

Abstract
Intracytoplasmic proteinaceous inclusions, primarily composed of tau or α-synuclein (α-syn), are predominant pathological features of Alzheimer's disease (AD) and Parkinson's disease (PD), respectively. However, the coexistence of these pathological aggregates is identified in many neurodegenerative disorders, including spectrum disorders of AD and PD. Whereas α-syn can spontaneously polymerize into amyloidogenic fibrils, in vitro, tau polymerization requires an inducing agent. The current study presents a human-derived cellular model, in which recombinant, preformed α-syn fibrils cross-seed intracellular tau to promote the formation of neurofibrillary tangle-like aggregates. These aggregates were hyperphosphorylated, Triton insoluble, and thioflavin-S positive, either comingling with endogenously expressed α-syn aggregates or induced by only exogenously applied recombinant α-syn fibrils. Furthermore, filamentous, amyloidogenic tau took over the cellular soma, displacing the nucleus and isolating or displacing organelles, likely preventing cellular function. Although a significant proportion of wild-type tau formed these cellular inclusions, the P301L mutation in tau increased aggregation propensity resulting from α-syn seeds to over 50% of total tau protein. The role of phosphorylation on the development of these tau aggregates was investigated by coexpressing glycogen synthase kinase 3 β or microtubule-associated protein/microtubule affinity-regulating kinase 2. Expression of either kinase inhibited the formation of α-syn-induced tau aggregates. Analyses of phosphorylation sites suggest that multiple complex factors may be associated with this effect and that Triton-soluble versus Triton-insoluble tau may be independently targeted by kinases. The current work not only provides an exceptional cellular model of tau pathology, but also examines α-syn-induced tau inclusion formation and provides novel insights into hyperphosphorylation observed in disease.
Introduction
Accumulations of proteinaceous, hyperphosphorylated inclusions, composed of amyloidogenic misfolded protein in the brain, are major pathological features of Alzheimer's disease (AD) and Parkinson's disease (PD). Intracellular localization of these pathological aggregates can be identified in these disorders, primarily composed of tau-forming neurofibrilliary tangles (NFTs) in AD or of α-synuclein (α-syn)-forming Lewy pathology in PD. The aberrant polymerization and accumulation of tau or α-syn additionally classifies a host of other neurodegenerative disorders, termed tauopathies and synucleinopathies, respectively (Goedert et al., 1998; Buee et al., 2000; Duda et al., 2000; Lee et al., 2001).
Familial mutations in MAPT and SCNA (the genes for tau and α-syn, respectively) that are causative of disease have also been identified. Genetic mutations in tau are causative of the tauopathy frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) (Hutton et al., 1998; Lee et al., 2001; Goedert and Spillantini, 2006), and mutations in α-syn result in familial forms of PD (Polymeropoulos et al., 1997). Mutations in these proteins increase the propensity of filamentous aggregate formation in vitro and in transgenic animal models (Conway et al., 1998; Arrasate et al., 1999; Giasson et al., 1999, 2002; Lewis et al., 2000), further supporting the association between these proteins and disease.
Tau and α-syn amyloidogenic fibrils are both nucleation dependent (Wood et al., 1999; Congdon et al., 2008) and hyperphosphorylated in their pathological forms (Billingsley and Kincaid, 1997; Buee et al., 2000; Fujiwara et al., 2002; Kahle et al., 2002; Neumann et al., 2002; Nishie et al., 2004; Anderson et al., 2006; Waxman and Giasson, 2008). Antibodies recognizing phosphoepitopes S396/S404 (PHF1) and S202/T205 (AT8) on tau are among the most characterized identifiers of NFTs (Otvos et al., 1994; Goedert et al., 1995), and phosphorylation of these sites by glycogen synthase kinase 3 β (GSK3β) may promote tau aggregate formation (Sato et al., 2002; Liu et al., 2007; Rankin et al., 2007). However, microtubule-associated protein/microtubule affinity-regulating kinase 2 (MARK2) targets the core, repeat region required for tau filament formation, potentially inhibiting aggregation (Crowther et al., 1989, 1992; Goode and Feinstein, 1994; Drewes et al., 1995, 1997; Schneider et al., 1999).
Whereas α-syn can spontaneously self-polymerize in vitro (Conway et al., 1998; Giasson et al., 1999), tau requires the presence of an inducing agent (Goedert et al., 1996). α-Syn induces tau polymerization in vitro (Giasson et al., 2003), and tau and α-syn pathology coexist in patients with familial forms of PD and in spectrum diagnoses between PD and AD, termed dementia with Lewy bodies and Lewy body variant of Alzheimer's disease (LBVAD) (Hansen et al., 1990; McKeith, 1996; Duda et al., 2002; Giasson et al., 2003). However, the manner in which these proteins interact to promote aggregation has yet to be modeled in vivo.
The current work provides the first cellular modeled system, mimicking coexisting tau and α-syn pathology. Using previously described methods for cellular seeding (Waxman and Giasson, 2010), recombinant, preformed α-syn fibrils robustly promoted intracellular tau aggregation with filamentous morphology with similar characteristics to that observed postmortem. We further characterized the hyperphosphorylation of tau and the effects of kinase overexpression within this model system.
Materials and Methods
Expression and purification of recombinant α-syn and tau.
The human α-syn cDNA was cloned into the bacterial expression vector pRK172, using NdeI and HindIII restriction sites. The pRK172 DNA construct expressing N-terminal truncated 21–140 α-syn (with a Met codon added before amino acid 21) was generously provided by Dr. Virginia Lee (University of Pennsylvania, Philadelphia, PA). α-Syn proteins were expressed in Escherichia coli BL21 (DE3) and purified as described previously (Giasson et al., 2001; Greenbaum et al., 2005). Human full-length tau cDNA (2N/4R isoform) was cloned into the bacterial expression vector pRK172 using NdeI and EcoRI restriction sites (kindly provide by Dr. Michel Goedert, University of Cambridge, Cambridge, UK). pRK172 plasmid expressing human tau with the P301L mutation was created with oligonucleotides corresponding to the amino acid substitutions by QuickChange site-directed mutagenesis (Stratagene). Recombinant full-length wild-type (WT) or P301L tau was expressed in E. coli BL21 and purified as described previously (Hong et al., 1998; Giasson et al., 2003).
Fibril preparation of recombinant α-syn for in situ experimentation.
For cellular experiments, α-syn proteins were assembled into filaments by incubation at 37°C at concentrations >5 mg/ml in sterile PBS (Invitrogen) with continuous shaking at 1050 rpm (Thermomixer R; Eppendorf). Experimentation was planned so that α-syn would be visibly assembled (by filamentous clusters observed in the solution) by the day of cellular experimentation. α-Syn fibrils were diluted to a concentration of 1–3 mg/ml in sterile PBS and treated by water bath sonication for a minimum of 2 h. In experiments where large fibrils were separated from small oligomers, diluted fibrillized α-syn (before sonication) was centrifuged at 16,000 × g for 5 min, the supernatant was removed, and the pellet was resuspended in sterile PBS. Protein concentrations of the original diluted fibrils and the supernatant were measured by BCA protein assay (Pierce). Concentrations of the large fibrils were determined as the concentrations of the total minus the supernatant. All samples were sonicated in a water bath before addition to cell culture media. Cells expressing both α-syn and tau were treated with 1 μm of recombinant 21–140 α-syn fibril mix, and cells expressing only tau were treated with 1 μm of WT α-syn fibril mix unless specified otherwise.
Cell culture and transfection.
QBI293 cells, derived from human embryonic kidney cells, were maintained using DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and penicillin (100 U/ml)/streptomycin (100 μg/ml). The mammalian expression vector pcDNA3.1 cloned with WT human α-syn cDNA was described previously (Paxinou et al., 2001). The pcDNA3.1 expression plasmids containing cDNA for human WT full-length tau (2N/4R) or tau containing the P301L mutation were generously provided by Dr. Virginia Lee. The S262A and S356A mutations in WT or P301L tau were created with oligonucleotides corresponding to the amino acid substitutions by QuickChange site-directed mutagenesis (Stratagene). The following amino acid substitutions were completed by serial QuickChange reactions: S262A/S356A, S202A/T205A, S202E/T205E. All mutations and the absence of copy errors were confirmed by sequencing the entire length of the tau cDNA. The mammalian expression vector pCMV, cloned with myc-tagged MARK2 cDNA, was generously provided by Dr. Marina Picciotto (Yale University, New Haven, CT). Human HA-tagged GSK3β cDNA in pcDNA3 was generated by Dr. Jim Woodgett (He et al., 1995) and acquired from Addgene.
Cells were plated onto poly-d-lysine-coated six-well plates and transfected at ∼30% confluency using calcium phosphate precipitation as described previously (Waxman and Giasson, 2010). Cells were transfected with an equal proportion of tau-containing expression plasmid and α-syn-containing expression plasmid or pcDNA3.1 (empty vector), except in experiments where MARK2 overexpression was evaluated. In MARK2 experiments, cells were transfected with a 3:1 ratio of tau to MARK2 expression plasmids. Four hours after transfection, 1 μm (final concentration) sonicated α-syn fibrils were added dropwise to media, after which cells were incubated over night at 37°C, 5% CO2.
Approximately 16 h after transfection, calcium phosphate was washed two-times with PBS, and media was replaced with warm DMEM containing 3% FBS and penicillin (100 U/ml)/streptomycin (100 μg/ml) (reduced serum). Cells were harvested for biochemical fractionation or fixed 72 h after transfection unless specified otherwise.
Biochemical cellular fractionation.
