

Abstract
Mitochondrial proton and electron leak have a major impact on mitochondrial coupling efficiency and production of reactive oxygen species. In the first part of this chapter, we address the molecular nature of the basal and inducible proton leak pathways, and their physiological importance. The basal leak is unregulated, and a major proportion can be attributed to mitochondrial anion carriers, while the proton leak through the lipid bilayer appears to be minor. The basal proton leak is cell-type specific and correlates with metabolic rate. The inducible leak through the adenine nucleotide translocase (ANT) and uncoupling proteins (UCPs) can be activated by fatty acids, superoxide, or peroxidation products. The physiological role of inducible leak through UCP1 in mammalian brown adipose tissue is heat production, whereas the roles of non-mammalian UCP1 and its paralogous proteins, in particular UCP2 and UCP3, are not yet resolved. The second part of the chapter focuses on the electron leak that occurs in the mitochondrial electron transport chain. Exit of electrons prior to the reduction of oxygen to water at cytochrome c oxidase causes the production of superoxide. As the mechanisms of electron leak are crucial to understanding their physiological relevance, we summarize the mechanisms and topology of electron leak from Complex I and III in studies using isolated mitochondria. We also highlight recent progress and challenges of assessing electron leak in the living cell. Finally, we emphasise the importance of proton and electron leak as therapeutic targets in body weight regulation and insulin secretion.
Keywords: Proton leak, electron leak, uncoupling, reactive oxygen species, superoxide, basal proton leak, adenine nucleotide translocase, uncoupling protein, ucp, superoxide, brown adipose tissue, mitochondrial anion carriers, insulin secretion, obesity, aging, semiquinone radical, DNP, GSIS, beta cells
Introduction
The chemiosmotic theory
Prior to Peter Mitchell’s development of the chemiosmotic hypothesis in 1961, the link between respiration and ATP synthesis was unknown and highly debated. The prevailing view at the time was that a high-energy intermediate, derived via the electron transport chain, is responsible for ATP synthesis. Mitchell’s scheme, however, proposed that energy harvested from the respiratory chain is used to establish an electrochemical proton gradient to drive mitochondrial ATP production [1]. This was later substantiated and is now referred to as the chemiosmotic theory. In this theory, a proton circuit across the mitochondrial inner membrane is the mechanism that drives oxidative phosphorylation, coupling substrate oxidation and ADP phosphorylation (Fig. 1). Oxidation of substrates releases electrons to cofactors such as NADH or FADH2. These electrons are passed through electron carriers in respiratory chain complexes with increasing oxidation potentials, ultimately reducing molecular oxygen to water. This exergonic process is used to pump protons from the mitochondrial matrix into the intermembrane space, creating an electrochemical gradient known as the protonmotive force, Δp. Δp drives protons back into the matrix through the ATP synthase, driving the conversion of ADP and inorganic phosphate to ATP and thereby coupling substrate oxidation and ADP phosphorylation.
Figure 1. Proton pumping and leak across the mitochondrial inner membrane.
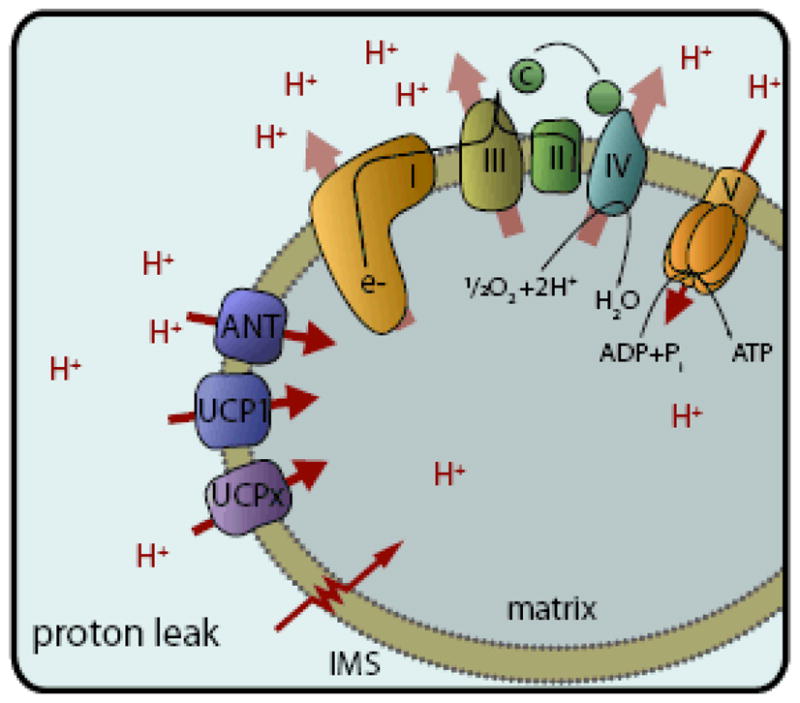
Protons are pumped out of the matrix into the intermembrane space (IMS) by complexes I, III, and IV of the electron transport chain. This establishes a proton motive force ( Δp) across the inner membrane. Proton re-entry through the ATP synthase (complex V) couples the release of Δp to ATP synthesis. All other means of proton re-entry constitute proton leak, as Δp derived from substrate oxidation is depleted without catalysing ATP synthesis. Mechanisms of proton leak include direct movement of protons across the phospholipid membrane (the “water wires” model), diffusion through or around integral membrane proteins, or inducible transport through the adenine nucleotide translocase (ANT) or uncoupling proteins (UCP1, UCPx).
