028–1035 [DOI] [PubMed] [Google Scholar]
- 17. Kristian S. A., et al. 2003. Alanylation of teichoic acids protects Staphylococcus aureus against Toll-like receptor 2-dependent host defense in a mouse tissue cage infection model. J. Infect. Dis. 188:414–423 [DOI] [PubMed] [Google Scholar]
- 18. Kuehl R., et al. 2009. Furanone at subinhibitory concentrations enhances staphylococcal biofilm formation by luxS repression. Antimicrob. Agents Chemother. 53:4159–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Learmonth I. D., Young C., Rorabeck C. 2007. The operation of the century: total hip replacement. Lancet 370:1508–1519 [DOI] [PubMed] [Google Scholar]
- 20. Lee J. H., Kaplan J. B., Lee W. Y. 2008. Microfluidic devices for studying growth and detachment of Staphylococcus epidermidis biofilms. Biomed. Microdevices 10:489–498 [DOI] [PubMed] [Google Scholar]
- 21. Mann E. E., et al. 2009. Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS One 4:e5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mascio C. T., Alder J. D., Silverman J. A. 2007. Bactericidal action of daptomycin against stationary-phase and nondividing Staphylococcus aureus cells. Antimicrob. Agents Chemother. 51:4255–4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murray P. R., Baron E. J., Jorgensen J. H., Pfaller M. A., Yolken R. H. (ed.). 2003. Manual of clinical microbiology, 8th ed. American Society for Microbiology, Washington, DC [Google Scholar]
- 24. Novick R. P., Geisinger E. 2008. Quorum sensing in staphylococci. Annu. Rev. Genet. 42:541–564 [DOI] [PubMed] [Google Scholar]
- 25. Olson M. E., Slater S. R., Rupp M. E., Fey P. D. 2010. Rifampicin enhances activity of daptomycin and vancomycin against both a polysaccharide intercellular adhesin (PIA)-dependent and -independent Staphylococcus epidermidis biofilm. J. Antimicrob. Chemother. 65:2164–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Neill E., et al. 2008. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J. Bacteriol. 190:3835–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Otto M. 2008. Staphylococcal biofilms. Curr. Top. Microbiol. Immunol. 322:207–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otto M. 2009. Staphylococcus epidermidis–the ‘accidental’ pathogen. Nat. Rev. Microbiol. 7:555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peeters E., Nelis H. J., Coenye T. 2008. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J. Microbiol. Methods 72:157–165 [DOI] [PubMed] [Google Scholar]
- 30. Peschel A., et al. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 274:8405–8410 [DOI] [PubMed] [Google Scholar]
- 31. Qu Y., Daley A. J., Istivan T. S., Rouch D. A., Deighton M. A. 2010. Densely adherent growth mode, rather than extracellular polymer substance matrix build-up ability, contributes to high resistance of Staphylococcus epidermidis biofilms to antibiotics. J. Antimicrob. Chemother. 65:1405–1411 [DOI] [PubMed] [Google Scholar]
- 32. Rice K. C., et al. 2007. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 104:8113–8118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schaad H. J., Bento M., Lew D. P., Vaudaux P. 2006. Evaluation of high-dose daptomycin for therapy of experimental Staphylococcus aureus foreign body infection. BMC Infect. Dis. 6:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Silverman J. A., Perlmutter N. G., Shapiro H. M. 2003. Correlation of daptomycin bactericidal activity and membrane depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 47:2538–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steenbergen J. N., Alder J., Thorne G. M., Tally F. P. 2005. Daptomycin: a lipopeptide antibiotic for the treatment of serious Gram-positive infections. J. Antimicrob. Chemother. 55:283–288 [DOI] [PubMed] [Google Scholar]
- 36. Stewart P. S., Davison W. M., Steenbergen J. N. 2009. Daptomycin rapidly penetrates a Staphylococcus epidermidis biofilm. Antimicrob. Agents Chemother. 53:3505–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Straus S. K., Hancock R. E. 2006. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta 1758:1215–1223 [DOI] [PubMed] [Google Scholar]
- 38. Trafny E. A. 1998. Susceptibility of adherent organisms from Pseudomonas aeruginosa and Staphylococcus aureus strains isolated from burn wounds to antimicrobial agents. Int. J. Antimicrob. Agents 10:223–228 [DOI] [PubMed] [Google Scholar]
- 39. Van Oss C. J. 1995. Hydrophobic, hydrophilic and other interactions in epitope-paratope binding. Mol. Immunol. 32:199–211 [DOI] [PubMed] [Google Scholar]
- 40. Vuong C., et al. 2004. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 6:269–275 [DOI] [PubMed] [Google Scholar]
- 41. Xu L., et al. 2006. Role of the luxS quorum-sensing system in biofilm formation and virulence of Staphylococcus epidermidis. Infect. Immun. 74:488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang S. J., et al. 2009. Enhanced expression of dltABCD is associated with the development of daptomycin nonsusceptibility in a clinical endocarditis isolate of Staphylococcus aureus. J. Infect. Dis. 200:1916–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zimmerli W., Trampuz A., Ochsner P. E. 2004. Prosthetic-joint infections. N. Engl. J. Med. 351:1645–1654 [DOI] [PubMed] [Google Scholar]