Cells were washed one time in ice-cold PBS, and samples were harvested in 25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 20 mm NaF, and a mixture of protease inhibitors containing 1 mm phenylmethylsulfonyl and 1 mg/ml each of pepstatin, leupeptin, N-tosyl-l-phenylalanyl chloromethyl ketone, N-tosyl-lysine chloromethyl ketone, and soybean trypsin inhibitor. Samples were sedimented at 100,000 × g for 30 min at 4°C. Supernatants were removed, and pellets were sonicated in 1.5× Laemmli sample buffer (75 mm Tris-HCl, pH 6.8, 3% SDS, 15% glycerol, 3.75 mm EDTA, pH 7.4). Sonication in Laemmli buffer was sufficient to reduce Triton-insoluble tau to monomeric form. SDS sample buffer (10 mm Tris, pH 6.8, 1 mm EDTA, 40 mm DTT, 1% SDS, 10% sucrose) was added, and samples were heated to 100°C for 5 min before Western blot analysis. Equal proportions of supernatant (Triton-soluble protein) and pellet (Triton-insoluble protein) were loaded and analyzed as the percentage pelleted.
Microtubule-binding assay.
Microtubule-binding assays were performed as described previously (Vogelsberg-Ragaglia et al., 2000). Briefly, cells were harvested with homogenization into prewarmed 100 mm MES, pH 6.8, 1 mm EGTA, 0.5 mm MgSO4, 2 mm DTT, 0.75 m NaCl, 20 mm NaF, 0.1% Triton X-100, 20 μm taxol, 2 mm GTP, and a mixture of protease inhibitors at 37°C. After centrifugation for 20 min at 50,000 × g, the supernatant containing nonmicrotubule bound protein was removed from the microtubule bound fraction (pellet). SDS sample buffer was added and samples were heated to 100°C for 5 min before Western blot analysis. Equal proportions of supernatant and pellet were loaded and analyzed as percentage pelleted.
Antibodies.
SNL4 is a polyclonal rabbit antibody raised against a synthetic peptide corresponding to amino acids 2–12 of α-syn (Giasson et al., 2000). pSer129 is a mouse monoclonal antibody that specifically recognizes α-syn phosphorylated at S129 (Waxman and Giasson, 2008). Rabbit polyclonal anti-tau antibody 17025 (generously provided by Dr. Virginia Lee) or goat polyclonal anti-tau antibody (sc-1995; Santa Cruz Biotechnology) was used to examine general tau immunoreactivity. Antibody 17025 was used for Western blot analyses and colabeling with thioflavin-S reactivity; sc-1995 was used for all other anti-tau double-immunofluorescence immunolabeling. AT8 (Thermo-fisher) is specific toward phosphorylation sites S202 and T205 in tau (Goedert et al., 1995), PHF1 (generously provided by Dr. Peter Davies, Albert Einstein University, New York, NY) is specific toward phosphorylation sites S396 and S404 in tau (Otvos et al., 1994), and 12E8 (generously provided by Dr. Peter Seubert, Elan Pharmaceuticals, San Francisco, CA) specifically recognizes phosphorylation at S262 and S356 in tau (Seubert et al., 1995). Anti-myc antibody clone 9E10 (Sigma-Aldrich) was used to detect MARK2 overexpression. Additional antibodies include anti-vimentin (Sigma-Aldrich), anti-lamp1 (BD Biosciences), anti-γ-tubulin (Sigma-Aldrich), anti-β-tubulin (Sigma-Aldrich), and anti-mannosidase II (Millipore Bioscience Research Reagents).
Western blot analysis.
Protein samples were resolved by SDS-PAGE on 8% gels for tau or MARK2 and 15% polyacrylamide gels for α-syn, followed by electrophoretic transfer onto nitrocellulose membranes. Membranes were blocked in Tris-buffered saline (TBS) with 5% dry milk and incubated overnight with antibodies SNL4, 9E10, or 17025 diluted in TBS/5% dry milk or phosphospecific antibodies diluted in TBS/5% bovine serum albumin (BSA). Each incubation was followed by goat anti-mouse conjugated horseradish peroxidase (HRP) (GE Healthcare) or goat anti-rabbit HRP (Cell Signaling Technology), and immunoreactivity was detected using chemiluminescent reagent (NEN) followed by exposure onto x-ray film.
Cell culture and tissue immunofluorescence.
Double immunofluorescence of transfected cells was completed as described previously (Mazzulli et al., 2006; Waxman et al., 2009). Cells were fixed at −20°C with 100% MeOH for 20 min, followed by 50% MeOH and 50% acetone for 5 min. For thioflavin-S immunostaining, coverslips were fixed in 4% paraformaldehyde for 8 min, followed by PBS/0.1% Triton X-100 for 10 min. After washes with PBS, coverslips were blocked with PBS containing 3% BSA, 1% dry milk, and 1% fish gelatin, and primary antibodies were diluted into blocking solution and incubated for 1–2 h at room temperature. After PBS washes, coverslips were incubated in secondary antibodies conjugated to Alexa 488 or Alexa 594 for 1 h. For double immunofluorescence between pSer129 and PHF1 or AT8, secondary antibodies goat anti-mouse IgG1 conjugated to Alexa 488 (for PHF1 or AT8) and IgG2A conjugated to Alexa 594 (for pSer129) were used. Thioflavin-S (Sigma-Aldrich) immunostaining was performed after secondary antibody incubation at a concentration of 0.05%, after which coverslips were washed three times in 70% ethanol followed by washes in water. Immunofluorescence of postmortem brain samples was completed with paraffin-embedded brain sections as described previously (Giasson et al., 2006). Nuclei were counterstained with Hoechst trihydrochloride trihydrate 33342 (Invitrogen), and coverslips were mounted using Fluoromount-G (Southern Biotech).
Fluorescence microscopy.
Low-magnification double-immunofluorescence pictures were captured on an Olympus BX51 fluorescence microscope mounted with a DP71 digital camera (Olympus). Confocal microscopy images were captured on a Zeiss Axiovert 200M inverted confocal microscope mounted with a Zeiss LSM510 META NLO digital camera using Zeiss LSM510 META 3.2 confocal microscope software. Confocal images were captured with 63× oil optics. Representative images were of one Z-plane of <0.7 μm, or 10–12 stacked Z-planes, where indicated for three-dimensional (3D) projection images. Three-dimensional projection images were rotated on a central axis for optimal representation.
Ultrastructural electron microscopy cellular analyses.
Cells that were transfected and, in some experiments, treated with recombinant α-syn fibrils as described above were fixed with 2% paraformaldehyde/2.5% glutaraldehyde/0.1 m sodium cacodylate buffer, pH 7.4, at 4°C overnight. Cells were harvested, pelleted, and washed in the same buffer before secondary fixation in 2% osmium tetroxide. After rinses to remove osmium tetroxide, cells were stained en bloc in 2% aqueous uranyl acetate and then dehydrated in a graded ethanol series before infiltration and embedding in EMbed-812 (Electron Microscopy Sciences). Epone blocks were cut, and sections were stained with uranyl acetate and lead citrate. Sections were examined with a JEOL 1010 transmission electron microscope fitted with a Hamamatsu digital camera and Advanced Microscopy Techniques Advantage image capture software. For immunoelectron microscopy, cells were fixed for 60 min in 4% paraformaldehyde/0.25% glutaraldehyde in PBS, permeabilized with ethanol, blocked with 5% horse serum/1% BSA/0.2% cold-water fish skin gelatin in PBS, incubated with anti-tau antibody 17025 or anti-α-syn antibody SNL4 diluted in blocking solution, followed by incubation with a goat anti-rabbit HRP-conjugated antibody (Santa Cruz Biotechnology), and developed with 3,3′-diaminobenzidine. The chemical product was enhanced using a modification of the Rodriguez silver/gold enhancement method (Teclemariam-Mesbah et al., 1997). Cells were then further fixed in 2% paraformaldehyde/2.5% glutaraldehyde/0.1 m sodium cacodylate buffer, pH 7.4, and prepared for ultrastructural analysis as described above, with the omission of lead citrate staining.
In vitro filament assembly and centrifugal sedimentation.
Recombinant-generated α-syn proteins were preassembled into filaments by incubation at 37°C in 100 mm sodium acetate, pH 7.4, with continuous shaking at 1050 rpm (Thermomixer R; Eppendorf). After assembly, α-syn protein was treated by water-bath sonication for a minimum of 2 h, after which prefibrillized, sonicated α-syn was diluted to 0.25 mg/ml in 100 mm sodium acetate, pH 7.4, and incubated with tau (WT or P301L) at 1 mg/ml or recombinant nonfibrillized α-syn at 0.3 mg/ml for 3 d at 37°C, 1050 rpm. A fraction of each sample was set aside for K114 fluorometry. The remainder of each sample was centrifuged at 100,000 × g for 30 min. SDS sample buffer was added to pellets and supernatants, which were heated to 100°C for 10 min. Equal volumes of supernatants and pellets were separated by SDS-PAGE and were quantified by densitometry of Coomassie blue R-250-stained gels.
Negative staining and electron microscopic ultrastructural analyses of recombinant α-syn polymers.
Assembled α-syn polymers were absorbed onto 300 mesh carbon-coated copper grids, stained with 1% uranyl acetate and visualized with a JEOL 1010 transmission electron microscope. Images were captured with a Hamamatsu digital camera using Advanced Microscopy Techniques software.
K114 fluorometry.
α-Syn and tau fibrils are amyloidogenic, and their formation can be quantified using the fluorescent amyloid-binding dye K114 (Conway et al., 2000; Crystal et al., 2003). K114 is derived from the structure of Congo red and demonstrates a tremendous increase in fluorescence in solution assays with binding to amyloidogenic fibrils (Crystal et al., 2003). This assay was conducted as described previously (Crystal et al., 2003) by incubating a fraction of each sample with the K114 (50 μm) in 100 mm glycine, pH 8.6, and measuring fluorescence (λex = 380 nm; λem = 550 nm; cutoff, 530 nm) with a SpectraMax Gemini fluorometer and SoftMax Pro 4.0 software.