Proton leak
This coupling of ATP synthesis and substrate oxidation is not complete, as protons can return to the matrix independently of ATP synthase. The processes by which this occurs are collectively termed “proton leak”. Proton leak can be demonstrated as the depletion of Δp in the presence of the ATP synthase inhibitor oligomycin. The proton leak behaves in a non-ohmic manner (Fig. 2). This can be seen as an approximately exponential increase of the proton leak rate (usually measured indirectly as the oxygen consumption used to drive it) as Δp rises in isolated mitochondria. Importantly, this behaviour is also apparent in intact cells, demonstrating that the leak is not caused by damage to the mitochondrial inner membrane during the isolation process [2].
Figure 2. The kinetics of the proton leak.
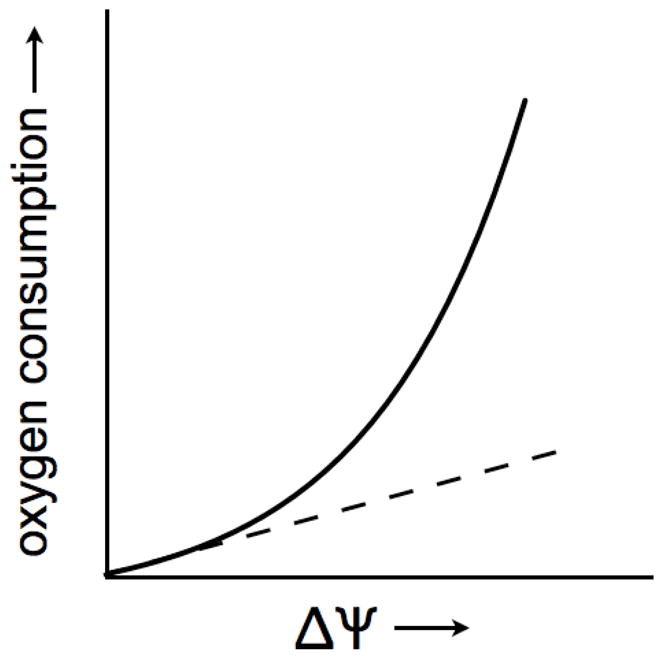
The rate of oxygen consumption as a function of membrane potential increases approximately exponentionally. This can be mistaken for pseudo-linearly when Δp is low, as indicated by the dashed line. As the membrane potential increases, however, a disproportionately large rate of oxygen consumption becomes apparent to defend the membrane potential ( ΔΨ). Therefore, proton leak across the mitochondrial inner membrane is non-ohmic (full line).
Another explanation put forward to explain the disproportionate increase in oxygen consumption at high Δp is “electron slip,” where electrons are transferred through the respiratory complexes, in particular cytochrome c oxidase, without pumping protons into the intermembrane space [3]. This alternative process, however, has not been convincingly demonstrated experimentally to occur under physiological conditions, and is reviewed elsewhere [4, 5].
The first section of this review will discuss the mechanisms of proton leak and its physiological importance for metabolic rate, adaptive non-shivering thermogenesis and body mass regulation.
Electron leak
Mitochondrial superoxide production, caused by electron leak from the respiratory chain, is reviewed in the second part of this chapter. Electron leakage occurs when electrons passed down the respiratory chain exit prior to the reduction of oxygen to water at cytochrome c oxidase, reacting instead with oxygen to form superoxide (O2•−). Although the generation of superoxide by respiratory complexes is a well-established phenomenon, it is poorly understood mechanistically. Superoxide can be dismutated to hydrogen peroxide, or the lipid-soluble hydroperoxyl radical reacts with polyunsaturated fatty acyl groups forming carbon-centred fatty acyl radicals.. At low concentrations, superoxide production may be involved in cellular signal transduction, but at high concentrations the radicals cause oxidative damage due to their high reactivity towards other cellular compounds [6].
Proton and electron leak are intricately linked, as superoxide production is highly sensitive to the decrease in Δp due to proton leak [7]. Referred to as mild uncoupling, increasing proton leak is an exciting therapeutic target to alter pathophysiology associated with mitochondrial superoxide production.
Proton leak
The total proton leak of a mitochondrion can be thought of as the sum of two processes: basal leak, which is unregulated, and inducible leak, which is catalysed by specific mitochondrial inner membrane proteins and can be inhibited and activated.
Molecular nature of the basal proton leak
The precise mechanism of basal proton leak is not fully understood. The model of proton translocation across lipid bilayers posits its occurrence through “water wires”. The magnitude of proton conductance correlates with the phospholipid fatty acyl composition of the mitochondrial membrane, but this effect disappears in liposomes derived from mitochondrial membranes. For example, liver mitochondria from a range of vertebrate species can have a 10-fold difference in basal proton leak, but there is no difference in proton conductance in liposomes prepared from these mitochondria, despite a three-fold difference in the unsaturation index of the phospholipid fatty acyl groups. The proton conductance through the lipid bilayer, however, only accounts for about 5% of total proton leak in rat liver mitochondria. The magnitude of proton conductance also correlates with the abundance of mitochondrial anion carrier proteins such as the adenine nucleotide translocase (ANT) [8; 9]. The interface of these integral membrane proteins and the lipid bilayer may be responsible for the majority of the basal proton leak. Up to two-thirds of the basal leak is attributable to the ANT, a transmembrane protein responsible for shuttling ATP and ADP across the mitochondrial inner membrane [9]. In brown adipose tissue, the high abundance of uncoupling protein 1 (UCP1), which can comprise up to 8% of total mitochondrial protein in mice, also appears to contribute to basal proton leak [10].