Quantitative analysis.
Cells were counted by capture and counting of one to four random fields per experiment, independently assessing immunofluorescence of AT8, PHF1, or anti-tau antibodies. Western blot data were quantified by ImageJ software (NIH). Changes in Triton-insoluble protein were calculated as [Triton insoluble/(soluble + Triton insoluble)] * 100, and direct comparisons were performed by paired t tests using GraphPad InStat software. Additional data between two groups were analyzed by two-way, parametric t tests or by nonparametric t tests or U tests (in cases of significantly different variances). Analyses of phosphospecific immunoreactivity were performed by standardizing densitometry to total anti-tau immunoreactivity (17025) and the control (normally WT tau in the absence of fibril treatment) condition. Comparisons to the control condition after standardization were performed by one-sample t tests. Comparisons between multiple groups were performed by one-way ANOVA followed by Bonferroni post-test analyses. Each experiment was performed a minimum of three independent times.
Results
α-Synuclein promotes aggregation of cellular tau
Recent evidence suggests that α-syn is capable of membrane translocation and recycling (Ahn et al., 2006; Liu et al., 2009), and fibrillized α-syn may be transferred between neurons (Desplats et al., 2009; Lee et al., 2008). Based on this paradigm, we recently developed a model system of cellular α-syn aggregation, using preformed, recombinant α-syn fibrils to seed intracellular α-syn aggregate formation (Waxman and Giasson, 2010). We hypothesized that a similar mechanism may be involved in the formation of tau-containing NFTs that are observed concurrently with Lewy pathology in vivo.
To examine the ability of α-syn aggregation to induce cellular tau aggregation, QBI293 cells were initially cotransfected with expression plasmids for α-syn and full-length tau (2N/R4). Under basal conditions using methanol fixation, α-syn staining was diffuse, and tau appeared bundled with some diffuse immunoreactivity (Fig. 1A,B). Colocalization of the two proteins seemed incidental. To examine the effects of recombinant fibril seeds on tau localization, cells were treated with recombinant, prefibrillized 21–140 α-syn protein after transfection. Previous studies show that 21–140 α-syn is capable of seeding intracellular amyloid fibrils similarly to full-length α-syn in cellular seeding experiments (Waxman and Giasson, 2010), and 21–140 α-syn was used so that an anti-α-syn antibody directed to the N terminus of the protein (SNL4) could be used to specifically identify changes to intracellularly expressed full-length α-syn. Using double immunofluorescence between anti-α-syn and anti-tau antibodies, large intracellular aggregates were formed, composed of both α-syn and tau proteins and in most cases encompassing the entire cell (Fig. 1C–E). Colocalization between α-syn and tau was observed in these cells, and in some cases these aggregates appeared round with Lewy-body-type morphology containing a tau immunopositive “tail” (Fig. 1E).
Figure 1.
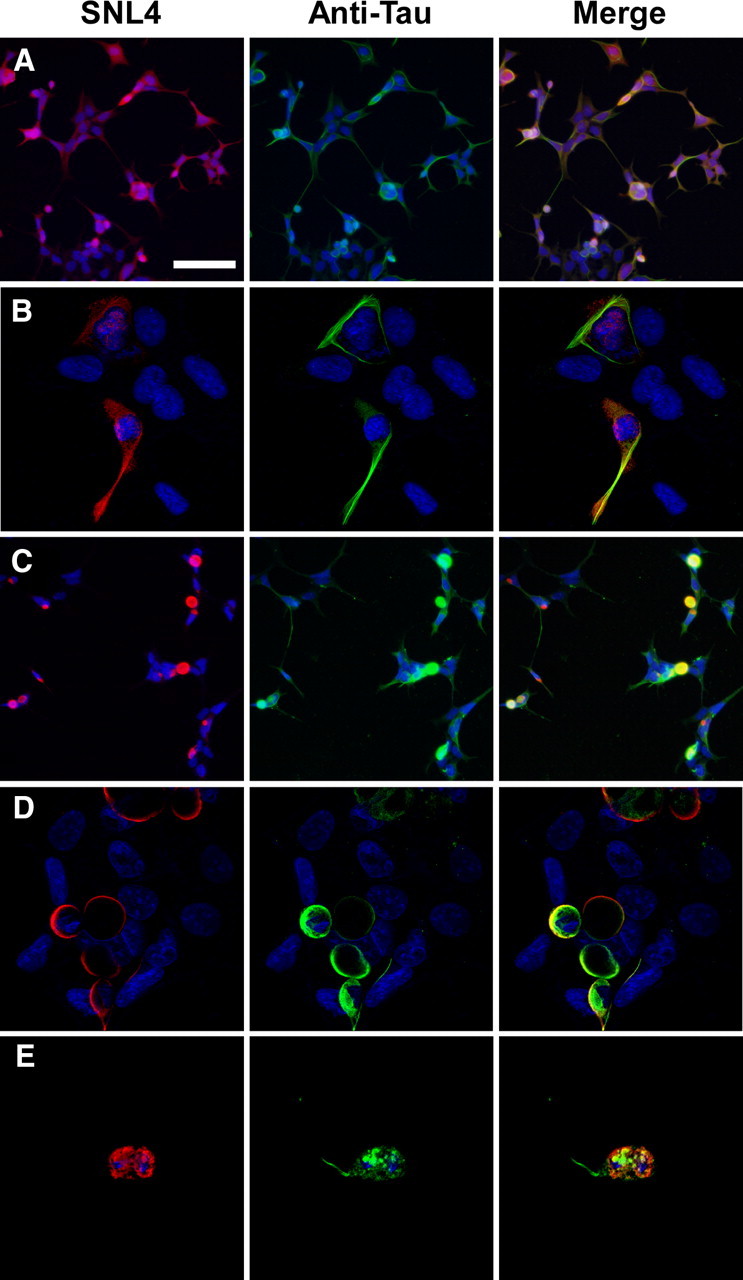
Tau colocalized with α-syn in cells that form cellular aggregates. Representative double immunofluorescence between SNL4 (anti-α-syn antibody) and an anti-tau antibody was performed on QBI293 cells cotransfected with expression plasmids for human wild-type α-syn and tau. A, B, In the absence of fibril treatment, α-syn appeared diffuse, and tau appeared diffuse and bundled. C–E, For cultures treated with recombinant, prefibrillized 21–140 α-syn protein, intracellular, endogenously generated α-syn formed large cellular aggregates that sometimes contained tau. Colocalization of α-syn and tau was assessed by confocal microscopy (B, D, E). Overlay between a portion of SNL4 and anti-tau immunoreactivity was observed in aggregate-containing cells. Many of these cells appeared rounded in morphology, and occasional tau “tails” were observed attached to aggregated α-syn proteins. Representative images are of cells fixed 72 h after transfection. Equivalent exposures were provided for all representative samples. Scale bar: A (for A, C) 100 μm, (for B, D, E) 30 μm.
Since pathological aggregates can be more specifically recognized with antibodies specific to phosphoepitopes on α-syn or tau, we performed double immunofluorescence between pSer129 (recognizing phospho-S129 on α-syn) (Waxman and Giasson, 2008) and PHF1 or AT8 [recognizing phospho-S396/S404 (Otvos et al., 1994) or phospho-S202/T205 (Goedert et al., 1995), respectively, on tau]. In the absence of recombinant α-syn fibril treatment, a paucity of phosphospecific immunoreactivity was observed (Fig. 2A, pSer129, PHF1) (data not shown for AT8). However, in the presence of recombinant, prefibrillized α-syn, robust immunopositive pSer129 α-syn aggregates were observed on the edge with some overlay of PHF1 (Fig. 2B,C) or AT8 (D) immunoreactivity. In addition, some cellular morphology bared remarkable similarity to α-syn- and tau-containing aggregates observed in the brain of patients with the LBVAD diagnosis (Fig. 2, compare D, E).
Figure 2.
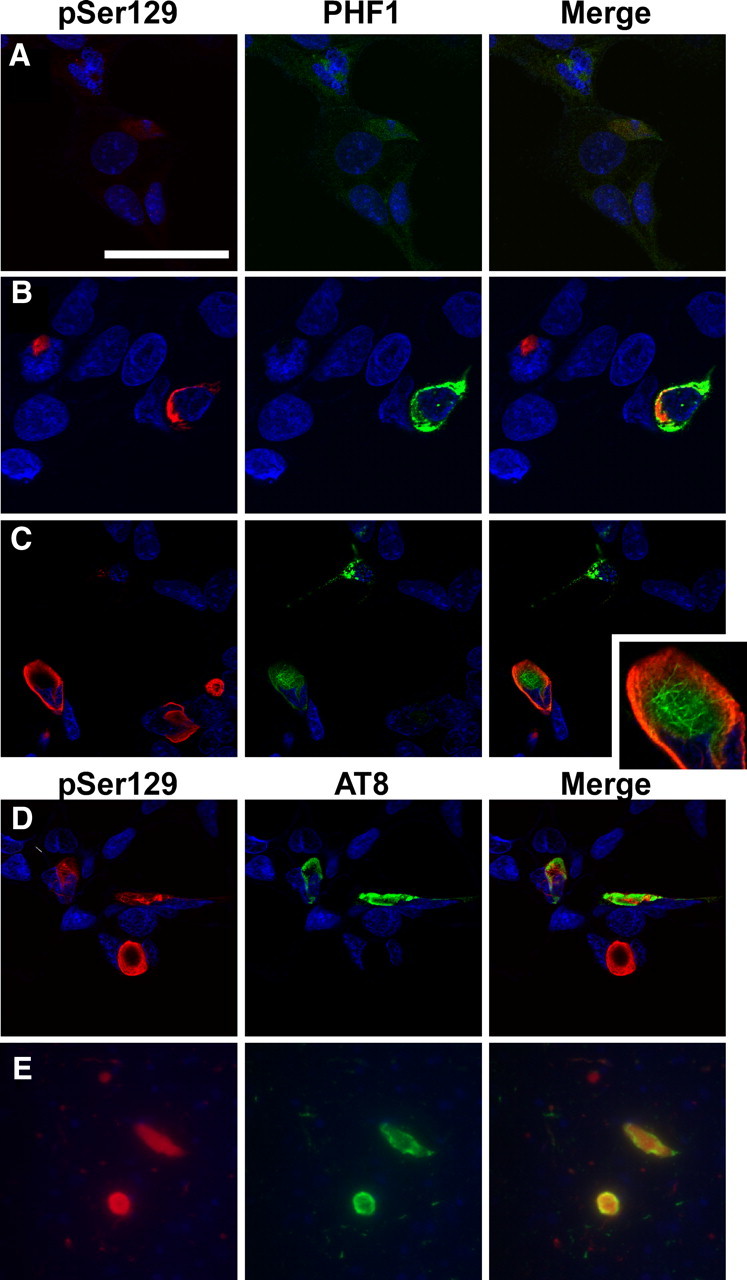
Representative confocal microscopy of double immunofluorescence with phosphospecific antibodies on α-syn and tau show mingling of α-syn and tau aggregates. A, Cells that were cotransfected with expression plasmids for α-syn and tau display a paucity of immunostaining with pSer129 or PHF1. B–D, Transfected cells treated with recombinant, prefibrillized 21–140 α-syn protein presented pSer129- positive and PHF1- (B, C) or AT8-positive (D) aggregates. Aggregates sometimes appeared intertwined or with phosphorylated α-syn circling phosphorylated tau. Representative images are of cells fixed 72 h after transfection. E, Double immunofluorescence between pSer129 and AT8 on the amygdala of a patient diagnosed with LBVAD shows similar morphology in vivo. Equivalent exposures were provided for all representative samples. Scale bar: A (for A, C, D) 60 μm, (for B) 40 μm, (for E) 100 μm, (for inset in C) 20 μm.
Although α-syn and tau pathology can localize and intermingle within the same aggregate in vivo, most Lewy pathology and NFTs coexist in the same brain region, but in different neurons (Giasson et al., 2003; Lee et al., 2004). We therefore hypothesized that preformed α-syn fibrils may seed tau aggregation even in the absence of α-syn overexpression. QBI293 cells transfected with only the expression plasmid for full-length tau displayed rare PHF1 (data not shown) or AT8 (Fig. 3A) immunoreactivity and typically only in cells that were rounded in morphology. Treatment with recombinant, prefibrillized full-length α-syn significantly increased the number of tau-expressing cells that were PHF1 and AT8 immunopositive (Fig. 3B,E,F). In fact, by confocal microscopy, these PHF1- and AT8-immunoreactive cells often appeared NFT-like (Fig. 3C). We also found that the propensity of these cells to form these AT8- or PHF1-positive, NFT-like aggregates was further promoted when QBI293 cells were transfected with an expression plasmid containing full-length tau with the FTDP-17 mutation P301L (Fig. 3D–F).
Figure 3.
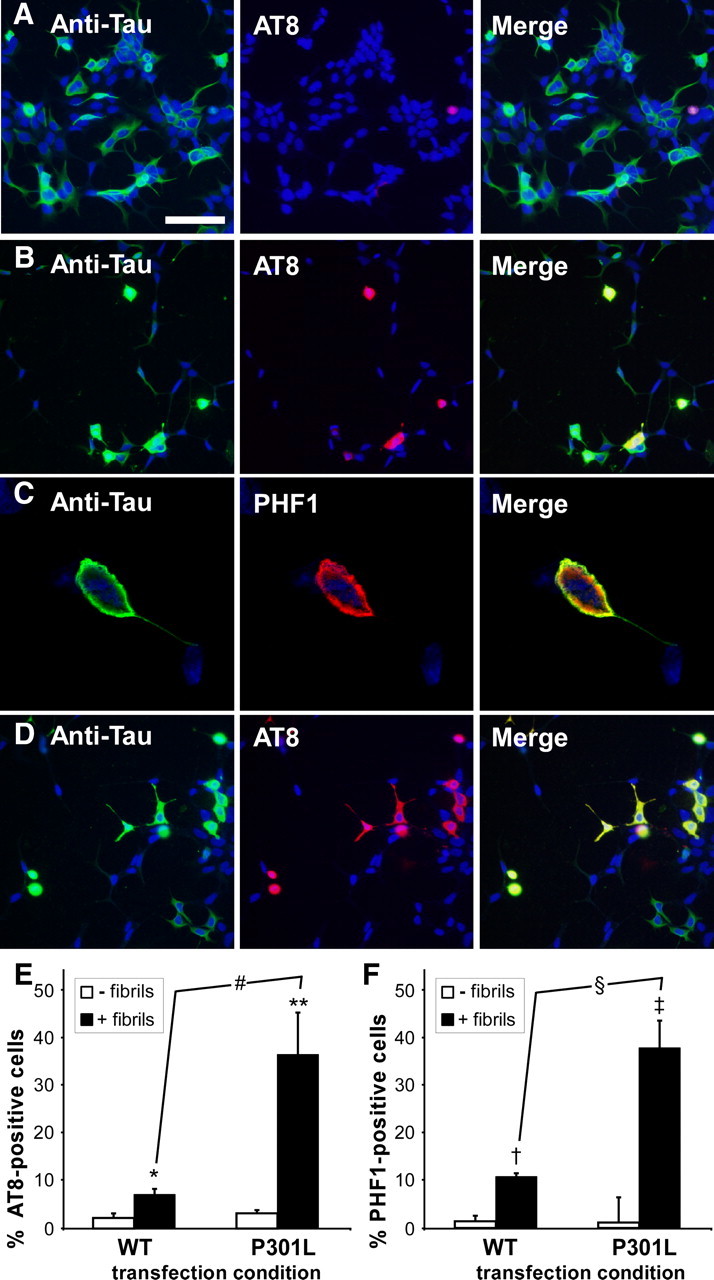
Recombinant, prefibrillized α-syn promotes the formation of tau aggregates in cells only overexpressing tau. A–D, QBI293 cells were transfected with expression plasmids for WT tau (A–C) or tau containing the P301L mutation (D), and double immunofluorescence was performed between an anti-tau antibody (green) and AT8 (A, B, D) or PHF1 (C) (red). Representative images are of cells fixed 72 h after transfection. In the absence of recombinant, prefibrillized α-syn treatment, rare AT8 positive cells were observed, and tau appeared mostly bundled (A). After recombinant, prefibrillized α-syn treatment, AT8 immunoreactivity increased for both wild-type (B) and P301L (D) tau-expressing cells. These aggregates displayed greater anti-tau immunoreactivity, and a greater percentage of transfected cells were AT8-immunopositive in the presence of the P301L mutation. C, Cellular tau aggregates were also positive for PHF1, and were often observed as encompassing the entire cell body, as observed by confocal microscopy. Equivalent exposures were provided for all representative samples. Scale bar: A (for A, B, D) 100 μm, (for C) 20 μm. E, F, Summary of the percent of tau-positive cells that displayed AT8 (E) or PHF1 (F) immunoreactivity in the absence (−) or presence (+) of fibril treatment (for AT8, *p = 0.0005 by two-way, parametric t test; **p = 0.005; #p = 0.007 by nonparametric Welsh t test; for PHF1, †p = 0.04; ‡p = 0.008; §p = 0.001 by nonparametric Welsh t test; n = 4 for all sets). Data represent the meant ± SD.
Intracellular, aggregated tau is composed of Triton-insoluble, hyperphosphorylated, thioflavin-S-positive fibrillar aggregates
Biochemical cellular fractionation further supported the aggregation of intracellular tau, promoted by the addition of recombinant, preformed α-syn fibrils (Fig. 4). Both in the absence (pcDNA) and presence (α-syn) of α-syn coexpression, recombinant 21–140 α-syn fibrils induced the formation of Triton-insoluble tau, with an increased propensity of P301L tau over that of WT tau. In cells expressing both α-syn and tau and treated with recombinant, prefibrillized 21–140 α-syn protein, both tau and full-length α-syn (intracellularly expressed α-syn protein) developed Triton-insoluble aggregates (Fig. 4A). However, intracellular α-syn coexpression was not required for significant development of Triton-insoluble tau, indicating that small amounts of preformed, exogenously applied fibrils were sufficient to seed aggregation of tau. Phosphospecific antibodies were efficient at detecting α-syn-induced Triton-insoluble tau, with PHF1 and AT8 providing the most robust phosphospecific signals. The percentage of Triton-insoluble tau also increased over the course of time (Fig. 4B). Interestingly, when both tau and α-syn were concurrently expressed and treated with preformed, recombinant fibrils to form intracellular aggregates, the P301L mutation of tau displayed a significantly greater propensity to aggregate than α-syn in the same cells (at 48 h, p = 0.003 by two-tailed, parametric t test; n = 5), indicating that cellular seeding of P301L tau could be promoted more readily than α-syn. Furthermore, α-syn aggregation propensity was not affected by tau coexpression (data not shown).
Figure 4.