Importantly, the proton leak catalysed by these two mitochondrial carriers is not due to protein activity, as proton leak occurs in the presence of the highly specific ANT inhibitor carboxyatractylate and the UCP1 inhibitor GDP. Contribution to basal leak may be a general property of the all mitochondrial inner membrane carrier proteins, but they are generally present in such low abundance that any significant proportion of leak cannot by attributed to other particular transporters. In fact, over twenty mitochondrial carriers were shown to have no significant effect individually on basal proton conductance in yeast mitochondria [11].
The physiological significance of basal proton leak
The basal leak accounts for ~20–30% of the resting metabolic rate of hepatocytes and up to ~50% of the respiration of skeletal muscle of a rat [8]. Considering the high metabolic activity of the liver and the large proportion of skeletal muscle relative to body mass, basal proton leak contributes significantly to basal metabolic rate (BMR), of a resting mammal at thermoneutrality in the postabsorptive state. BMR, which varies with phylogeny, body mass or thyroid status, correlates with basal proton conductance. The phylogenetic relationship between proton leak and metabolic rate was initially studied by comparing proton leak in liver mitochondria between a mammal and a reptile of the same body size acclimated to 37°C. Oxygen consumption of hepatocytes in the reptile was about four times slower, correlating with a 4–5 fold lower proton permeability. This indication of a phylogenetic relationship with increasing proton conductance during evolution was later corroborated in studies using a broader range of ectothermic and endothermic species. It may represent a mechanism to link higher proton conductance through the ANT to higher oxidative phosphorylation rates (and therefore higher ANT concentrations), resulting in protection by mild uncoupling against excessive ROS production in active tissues and organisms [9].
Methodological considerations assaying leak in vivo
In perfused resting rat skeletal muscle, futile proton cycling contributes to ~50% of the respiratory rate [12]. Other studies using magnetic resonance find a smaller contribution (<10%), but this proportion increases during aging [13]. The discrepancy between these methods illustrates that further work is required to resolve the physiological contribution of proton leak.
The proportion of mitochondrial respiration that is used for ATP synthesis is the coupling efficiency, estimated as the proportion of mitochondrial respiration that is sensitive to inhibition of the ATP synthase by oligomycin in living cells. This estimate ignores the small increase in Δp that occurs when ATP synthesis stops. Values corrected for membrane potential are generally ~10% higher [14].
The molecular nature of the inducible proton leak
Inducible proton leak requires activation of mitochondrial anion carrier protein function, and is catalysed by the ANT and UCPs. The uncoupling function of the ANT can be activated by fatty acids and reactive alkenals, such as hydroxynonenal (HNE), and potently inhibited with carboxyatractylate [15].
Uncoupling protein 1
In brown adipose tissue, UCP1 activity leads to a net transport of protons back to the matrix, dissipating Δp and increasing heat production. The high abundance of UCP1, along with mitochondrial biogenesis and increased oxidative capacity, allows small rodents, hibernators and human infants to defend their body temperature in the cold [16]. The UCP1-catalysed proton transport is inhibited by purine nucleoside di- and tri-phosphates and activated by free fatty acids. There are three competing models for the mechanism of proton translocation by UCP1: (a) fatty acids are obligatory cofactors facilitating transport of protons [17]; (b) fatty acid cycling by UCP1 is required for proton transport [18]; (c) UCP1 remains inhibited in the presence of purine nucleotides until fatty acids overcome inhibition by simple competitive kinetics [19; 20]. The molecular activation of UCP1 can be regulated by cellular signal transduction, as noradrenergic stimulation activates lipolysis in brown adipocytes and releases free fatty acids to activate UCP1. UCP1 is not found exclusively in mammalian brown adipose tissue, as a functional UCP1 was recently found in rodent thymocytes, though its physiological relevance there is unclear [21].
Evolution and definition of the core UCP family
The UCP1 gene is present in almost all eutherians (modern mammals), but was lost in pigs by disruption of the UCP1 gene during evolution. It was long assumed that UCP1 is only present in eutherians and promoted the radiation of mammals to cold climates. Surprisingly, orthologues of UCP1 have been identified in fish, amphibians, monotremes and marsupials [22]. While it is unlikely that UCP1 contributes to heat production in ectothermic fish, its function has yet to be determined in these species.
Based on sequence identity, other mitochondrial proteins were grouped into the core UCP family and named UCP2 and UCP3. In vertebrates, UCP2 mRNA appears to be ubiquitously expressed but the protein has only been detected in a few cell types, including macrophages, thymocytes and pancreatic βcells. UCP3 gene expression is restricted to muscle and eutherian brown adipose tissue. In contrast to mammals, birds only possess one UCP, which, like UCP3, appears to be upregulated upon cold exposure, fasting and high fat diets. Phylogenetic inference classifies avian UCP as a UCP3 orthologue [23].
The function of novel uncoupling proteins
The identification of these paralogues prompted speculation of a thermogenic role, but the lack of adaptive non-shivering thermogenesis in UCP1-ablated mice excludes this idea. Their ability to move protons is debated, as the ablation of UCP2 and UCP3 in mice does not result in a changed basal proton conductance. They may, however, catalyse proton leak when appropriately activated [24].