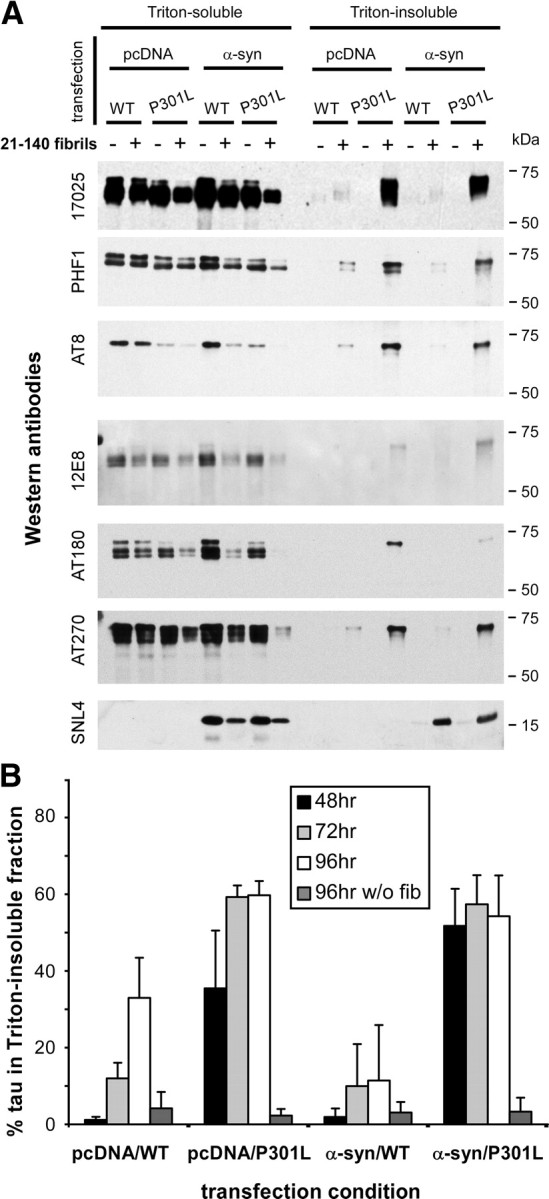
Biochemical cellular fractionation performed on tau-expressing cells after treatment with recombinant, prefibrillized α-syn protein. QBI293 cells were cotransfected with expression plasmids for WT or P301L tau and α-syn or pcDNA3.1 (pcDNA) empty vector. Parallel samples either received no treatment or were treated with recombinant prefibrillized 21–140 α-syn protein. Cells were harvested 72 h after transfection, and biochemical cellular fractionation was performed as described in Materials and Methods. A, Representative immunoblots of biochemical fractionation with anti-tau antibody 17025, with phosphospecific antibodies PHF1, AT8, 12E8, AT180, and AT270, and with anti-α-syn antibody SNL4. Triton-insoluble tau and α-syn were observed only after treatment with recombinant, prefibrillized α-syn treatment, and robust increases in Triton-insoluble tau were observed with cells expressing P301L tau. Representative immunoblots are from the same experiment. B, Quantitative summary of time course of the formation of Triton-insoluble tau, quantified by densitometry of anti-tau (17025) immunoreactivity. Indicated time is hours after transfection. The percentage of Triton-insoluble tau was calculated as Triton-insoluble tau/(Triton-soluble + Triton-insoluble tau) * 100. Data represent the meant ± SD (n = 4).
To further characterize the ability of α-syn to promote cellular tau aggregation, populations of polymers of recombinant α-syn were separated and compared in this cellular assay. After fibrillization of recombinant α-syn protein, large α-syn fibrils were isolated from small α-syn oligomers by centrifugation, and each population was subjected to sonication. Electron microscopy revealed various morphologies of α-syn polymers after sonication. Total recombinant α-syn fibrils (fibril mix) displayed a combination of larger fibrils and smaller oligomers (Fig. 5A). Isolated small oligomers appeared similar to the fibril mix; however, fewer larger fibrils were observed (Fig. 5B). Isolated large fibrils were composed of larger aggregated structures after sonication (Fig. 5C). However, they often had an amorphous appearance, and smaller circular structures were observed (Fig. 5C, arrowheads). The polymerized 21–140 (N-terminal truncated) α-syn protein appeared more resistant to sonication, as the majority of polymers observed were longer fibrils (Fig. 5D). Although there were differences between experiments depending on α-syn recombinant preparation and experimental variability, each polymer type was capable of inducing intracellular tau aggregation with similar efficiency, whereas recombinant, soluble (monomeric, nonfibrillized) α-syn did not typically augment tau aggregation propensity (Fig. 5E).
Figure 5.

Polymer subpopulations of fibrillized, recombinant α-syn can promote cellular tau aggregation. Recombinant full-length or 21–140 α-syn was prefibrillized. Before sonication, polymers of full-length α-syn were separated by centrifugation as described in Materials and Methods, and all populations were subjected to sonication. A–D, Ultrastructural electron microscopic analyses were performed on full-length α-syn that had not been separated by centrifugation (mix) (A), small α-syn oligomers (B), large α-syn fibrils (C), or 21–140 α-syn mix (D). Small circular structures were observed in large α-synuclein fibril condition (C, arrowheads). E, Cells expressing WT or P301L tau were treated with each of these polymer types at a final concentration of 1 μm. Cells were harvested 72 h after transfection and biochemical cellular fractionation was performed, separating the Triton X-100 (TX)-soluble from the TX-insoluble fraction. No differences were noted in aggregation propensity between these polymer types, whereas soluble, recombinant, nonfibrillized α-syn did not promote cellular tau aggregation.
Confocal microscopy analyses were then used to evaluate the formation of amyloidogenic aggregates in QBI293 cells expressing WT or P301L tau and treated with α-syn fibrils. AT8- and anti-tau-positive (17025) aggregates colocalized with thioflavin-S reactivity (Fig. 6A,B), whereas recombinant, prefibrillized α-syn protein, as visualized with antibody SNL4, rarely colocalized with a robust thioflavin-S signal (Fig. 6C). Furthermore, SNL4 immunoreactivity that was thioflavin-S positive was only identified extracellularly, rather than colocalizing with the large Thioflavin-S-positive intracellular aggregates (arrow). Surprisingly, thioflavin-S reactivity was often observed throughout the entire cell, suggesting widespread, rather than localized, intracellular tau amyloidogenic inclusions. Although the core of the tau aggregates typically exhibited reduced antibody immunoreactivity, likely because of reduced antibody penetrance, thioflavin-S, a chemical dye, was capable of completely labeling the entire cellular aggregate. Thioflavin-S reactivity was not observed with cells that were transfected with WT or P301L tau, but not treated with recombinant α-syn fibrils (data not shown).
Figure 6.

Thioflavin-S reactivity assessed by confocal microscopy on tau-expressing cells treated with recombinant, prefibrillized α-syn. A–C, Representative images of thioflavin-S (ThioS; green) immunoreactivity of QBI293 cells that were transfected with expression plasmids for WT (A) or P301L (B, C) tau and treated with recombinant, prefibrillized full-length α-syn. Representative images are of cells fixed 72 h after transfection. Intracellular, ThioS-positive aggregates were double labeled with AT8 (A) or 17025 (B) (red). Recombinant α-syn (SNL4 immunoreactivity; C, red) was observed sticking to the outside of cells, and only larger extracellular aggregates displayed robust ThioS reactivity. Anti-α-syn antibody SNL4 did not immunolabel large, intracellular ThioS aggregates (C, arrow). Scale bar, 20 μm.
Ultrastructural analyses demonstrated that the intracellular tau inclusions were comprised of 16.6 ± 3.8 nm (SD) wide, predominantly straight fibrils, similar in size and morphology to tau fibrils observed in human brain inclusions (Terry et al., 1964; Iqbal et al., 1975; Goedert, 1999) (Fig. 7). In addition, these studies provided insights into the effects of this amyloidogenic, aggregated tau. Cellular morphology was significantly disrupted by the formation of these intracellular tau aggregates with the cellular soma packed throughout with fibrous protein (Fig. 7A–E). This resulted in displacement or retraction of the nucleus (Fig. 7A,B). In addition, organelles were displaced and/or isolated amid the fibrous network with mitochondria appearing “trapped” and “sick” (Fig. 7A,B, arrowheads). Confocal microscopy analyses between tau and cellular markers lamp1 (for lysosomes) or mannosidase II (for Golgi) indicated that these organelles were also frequently trapped within these large cytoplasmic aggregates (data not shown).
Figure 7.
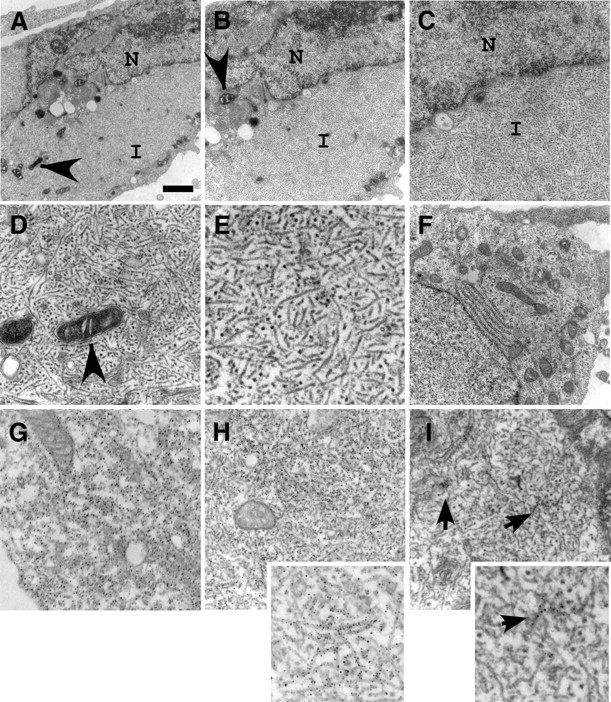
Ultrastructural analyses of cells forming tau aggregates. Electron microscopy was performed on transfected cells expressing P301L tau and treated with recombinant, prefibrillized α-syn protein. Representative images are cells fixed 72 h after transfection. A–C, Progressively increasing magnification on the center of the same cell to show cellular morphology and the cytoplasmic localization of fibrous protein aggregates. Examples of damaged mitochondria were observed (arrowheads). D, Higher magnification of cytoplasmic fibrils isolating organelles. E, High magnification of intracellular tau fibril morphology. F, Electron microscopy of a normal cell is provided for comparison. G, H, Immunolabeling with anti-tau antibody 17025 of P301L tau transfected cells in the absence (G) or presence (H) of recombinant, prefibrillized α-syn. I, Immunolabeling with anti-α-syn antibody SNL4 on P301L-transfected cells treated with recombinant, prefibrillized α-syn. Clusters of SNL4-positive fibrils were noted (arrows). Insets in H and I provide higher magnification of cellular fibrils within each frame. N, Nucleus; I, inclusion. Scale bar: A, 1.5 μm, (for B, F) 1 μm, (for C) 500 nm, (for D) 300 nm, (for E) 185 nm, (for G) 416 nm, (for H, I) 333 nm, (for H, inset) 166 nm, (for I, inset) 100 nm.
To identify the composition of these large cellular aggregates, we immunolabeled P301L tau-transfected cells with anti-tau antibodies before ultrastructural analyses. In the absence of recombinant fibril treatment, tau immunolabeling appeared diffuse and cytosolic (Fig. 7G), whereas in the presence of recombinant fibril treatment, cellular fibrillar aggregates were ubiquitously labeled (Fig. 7H). Similar findings were observed in cells transfected with WT tau (data not shown). Immunolabeling with anti-α-syn antibodies identified only small clusters of intracellular fibrils among the large cellular aggregates (Fig. 7I, arrows). In cells that were transfected with α-syn instead of tau, recombinant α-syn fibrils induced the formation of fibrillar α-syn inclusion (Waxman and Giasson, 2010), with robust, ubiquitous immunolabeling of intracellular fibrils with anti-α-syn antibodies (data not shown).
Cellular tau aggregation and the cytoskeleton
Under nonpathological conditions, tau binds to and participates in microtubule assembly (Gustke et al., 1994; Hong et al., 1998; Lee et al., 2001; Goedert and Spillantini, 2006). Confocal microscopy analyses of tau versus cytoskeletal markers suggested the presence of β-tubulin and vimentin inside the fibrous tau aggregate (Fig. 8A,C). Large, bulbous tau aggregates were observed, using 3D reconstruction of confocal microscopy images, and these aggregates often displaced γ-tubulin outside the center of cellular plane (Fig. 8B).
Figure 8.

Confocal microscopy analyses of cytoskeletal markers on cells containing tau aggregates. A–C, QBI293 cells were transfected with expression plasmids for WT (A, C) or P301L (B) tau and treated with recombinant, prefibrillized full-length α-syn. Cells containing tau aggregates were identified by the characteristic increased anti-tau immunoreactivity and displacement of the nuclear membrane. Representative images are cells fixed 72 h after transfection. Double immunofluorescence was performed between anti-tau antibodies (green) and anti-β-tubulin (A), anti-γ-tubulin (B), or anti-vimentin (C) (red). Three-dimensional projections of cells were composed by stacking 10–12 confocal images and rotating images 15–30° on a central axis (first three columns). A single, merged Z-plane (<0.7 μm) of each representative image is provided at the far right column. Anti-β-tubulin (A) and anti-vimentin immunoreactivity (C) were observed around and inside some tau aggregates. Anti-γ-tubulin immunoreactivity (B) was observed outside tau aggregates, often displayed outside of the center of the cellular plane. Similar morphology of tau aggregates were observed for wild-type and P301L-expressing cells. Scale bar, 40 μm.
We then performed biochemical analyses to identify the percent of tau protein bound to microtubules under basal conditions and to identify the propensity of β-tubulin to aggregate under conditions forming cellular tau aggregates. Approximately 40% of cellular tau bound to microtubules, as determined through biochemical microtubule-binding assays (Fig. 9). Surprisingly, no differences in microtubule-bound proteins were identified between WT and P301L tau proteins. Furthermore, under biochemical cellular fractionation conditions to identify aggregated protein, no Triton-insoluble β-tubulin was identified.
Figure 9.

Binding of tau to microtubules is not altered by the P301L mutation. QBI293 cells transfected with expression plasmids for WT or P301L tau were harvested 48 h after transfection for microtubule (MT) binding assays, as described in Materials and Methods. Representative immunoblots with anti-tau (17025) or anti-β-tubulin antibodies of unbound fraction (supernatant; S) or microtubule-bound fraction (pellet; P) and a concurrent experiment on cells with or without fibril treatment and harvested by biochemical cellular fraction (Triton-soluble and Triton-insoluble fractions) are shown. Representative immunoblots are from the same Western blot per antibody, but rearranged for presentation [41 ± 6% (SD) of WT and 40 ± 14% (SD) of P301L tau were bound to microtubules; n = 3].
The P301L tau mutation promotes aggregation propensity resulting from α-syn seeds in vitro
Based on microscopy analyses and biochemical data, recombinant, prefibrillized α-syn caused a profound increase in P301L tau aggregation compared to WT tau. Since P301L tau did not differentially bind to microtubules in our system, we examined the direct effects of α-syn seeding on tau aggregation in vitro. Recombinant, prefibrillized, sonicated α-syn was incubated with recombinant-generated WT or P301L tau under assembly conditions in vitro. After three days, α-syn significantly increased the sedimentation and K114 fluorometry of P301L tau over that of WT tau (Fig. 10A–C), whereas in the absence of α-syn neither WT nor P301L tau aggregated after 7 d (Fig. 10A, right). We then aimed to identify any potential changes in rate of aggregation between WT tau, P301L tau, and α-syn. In these experiments, soluble, recombinant α-syn was used at a concentration of 0.3 mg/ml such that α-syn and tau were approximately equimolar. At this concentration, α-syn only minimally begins to aggregate after 7 d of agitation (Fig. 10A, left). Each of these soluble recombinant proteins were incubated with recombinant, prefibrillized, sonicated α-syn, and the amount of sedimented protein was assessed after 1, 3, or 7 d. WT tau and α-syn reached maximum sedimentation after 1 d of incubation with prefibrillized α-syn (Fig. 10D). However, P301L tau sedimentation continued to increase over the course of 7 d. Under these in vitro conditions, P301L tau aggregation propensity resulting from α-syn fibril seeds was reduced when compared to α-syn.
Figure 10.

The P301L mutation in tau enhances the ability of prefibrillized α-syn to promote tau fibrillization in vitro. A, Left, Representative Coomassie blue R-250-stained SDS-polyacrylaminde gel of sedimentation analyses of preformed, sonicated recombinant α-syn fibrils (0.25 mg/ml) and tau (1 mg/ml) after 3 d under assembly conditions in vitro, as described in Materials and Methods. Right, Representative Coomassie blue R250-stained gel of soluble, recombinant α-syn (0.3 mg/ml) and tau (1 mg/ml) after 7 d of incubation in the absence of recombinant α-syn fibril seeds show an inability of tau to sediment. B, Quantitative summary of the ability of recombinant α-syn fibril seeds to promote tau aggregation at 3 d of incubation. P301L tau sedimented to a greater extent than WT tau (*p < 0.0001 by Mann–Whitney nonparametric U test). C, K114 fluorometry of assembled α-syn and tau under the above indicated conditions provided in arbitrary fluorescence units (AFU). α-Syn promoted increased K114 fluorometry in the presence of P301L tau (*p < 0.0001 by one-way ANOVA; p < 0.001 for P301L tau when directly compared to other conditions by Bonferroni post-test analyses). All data represent the mean ± SD (n = 9 in 3 independent experiments). D, The ability of recombinant, prefibrillized, sonicated α-syn to promote sedimentation of soluble α-syn (0.3 mg/ml), WT tau, or P301L tau was compared at 1, 3, and 7 d of incubation. Maximum sedimentation of WT tau and soluble α-syn was observed after 1 d of incubation, whereas sedimentation of P301L tau increased over the course of 7 d. Significant differences between all three protein types were observed at 1 and 3 d (1 d, p < 0.0001 by one-way ANOVA, p < 0.05 between WT and P301L tau, p < 0.001 between α-syn and WT or P301L tau, Bonferroni post-test analyses; 3 d, p < 0.0001 by one-way ANOVA, p < 0.001 between all conditions, Bonferroni post-test analyses; 7 d, p < 0.0001 by one-way ANOVA, p < 0.001 between WT tau and P301L or α-syn, Bonferroni post-test analyses; n = 5 to 13, depending on condition, each performed in a minimum of 3 independent experiments). Data represent the mean ± SD.
The role of phosphorylation on cellular aggregate formation
Tau contains many phosphorylation sites, many of which have been implicated in the formation of NFTs (Grundke-Iqbal et al., 1986; Goedert et al., 1992; Billingsley and Kincaid, 1997; Buee et al., 2000; Alonso et al., 2001; Hanger et al., 2007). Based on analyses of the hyperphosphorylation of tau cellular aggregates (Fig. 4, immunoblots), AT8 immunoreactivity was overrepresented in the Triton-insoluble fraction of tau (when compared to 17025; P301L plus fibrils, p = 0.003 by one-sample t test), and 12E8 was underrepresented (when compared to 17025; P301L plus fibrils, p = 0.05 by one-sample t test; n = 5). Kinases such as GSK3β and MARK2 may therefore alter propensities of tau aggregate formation (Schneider et al., 1999; Sato et al., 2002; Liu et al., 2007; Rankin et al., 2007).