Superoxide, and derived lipid peroxidation products such as HNE, activate UCP-mediated proton conductance. These may act in a negative feedback loop to induce mild uncoupling that subsequently lowers ROS production [24]. These compounds can also induce uncoupling via the ANT. Further complicating matters is the promiscuity of GDP, which is often used to diagnose UCP-catalyzed uncoupling but also decreases proton conductance via the ANT [25].
Other UCP functions have been hypothesised based on physiological observations. Although the transport of fatty acids from the mitochondrial matrix was implicated as a physiologically relevant reaction catalysed by UCP3 during increased lipid metabolism, this hypothesis has been recently refuted [26]. Avian and plant UCP also uncouple in response to superoxide and HNE but have not been further investigated mechanistically.
A recent theoretical approach to predict the substrates of all mitochondrial anion carrier proteins analysed their amino acid sequences. Deviation from the 3-fold pseudosymmetrical structure of these proteins was used to predict substrate specificity [27]. The UCPs were predicted to transport small carboxylic or keto acids. In proteoliposomes, reconstituted UCPs transport protons, fatty acids and a variety of anions including chloride and pyruvate [28].
Electron leak
The initial site of electron loss from the electron transport chain to form superoxide is generally considered to be a semiquinone radical (QH•) or reduced flavin (FMN and FAD) [6]. Identifying the specific redox centre responsible for ROS generation from a particular enzyme is especially pertinent because the rate of ROS production will depend on the degree of reduction of that particular site.
Under normal electron transport, complex IV reduces oxygen, which acts as the terminal electron acceptor, forming H2O (Fig. 1). However, single electron transfer to dioxygen at other points in the chain results in the formation of superoxide (O2•−). Under aqueous conditions at physiological pH, superoxide exists mostly as the anion, Because of its pK, however, the membrane-soluble protonated form, hydroperoxyl radical (HO2•) is also present at non-negligible concentrations. Coordinated loss of a pair of electrons to oxygen results in the formation of hydrogen peroxide, but superoxide is the primary ROS formed by the electron transport chain. Complexes I and III of the electron transport chain can generate superoxide (Fig. 2) but other mitochondrial enzyme complexes have also been reported or demonstrated to produce ROS. Examples include the dihydrolipoamide dehydrogenase-containing FAD-linked pyruvate and α-ketoglutarate dehydrogenase complexes [29], as well as the flavoenzymes α-glycerophosphate dehydrogenase [30], and the electron-transferring flavoprotein:Q oxidoreductase (ETFQOR) of fatty acid β-oxidation [31]. Complexes I and III have been most extensively studied and will be expanded on here.
Complex I
There are two current theories on the site(s) of superoxide production from complex I; these theories are not mutually exclusive. Although Fe-S centres were also once implicated in complex I superoxide production, they have largely fallen out of favour. Currently, debate over the contributions from the flavin of the FMN moiety and from QH• in the ubiquinone binding site, designated in Fig. 2 as IF and IQ respectively, continues. Although not a consensus view, assignment of two distinct sites of superoxide production by complex I can be made based on markedly different maximal rates of production, responses to inhibitors and mitochondrial bioenergetic status as well as substrate requirements for electron entry. During forward electron flow with isolated mammalian complex I, redox titrations support the fully reduced flavin of the FMN moiety as the source of O2•− [32; 33]. Inhibitors of electron transfer to Q (IQ inhibitors), such as rotenone and piericidin, prevent electron escape from the complex and increase superoxide production by complex I under conditions of high NADH/NAD+. Thus, electrons from NADH entering the complex, combined with diminished capacity to leave via Q, results in an increased reduction state of the flavin and subsequently an increase in the rate of superoxide formation. The maximal rates from the IF site with IQ inhibitors are increased by the generation of a Δp by the addition of exogenous ATP [34]. This Δp dependent increase is abolished by nigericin, a H+/K+ antiporter that converts ΔpH into ΔΨ, suggesting that ΔpH is also important to the NADH-derived complex I ROS generation [34].
Under conditions of high Δp and a reduced Q pool, complex I can catalyze the unfavourable reduction of NAD+ to NADH by reverse electron transport. The superoxide produced by reverse electron transport declines markedly with small decreases in Δp, and is even more sensitive to ΔpH than to membrane potential [35]. Succinate is commonly used as a substrate in studies of mitochondrial O2•− production and often gives high rates in well-coupled systems in state 4 respiration. This may have led to the idea that complex II is a significant source of ROS in mitochondria; however, it is now established that most of the ROS produced by succinate-driven respiration is sensitive to IQ inhibitors, implicating complex I. Since the ROS generation from succinate is highly sensitive to Δp, poorly coupled mitochondria or the presence of ΔpH and Δp-diminishing substances (i.e. phosphate, ADP, nigericin or uncoupling agents) all result in diminished complex I IQ site ROS production from succinate and other Q-reducing substrates.
Complex III
Electrons from substrate oxidation are carried to complex III via the reduction of ubiquinone (Q) to ubiquinol (QH2). Under conditions of uninhibited electron flow through complex III, QH2 binds to the Qo site, then one electron is transferred to the Rieske Fe-S centre of the high potential chain, reducing cytochrome c before passing to complex IV and dioxygen [36]. The other electron is transferred to the Qi site by the low potential chain, consisting of cytochrome b566 and b562. This reaction is rapid, preventing significant accumulation of QH• in the Qo site. However, Qi site inhibitors prevent the electrons from leaving the complex during the normal reduction of Q during the Q-cycle. Under these conditions, QH2 can still pass an electron to the high potential chain, but the redox centres of the low potential chain become highly reduced, resulting in the formation of QH• in the Qo site and subsequent O2•− production [36]. Inhibitors of complex III that prevent Q binding at the Qo site, such as stigmatellin and myxothiazol, block electron entry into the complex and prevent ROS production even in the presence of Qi inhibitors. Paradoxically, some Qo site inhibitors such as myxothiazol may also induce complex III ROS generation, albeit to a much lesser degree than Qi site inhibitors [37]. While the mechanism for ROS production from complex III by myxothiazol is unclear, this is an important consideration, as myxothiazol has been used to define rates from the Qo site.