To examine the role of kinases and phosphorylation on tau aggregate formation, QBI293 cells were cotransfected with expression plasmids for WT or P301L tau and GSK3β or pcDNA. Overexpression of GSK3β robustly increased phospho-tau immunoreactivity with antibodies PHF1, AT8, AT180, and AT270, but not 12E8 (data not shown) [consistent with Godemann et al. (1999)]. These changes in immunoreactivities were immeasurable because of the profound increases resulting from GSK3β overexpression. We then examined the effects of GSK3β on tau by biochemical cellular fractionation. In the absence of α-syn fibril treatment, GSK3β overexpression increased the amount of Triton-insoluble tau; however, it also attenuated the propensity of tau to aggregate in the presence of recombinant α-syn fibrils (Fig. 11A,B).
Figure 11.
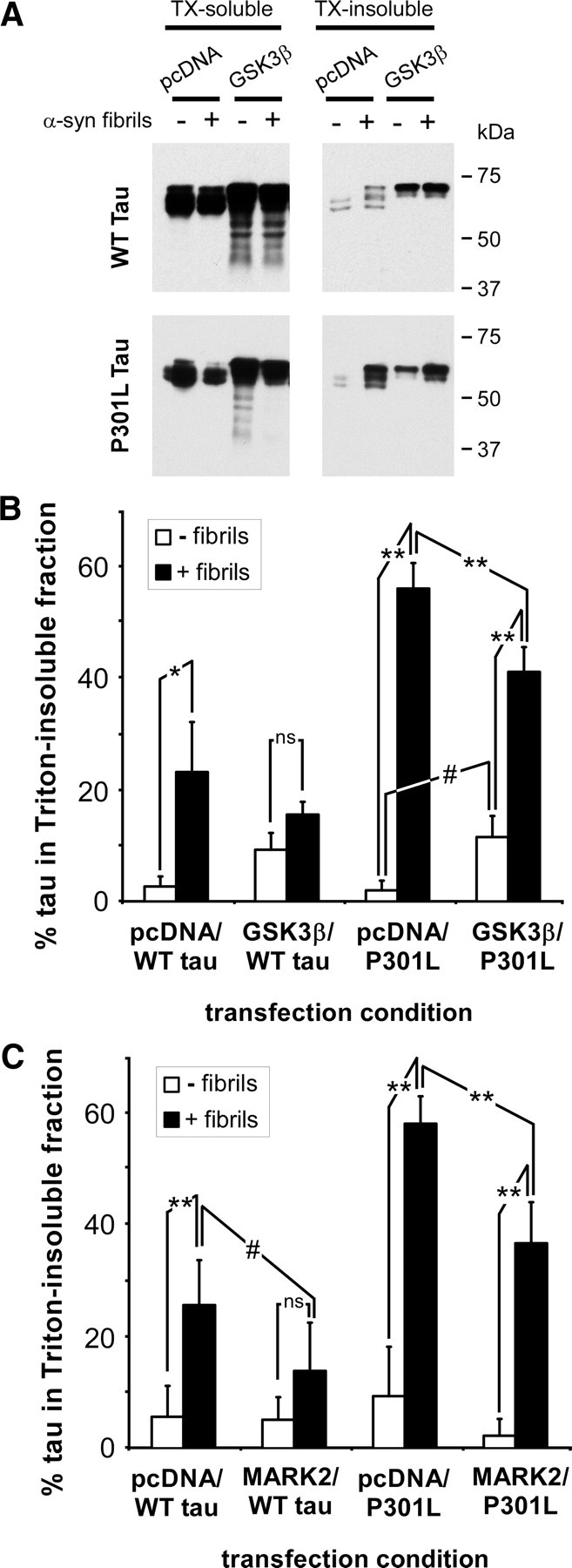
Overexpression of GSK3β or MARK2 inhibits the propensity of tau to form cellular aggregates. A–C, QBI293 cells were cotransfected with plasmids for the expression of WT or P301L tau and pcDNA (empty vector), GSK3β (A, B), or MARK2 (C). Cellular experimentation was performed in the absence (− fibrils) or the presence (+ fibrils) of recombinant, prefibrillized α-syn. Cells were harvested 72 h after transfection and biochemical cellular fractionation was performed, separating the Triton X-100 (TX)-soluble from the TX-insoluble fraction. A, Representative immunoblots of biochemical fractionation with anti-tau antibody 17025 on WT or P301L tau-expressing cells in the presence or absence of GSK3β overexpression. B, C, Quantitative summary of the percent of tau present in the Triton-insoluble fraction for each transfection and fibril treatment condition, as determined by densitometry of anti-tau (17025) immunoreactivity. With WT tau and GSK3β coexpression, no significant increase in Triton-insoluble tau was observed after the addition of α-syn fibrils, but a significant increase in Triton-insoluble tau was observed above samples with WT tau and pcDNA in the absence of fibrils (B) (p < 0.002 by one-way ANOVA; *p < 0.01 by Bonferroni post-test analyses; n = 4). With P301L tau, GSK3β coexpression resulted in a significant increase in Triton-insoluble in the absence of fibril treatment and a reduction of Triton-insoluble tau in the presence of treatment (p < 0.0001 by one-way ANOVA; **p < 0.001; #p < 0.05 by Bonferroni post-test analyses; n = 4). MARK2 coexpression significantly reduced the propensity of WT and P301L tau to aggregate after α-syn fibril treatment (C) (WT, p = 0.0006 by one-way ANOVA; P301L, p < 0.0001 by one-way ANOVA; **p < 0.001; #p < 0.05 by Bonferroni post-test analyses; n = 5). ns, Not significant. Data represent the mean ± SD.
To examine the effects of phosphorylation on the 12E8 epitopes S262/S356, QBI293 cells were cotransfected with expression plasmids for WT or P301L tau and MARK2 or pcDNA. In the presence of fibril treatment, MARK2 significantly inhibited the formation of Triton-insoluble WT and P301L tau (Fig. 11C). Concurrent with this effect, overexpression of MARK2 resulted in approximately a 10-fold increase in 12E8 immunoreactivity (Fig. 12A).
Figure 12.

Site-specific mutations of the 12E8 and AT8 epitopes alter the propensities of tau to form cellular aggregates. QBI293 cells were cotransfected with the indicated tau plasmids and pcDNA or MARK2. Cellular experimentation was performed in the absence (−) or presence (+) of recombinant, prefibrillized α-syn. A, Representative immunoblots of biochemical fractionation with antibodies 17025, 12E8, AT8, PHF1, or anti-myc (to identify MARK2 overexpression) were performed on cells expressing WT tau or tau containing S262A and/or S356A mutations in the presence or absence of MARK2 overexpression. B, Quantitative summary of the percentage of tau present in the Triton-insoluble fraction after treatment with recombinant α-syn fibrils. MARK2 coexpression significantly reduced the amount of Triton-insoluble tau in all conditions that did not contain the P301L mutation (p < 0.0001 by one-way ANOVA; *p < 0.05; **p < 0.001, Bonferroni post-test analyses; n = 4). Furthermore, tau S356A mutants coexpressed with MARK2 and treated with recombinant fibrils further reduced the propensity of tau to form cellular aggregates (#p < 0.01, Bonferroni post-test analyses; n = 4). No significant changes were observed between conditions expressing the P301L mutation of tau and treated with recombinant α-syn fibrils when multiple comparisons were factored into analyses. C, Quantitative summary of the percentage of tau present in the Triton-insoluble fraction after treatment with recombinant α-syn fibrils. Tau was mutated at the AT8 epitope residues S202 and T205 to either Ala or Glu. Both the S202A/T205A and the S202E/T205E mutations inhibited the propensity of tau to aggregate (p = 0.0004 by repeated-measures ANOVA; *p < 0.05; **p < 0.001, Bonferroni post-test analyses; n = 3). In the presence of the P301L mutation, only the additional S202E/T205E mutation significantly inhibited the propensity of cellular tau to aggregate (p = 0.02 by repeated-measures ANOVA; *p < 0.05 Bonferroni post-test analyses; n = 3). All data represent the meant ± SD.
To discern the effects of specific phosphorylation sites affected by MARK2 expression on the formation of tau aggregates, we coexpressed MARK2 in cells expressing tau with mutations S262A and/or S356A. In the absence of MARK2 overexpression, S262A and/or S356A mutations did not alter the propensities of tau aggregation in the presence of α-syn fibril treatment (Fig. 12A,B). Fibril treatment with S262A tau and MARK2 coexpression exhibited similar inhibition as that observed with WT tau and MARK2. However, surprisingly, S356A tau or S262A/S356A tau and MARK2 coexpression resulted in a significantly greater loss of tau aggregation when compared to WT tau and MARK2 coexpression. These results were only observed for tau without the P301L mutation. The P301L mutation in combination with the S262A and/or S356A mutations resulted in nonsignificant differences between fibril-treated samples when multiple comparisons were taken into account.
Interestingly, in the absence of MARK2, the S262A mutation reduced 12E8 immunoreactivity of Triton-soluble tau to 29 ± 18% (SD) when compared to WT tau, and P301L/S262A tau reduced 12E8 immunoreactivity of Triton-soluble tau to 45 ± 30% (SD), but only reduced 12E8 immunoreactivity of Triton-insoluble tau to 69 ± 11% (SD), when compared to the same conditions of P301L tau. Consistent with this finding, the S356A mutations reduced 12E8 immunoreactivity of Triton-soluble tau to 69 ± 27% (SD) and 56 ± 16% (SD) (when compared to WT and P301L tau, respectively), whereas 12E8 immunoreactivity of Triton-insoluble tau was reduced to 37 ± 31% (SD) of the same condition with P301L tau (n = 3 for all conditions). In the absence of MARK2 coexpression, Triton-insoluble WT tau was not observed with 12E8; therefore, no changes were observed in this fraction with S262A or S356A mutations. Presence of both S262A and S356A mutations completely abrogated 12E8 immunoreactivity in both fractions.