Topology of ROS formation
Phospholipid bilayer membranes are highly impermeable to the anionic O2•−. Although its protonated form, HO2•, is able to cross membranes, the pKa predicts that HO2•concentration is very small at physiological pH. With the mitochondrial inner membrane being a significant barrier to O2•−, it is important to appreciate that sidedness of production is important to the biological impacts of ROS. ROS formation from complex I is directed solely to the matrix, which is consistent with both sites of single electron escape, the flavin and Q binding pocket, being in or closely oriented to the matrix [38]. In support of this, recent modelling of complex I has suggested that electron transfer from the N2 Fe-S cluster to Q may occur in or on the boundary of the aqueous phase on the matrix side of the membrane [39]. Superoxide production by complex III in the presence of antimycin A results in release of ROS to both the matrix and intermembrane space [31; 38; 40]. Likewise, electron entry into α-glycerophosphate dehydrogenase results in ROS production to both sides of the mitochondrial inner membrane [40].
Relative contributions from each site
What are the major sites of mitochondrial ROS production in vivo? This remains of one of the physiologically most important yet insufficiently addressed questions regarding mitochondrial ROS metabolism. Complex I is viewed as the major contributor from the electron transport chain, with the uninhibited complex III considered a more minor source of superoxide [6], but the evidence is circumstantial at best. However, the relative contribution from α-glycerophosphate dehydrogenase [30], and the ETF:Q oxidoreductase of fatty acid β oxidation [31] are in need of better quantitative assessment, especially in the tissues where these enzymes are highly expressed. Although not a direct part of the electron transport chain, the pyruvate and α-ketoglutarate dehydrogenase complexes are also in need of consideration [29, 41].
Methodological complications for whole cells
The majority of knowledge on the mechanism(s) of mitochondrial ROS generation has come from studies using isolated enzyme complexes, mitochondrial membrane fractions, respiring/coupled submitochondrial particles and isolated mitochondria. Interpreting how these data should be translated to intact and functioning cells is challenging. For example, the concentrations of substrates, the ATP/ADP ratio, cytosolic and mitochondrial nicotinamide nucleotide redox couples (NADH/NAD+ and NADPH/NADP+) as well as allosteric and post-translational regulators of metabolic flux all vary (at times in opposing directions) in response to a number of variables. It is difficult to define the actual conditions that occur in vivo and elucidate which specific conditions are then important and need to be suitably mimicked in vitro.
Recently, fluorescent probes that display high sensitivity to superoxide have facilitated the capacity to make measurements with intact cells. Although methodological limitations make these predominantly relative rates, rather than direct or quantitative, fluorescent superoxide probes such as hydroethidine or the mitochondrially-targeted derivative mito-hydroethidine (mitoSOX Red) have opened up new areas of research on mitochondrial ROS production [42]. However, it is important to stress caution prior to the extension of inhibitor-enhanced mitochondrial ROS production to whole cell preparations. The inhibitors used to characterize the sites and mechanisms of ROS production with isolated mitochondria all potentially alter Δp, depending on the predominate substrate(s) being oxidized. Complex III inhibitors will be the most disruptive of Δp because they will impair proton translocation from all sites by fully reducing Q and starving complex IV of reduced cytochrome c (Fig. 1). Similarly, consider the addition of rotenone to cells: the flavin of complex I will become highly reduced and superoxide from this site is expected to increase. But rotenone will abolish the contribution of complex I to Δp, and substantial electron flow to complex III via Q reduction is also lost. Given the interplay between mitochondrial proton leak, Δp and electron leak (superoxide production), it is not reasonable to assert that in cells the uninhibited and inhibited states are directly comparable, greatly confounding interpretation.
Importance of proton and electron leak in disease
Modulation of proton and electron leak is a therapeutic target for many diseases including obesity, diabetes, and aspects of age-related diseases. Here, we discuss the role of proton leak in body weight regulation as well as the modulation of insulin secretion from β cells by UCP2.
Regulation of body weight by proton leak
Decreasing mitochondrial efficiency by pharmacologically increasing proton leak with chemical uncouplers was once used as a strategy for controlling body weight. The chemical uncoupler 2,4-dinitrophenol (DNP) was used extensively prior to 1938, but was withdrawn when overdoses caused illness or death. The major disadvantage of DNP is the narrow margin between therapeutic and toxic doses [43]. DNP also penetrates all tissues non-specifically. To study the effects of mitochondrial uncoupling in specific tissues, UCP1 was ectopically expressed in skeletal muscle of mice. The mice displayed reduced obesity, higher energy expenditure and less age-related disease [44, 45].
Two recent findings focus major interest on brown adipose tissue as a target for obesity treatment: the presence of UCP1 in mice prevents obesity even at thermoneutrality, and the identification of functional brown adipose tissue in adult humans [46, 47,48, 49].
Modulating the insulin response through mild uncoupling
Glucose-stimulated insulin secretion (GSIS) by pancreatic β cells is essential for maintaining euglycemia. Glucose stimulates respiration of β cell mitochondria, generating a high ATP/ADP ratio, which triggers the release of insulin-containing vesicles. Upon observation that UCP2 ablation increased GSIS, UCP2 was implicated as a negative regulator of insulin secretion, presumably by lowering the ATP/ADP ratio [50]. Although uncoupling mediated by UCP2 can explain the modulation of GSIS, this view of UCP2 activity in regulating glucose homeostasis is complicated by the role the protein may play in ROS attenuation. Recent investigation of UCP2 knockout mice of multiple background strains revealed chronic oxidative stress, as determined by a low proportion of reduced glutathione, and increased expression of other antioxidants [51]. To clarify the role of UCP2 in β cells, the determination of its function is required. The knock-down of UCP2 in INS-1E insulinoma cells is a useful model to investigate the function of UCP2 in β cell bioenergetics [14]. To fully understand whether UCP2 affects GSIS by uncoupling or other mechanisms, its contribution to proton leak in this system has to be further investigated.
Summary
Basal proton leak is unregulated and mediated mostly by the presence but not activity of mitochondrial anion carrier proteins.
Inducible proton leak is mediated by the adenine nucleotide translocase and uncoupling proteins.
The decrease in superoxide production by mild uncoupling, possibly mediated by mitochondrial anion carrier proteins, underline the interdependence between proton and electron leak
The sites of electron leak causing superoxide production in the respiratory chain are considered to be a semiquinone radical or a reduced flavin.
At complex I, superoxide may be either generated at the flavin or at the IQ site,
and is completely released to the matrix side.
At complex III, superoxide is produced at the Qo site, and released to
intermembrane space and matrix.
Figure 3. Sites of electron leak (loss) from electron transport chain complexes during electron transport.
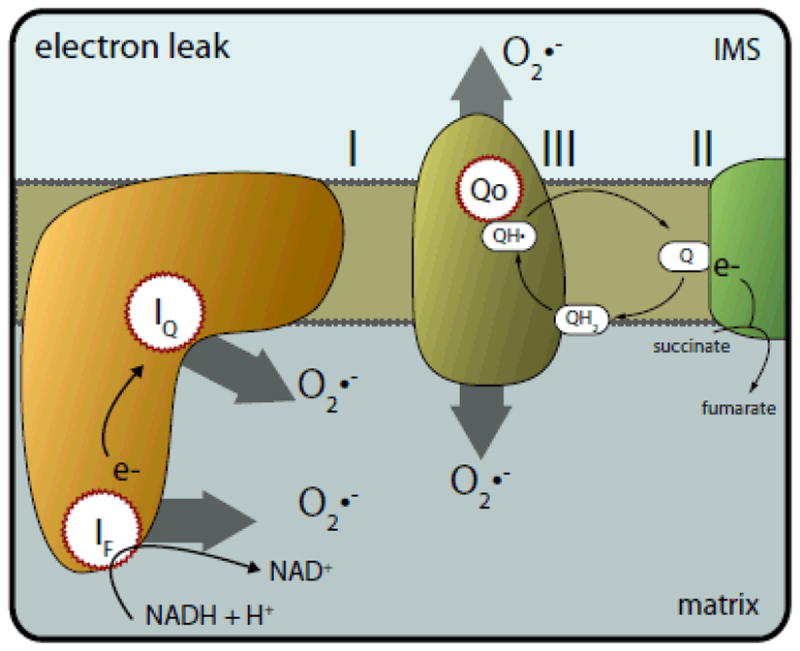
Electrons carried by NADH are transferred to the flavin mononucleotide(IF) site in complex I, where they normally pass down a chain of Fe-S centres to the ubiquinone binding site (IQ). At both the IF and IQ sites, these electrons react with O2, forming superoxide (O2•−) within the matrix. In complex III, QH2 binds to the QO site, where its electrons can bypass their normal transfer in the Q-cycle (see text) and react directly with oxygen to form superoxide that is released to both sides of the mitochondrial inner membrane.
Acknowledgments
Supported by grants from the National Institutes of Health (P01 AG025901, PL1 AG032118, P30 AG025708 and R01 AG033542), the W.M. Keck foundation, The Ellison Medical Foundation (AG-SS-2288-09), the Deutche Forschungsgemeinschaft (JA 1884/2-1) and a British Marshall Scholarship and National Science Foundation Graduate Research Fellowship to ASD.
Biographies
Martin Jastroch, Ph.D.
Dr Jastroch received his Ph.D. from the Philipps-Universität Marburg, studying the evolution of uncoupling proteins (UCPs) and brown adipose tissue. He performed comparative studies on proton leak and UCPs in a variety of vertebrate species at the University of Southern Queensland, and at the Medical Research Council in Cambridge. During his postdoctoral research with Dr Gerhard Heldmaier, he investigated the physiological adaptation of proton and electron leak in brown adipose tissue. In 2008, he joined the Brand Laboratory at the Buck Institute for Age Research to study the function of novel uncoupling proteins.
Ajit S. Divakaruni, B.S.
Ajit Divakaruni earned a B.S. (Honors) in Biochemistry and Mathematics from the University of Arizona and is currently a Ph.D. candidate in Biochemistry at the University of Cambridge. He is conducting research on the regulation of uncoupling proteins.
Shona Mookerjee, Ph.D.
Dr Mookerjee received her Ph.D. in 2005 from the University of Rochester where she investigated pathways of mitochondrial DNA damage and repair in the yeast S. cerevisiae. Shona Mookerjee is a postdoctoral fellow in the Brand Laboratory studying the regulation of the non-thermogenic uncoupling proteins UCP2 and UCP3. Her current focus is to understand the role of rapid degradation in the proper function of UCP2 and UCP3 in mammals.
Jason R. Treberg, Ph.D.
Following a B.Sc. in Marine Biology from the University of Guelph, Dr Treberg completed graduate studies in 2006 on the comparative physiology and biochemistry of cold adaption and organic osmolyte metabolism in fishes with Dr. William R. Driedzic at the Ocean Sciences Centre of Memorial University of Newfoundland. He conducted post-doctoral studies focusing on amino acid, methylamine and 1-carbon metabolism with Dr. J.T. Brosnan, Department of Biochemistry, Memorial University of Newfoundland. Dr. Treberg joined the Brand Lab at the Buck Institute in 2008 as a postdoctoral fellow and is currently studying mitochondrial ROS metabolism.
Martin D. Brand, Ph.D., F. Med. Sci.
Dr Brand holds a B.Sc. in Biochemistry from the University of Manchester Institute of Science and Technology and a Ph.D. in Biochemistry from the University of Bristol. He carried out postdoctoral research at Johns Hopkins University with Dr Albert Lehninger, and then took up a faculty position in the Biochemistry Department at the University of Cambridge, culminating in a Personal Readership in Cellular Biochemistry. Following ten years as a Group Leader at the Medical Research Council in Cambridge, he moved to the Buck Institute in 2008. Dr Brand is a Fellow of the Academy of Medical Sciences, a Life Fellow of Girton College, Cambridge, and the 2005 recipient of the Keilin Medal of the Biochemical Society. He is a member of these Editorial Boards: Aging Cell, Biochimica et Biophysica Acta Bioenergetics; Cell Metabolism; Physiological and Biochemical Zoology.
References
- 1.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 2.Nobes CD, Brown GC, Olive PN, Brand MD. Non-ohmic proton conductance of the mitochondrial inner membrane in hepatocytes. J Biol Chem. 1990;265:12903–12909. [PubMed] [Google Scholar]
- 3.Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- 4.Hafner RP, Brand MD. Effect of protonmotive force on the relative proton stoichiometries of the mitochondrial proton pumps. Biochem J. 1991;275:75–80. doi: 10.1042/bj2750075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy MP. Slip and leak in mitochondrial oxidative phosphorylation. Biochim Biophys Acta. 1989;977:123–141. doi: 10.1016/s0005-2728(89)80063-5. [DOI] [PubMed] [Google Scholar]
- 6.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papa S, Skulachev VP. Reactive oxygen species, mitochondria, apoptosis and aging. Mol Cell Biochem. 1997;174:305–319. [PubMed] [Google Scholar]
- 8.Brand MD, Brindle KM, Buckingham JA, Harper JA, Rolfe DF, Stuart JA. The significance and mechanism of mitochondrial proton conductance. Int J Obes Relat Metab Disord. 1999;23(Suppl 6):S4–11. doi: 10.1038/sj.ijo.0800936. [DOI] [PubMed] [Google Scholar]
- 9.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392:353–362. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker N, Crichton PG, Vidal-Puig AJ, Brand MD. Uncoupling protein-1 (UCP1) contributes to the basal proton conductance of brown adipose tissue mitochondria. J Bioenerg Biomembr. 2009 doi: 10.1007/s10863-009-9232-8. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 11.Roussel D, Harding M, Runswick MJ, Walker JE, Brand MD. Does any yeast mitochondrial carrier have a native uncoupling protein function? J Bioenerg Biomembr. 2002;34:165–176. doi: 10.1023/a:1016027302232. [DOI] [PubMed] [Google Scholar]
- 12.Rolfe DFS, Brand MD. Contribution of mitochondrial proton leak to skeletal muscle respiration and to standard metabolic rate. Am J Physiol. 1996;271:C1380–1389. doi: 10.1152/ajpcell.1996.271.4.C1380. [DOI] [PubMed] [Google Scholar]
- 13.Conley KE, Amara CE, Jubrias SA, Marcinek DJ. Mitochondrial function, fibre types and ageing: new insights from human muscle in vivo. Exp Physiol. 2007;92:333–339. doi: 10.1113/expphysiol.2006.034330. [DOI] [PubMed] [Google Scholar]
- 14.Affourtit C, Brand MD. Measuring mitochondrial bioenergetics in INS-1E insulinoma cells. Methods Enzymol. 2009;457:405–424. doi: 10.1016/S0076-6879(09)05023-X. [DOI] [PubMed] [Google Scholar]
- 15.Parker N, Vidal-Puig A, Brand MD. Stimulation of mitochondrial proton conductance by hydroxynonenal requires a high membrane potential. Biosci Rep. 2008;28:83–88. doi: 10.1042/BSR20080002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 17.Klingenberg M, Winkler E. The reconstituted isolated uncoupling protein is a membrane potential driven H+ translocator. EMBO J. 1985;4:3087–3092. doi: 10.1002/j.1460-2075.1985.tb04049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garlid KD, Orosz DE, Modriansky M, Vassanelli S, Jezek P. On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. J Biol Chem. 1996;271:2615–2620. doi: 10.1074/jbc.271.5.2615. [DOI] [PubMed] [Google Scholar]
- 19.Rial E, Aquirregoitia E, Jimenez-Jimenez J, Ledesma A. Alkylsulfonates activate the uncoupling protein UCP1: implications for the transport mechanism. Biochim Biophys Acta. 2004;1608:122–130. doi: 10.1016/j.bbabio.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Shabalina IG, Jacobsson A, Cannon B, Nedergaard J. Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem. 2004;279:38236–38248. doi: 10.1074/jbc.M402375200. [DOI] [PubMed] [Google Scholar]
- 21.Adams AE, Carroll AM, Fallon PG, Porter RK. Mitochondrial uncoupling protein 1 expression in thymocytes. Biochim Biophys Acta. 2008;1777:772–776. doi: 10.1016/j.bbabio.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 22.Jastroch M, Withers KW, Taudien S, Frappell PB, Helwig M, Fromme T, Hirschberg V, Heldmaier G, McAllan BM, Firth BT, Burmester T, Platzer M, Klingenspor M. Marsupial uncoupling protein 1 sheds light on the evolution of mammalian nonshivering thermogenesis. Physiol Genomics. 2008;32:161–169. doi: 10.1152/physiolgenomics.00183.2007. [DOI] [PubMed] [Google Scholar]
- 23.Jastroch M, Wuertz S, Kloas W, Klingenspor M. Uncoupling protein 1 in fish uncovers an ancient evolutionary history of mammalian nonshivering thermogensis. Physiol Genomics. 2005;22:150–156. doi: 10.1152/physiolgenomics.00070.2005. [DOI] [PubMed] [Google Scholar]
- 24.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 25.Khailova LS, Pridhodko EA, Dedukhove VI, Mokhova EN, Popov VN, Skulachev VP. Participation of ATP/ADP antiporter in oleate- and oleate hydroperoxide-induced uncoupling suppressed by GDP and carboxyatractylate. Biochim Biophys Acta. 2006;1757:1324–1329. doi: 10.1016/j.bbabio.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 26.Seifert EL, Bezaire V, Estey C, Harper ME. Essential role for uncoupling protein-3 in mitochondrial adaptation to fasting but not in fatty acid oxidation or fatty acid anion export. J Biol Chem. 2008;283:25124–25131. doi: 10.1074/jbc.M803871200. [DOI] [PubMed] [Google Scholar]
- 27.Robinson AJ, Overy C, Kunji ER. The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc Natl Acad Sci USA. 2008;105:17766–17771. doi: 10.1073/pnas.0809580105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jezek P, Garlid KD. New substrates and competitive inhibitors of the Cl-translocating pathway of the uncoupling protein of brown adipose tissue mitochondria. J Biol Chem. 1990;265:19303–19311. [PubMed] [Google Scholar]
- 29.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tretter L, Takacs K, Hegedus V, Adam-Vizi V. Characteristics of alpha-glycerophosphate-evoked H2O2 generation in brain mitochondria. J Neurochem. 2007;100:650–663. doi: 10.1111/j.1471-4159.2006.04223.x. [DOI] [PubMed] [Google Scholar]
- 31.St Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 32.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. 2008;36:976–980. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 34.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 35.Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crofts AR. The cytochrome bc1 complex: function in the context of structure. Ann Rev Physiol. 2004;66:689–733. doi: 10.1146/annurev.physiol.66.032102.150251. [DOI] [PubMed] [Google Scholar]
- 37.Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry. 2003;42:6493. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 38.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 39.Zickermann V, Kerscher S, Zwicker K, Tocilescu MA, Radermacher M, Brandt U. Architecture of complex I and its implications for electron transfer and proton pumping. Biochim Biophys Acta. 2009;1787:574–583. doi: 10.1016/j.bbabio.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miwa S, Brand MD. The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim Biophys Acta. 2005;1709:214–219. doi: 10.1016/j.bbabio.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Chinta SJ, Rane A, Yadava N, Andersen JK, Nicholls DG, Polster BM. Reactive oxygen species regulation by AIF-and complex I-depleted brain mitochondria. Free Radic Biol Med. 2009 doi: 10.1016/j.freeradbiomed.2009.01.010. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protocols. 2008;3:941–947. doi: 10.1038/nprot.2008.56. [DOI] [PubMed] [Google Scholar]
- 43.Harper JA, Dickinson K, Brand MD. Mitochondrial uncoupling as a target for drug development for the treatment of obesity. Obes Rev. 2001;2:255–265. doi: 10.1046/j.1467-789x.2001.00043.x. [DOI] [PubMed] [Google Scholar]
- 44.Li B, Nolte LA, Ju JS, Han DH, Coleman T, Holloszy JO, Semenkovich CF. Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat Med. 2000;6:1115–1120. doi: 10.1038/80450. [DOI] [PubMed] [Google Scholar]
- 45.Gates AC, Bernal-Mizrachi C, Chinault SL, Feng C, Schneider JG, Coleman T, Malone JP, Townsend RR, Chakravarthy MV, Semenkovich CF. Respiratory uncoupling in skeletal muscle delays death and diminishes age-related disease. Cell Metab. 2007;6:497–505. doi: 10.1016/j.cmet.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2008;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 47.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 48.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerbäck S, Nuutila P. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 50.Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 51.Pi J, Bai Y, Daniel KW, Liu D, Lyght O, Edelstein D, Brownlee M, Corkey BE, Collins S. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic beta-cell function. Endocrinology. 2009;150:3040–3048. doi: 10.1210/en.2008-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]