MARK2 overexpression also significantly decreased AT8 immunoreactivity of Triton-soluble tau [by 84 ± 11% (SD) for WT tau when standardized to 17025 immunoreactivity; p = 0.008 by one-sample t test; n = 4]. Furthermore, the S356A and the S262A/S356A mutations in tau reduced AT8 immunoreactivity to 51 ± 21% (SD) and 21 ± 14% (SD), respectively (compared to WT tau when standardized to 17025 immunoreactivity; p = 0.06, p = 0.01 by one-sample t test; n = 3). These effects appeared isolated, since relative AT8 immunoreactivity of Triton-insoluble tau was not altered when controlled for 17025 immunoreactivity, and PHF1 immunoreactivity was not affected by MARK2 or S356A mutations in the either fraction.
To discern potential effects caused by alterations of the residues associated with the AT8 epitope, we mutated residues S202 and T205 to either Ala or Glu. The Ala residue was used to remove any basal phosphorylation of these residues, whereas the Glu was used as a phosphomimetic. Cells transfected with S202A/S205A or S202E/S205E tau and treated with recombinant fibrils each demonstrated significant reductions in the propensity of tau to aggregate, but to varying degrees (Fig. 12C). With cells transfected to express P301L tau, only the additional mutations of S202E/S205E modestly reduced the propensity of tau to aggregate in the presence of recombinant fibrils.
Discussion
The current work provides in-depth analyses of the formation of NFT-like inclusions that develop in response to cellular seeding by recombinant, preformed α-syn fibrils. This study establishes an important connection between the formation of α-syn-containing Lewy pathology and the formation of NFTs, as observed in pathological brain. Hyperphosphorylated, Triton-insoluble, Thioflavin-S-positive, filamentous tau could be induced to intermingle with endogenously expressed aggregated α-syn or prompted to aggregate from the addition of only a small amount of α-syn fibril seeds. Furthermore, a significant number of tau-expressing cells (∼11%) and a significant proportion of total WT tau protein (up to ∼30%) formed aggregates in the presence of fibrillized α-syn seeds. These studies show that the entry of only a small amount of fibrillized α-syn is sufficient to induce the formation of tau fibrillar inclusions that encompass almost the entire cell, even in the absence of α-syn coexpression. This finding is consistent with the previously suggestions that only a minute amount of amyloidogenic α-syn may be necessary to induce tau aggregation with pathological consequences (Lee et al., 2004). Furthermore, this cellular model is uniquely capable of examining factors associated with tau inclusion formation, mimicking pathology observed postmortem.
Recent cellular studies have shown intracellular tau aggregation in response to treatment with recombinant, preformed tau fibrils, producing small intracellular, hyperphosphorylated tau aggregates (Frost et al., 2009; Nonaka et al., 2010). However, unlike α-syn that can polymerize by itself, tau aggregation requires an inducing agent (Goedert et al., 1996). This work suggests that α-syn polymers may initially induce tau aggregation, but once this process is initiated, aggregated forms of tau may propagate the aggregation process, further promoting amyloid formation.
In the cellular model described here, the intracellular tau inclusions developed to encompass the entire cellular soma, similar to NFTs in diseased human brains. This is unlike α-syn cellular aggregates (Waxman and Giasson, 2010), which typically expand around the γ-tubulin-positive centrosome and enlarge in a sphere-like manner, but remain relatively localized. These tau aggregates, instead, displaced the centrosome and isolated organelles, which likely ceased cellular activity. Based on cytoskeletal localization, these tau aggregates do not form aggresomes, but instead trap and mingle with cytoskeletal markers. Although these microscopic data are difficult to interpret, the overwhelming toxic appearance, including dying mitochondria, was apparent, indicating that these tau inclusions result in cellular injury. Furthermore, ultrastructural analyses of these pathological aggregates bare striking structural similarity to tau aggregates characterized in animal models of tauopathies (Lin et al., 2003) and to NFTs analyzed postmortem (Terry et al., 1964; Iqbal et al., 1975).
Interestingly, the P301L mutation in tau that is causal of FTDP-17 significantly promotes the cellular aggregation of tau. The P301L mutation enhances tau polymerization in vitro in studies performed with heparin (Goedert et al., 1999; Nacharaju et al., 1999; Barghorn et al., 2000). However, in vitro studies performed with prefibrillized α-syn show that WT tau aggregation reached its maximum after 1 d, whereas P301L tau continued to aggregate through 7 d of incubation. This difference could be attributable to differential protein interactions or differences in structural folding between WT and P301L tau. Furthermore, in cells expressing both P301L tau and α-syn, P301L tau augmented the propensity of tau aggregation in the presence of recombinant seeds more than that observed with α-syn in the same samples. This difference was not recapitulated in vitro, as α-syn seeds promoted α-syn aggregation more readily than P301L tau. This disconnect between in vitro and in situ may be attributable to the ability of recombinant α-syn to self-aggregate, whereas intracellular expressed α-syn requires seeding, or other cellular factors may promote P301L tau aggregation in vivo.
In vitro studies suggest that the P301L mutation reduces binding of tau to microtubules, thereby increasing the pool of tau available to aggregate (Hong et al., 1998; Sun and Gamblin, 2009). However, in this study, tau binding to microtubules was not altered by this mutation, similar to other reports (Vogelsberg-Ragaglia et al., 2000), suggesting a lack of microtubule involvement in the increased propensity of P301L tau to develop aggregates. Although the current study suggests that cytoskeletal markers may be present and potentially isolated within tau inclusions, biochemical cellular fractionation did not isolate β-tubulin in the Triton-insoluble fraction, indicating that microtubules are independent from tau inclusions.
The relationships between kinase activity, phosphorylation of tau, and NFT formation have been a long-standing debate. Tau is phosphorylated at an abundant number of sites, and most of these sites are hyperphosphorylated in the pathological form of tau. GSK3β and its phosphorylation of the PHF1 and AT8 epitopes have been widely implicated in the formation of pathological tau aggregates (Sato et al., 2002; Engel et al., 2006; Liu et al., 2007; Rankin et al., 2007; Jeganathan et al., 2008; Kwok et al., 2008). However, coexpression of GSK3β in this study resulted in two opposing processes: one in which basal expression of tau increased in the Triton-insoluble fraction, and another in which significantly decreased α-syn-induced tau aggregate formation. GSK3β phosphorylates tau at many sites, including but not limited to T181, S202/T205, T231/S235, and S396/S404 (Ishiguro et al., 1993; Dayanandan et al., 1999; Godemann et al., 1999; Hanger et al., 2007). Therefore, whereas phosphorylation at one site may increase the propensity of tau to aggregate, another site may induce inhibitory processes. However, structural forms of Triton-insoluble tau promoted by GSK3β expression may be independent from the tau fibrils created by recombinant α-syn seeds, which would suggest only inhibitory processes involved with the expression of GSK3β on tau inclusion formation. Nevertheless, these data are consistent with a previous report of GSK3β-mediated inhibition of tau fibrillization observed in vitro (Schneider et al., 1999).
MARK2 expression was also capable of significantly reducing tau aggregate formation. This is not surprising, since MARK2 primarily phosphorylates S262 and S356 within the tau repeat region (Drewes et al., 1995, 1997), the region that forms the core of tau filaments and is required for tau aggregation (Crowther et al., 1989, 1992; Goode and Feinstein, 1994), and MARK2 phosphorylation of tau can inhibit fibrillization in vitro (Schneider et al., 1999). However, during investigation of the potential residues involved in MARK2-mediated inhibition, single-point mutations did not identify either S262 or S356 as responsible. An uncharacterized phosphorylation site on tau or other biological effects may therefore be responsible. Interestingly, single-point mutation analyses identified S356 phosphorylation as potentially promoting tau aggregation, consistent with Triton-insoluble tau being enriched in phospho-S356 over phospho-S262.
An additional unexpected effect of MARK2 was that coexpression of MARK2 reduced AT8 immunoreactivity. This effect was not generalized to PHF1 immunoreactivity, suggesting a change in kinase specificity in regards to the S202/T205 sites. Studies suggest that AT8 phosphorylation promotes the formation of tau aggregates (Biernat et al., 1992; Rankin et al., 2007; Jeganathan et al., 2008). We therefore examined the role of the AT8 epitope by mutating these residues to block phosphorylation or add phosphomimetics. Mutations to either A or E both inhibited the propensity of tau to aggregate, with a stronger effect noted for E. These data suggest that any alterations to the S202/T205 residues may reduce the ability of tau to aggregate, rather than only abrogating or enhancing phosphorylation-like activity. Therefore, the role phosphorylation at the AT8 epitopes may not be adequately assessed by these means.
However, changes in AT8 immunoreactivity provided relevant information as to substrate targeting of tau by kinases. MARK2 coexpression or the P301L mutation in tau significantly decreased AT8 immunoreactivity of Triton-soluble tau (Boekhoorn et al., 2006; Han and Paudel, 2009) (Fig. 4) [AT8 immunoreactivity P301L, 66 ± 24% (SD) less than WT tau; p = 0.003; n = 5], but AT8 immunoreactivity in the Triton-insoluble fraction was not affected. These data suggest that tau is likely phosphorylated at the AT8 epitope after protein aggregation and that kinases may independently target soluble or aggregated tau. These studies suggest that investigations into factors that modulate the phosphorylation of the soluble forms of tau may not be generalized to the aggregated, NFT-like form of tau.
Investigations into the formation of pathological tau aggregates have been limited to date by the availability of adequate cellular models. Although in vitro studies are informative, the cellular milieu needs to be considered for potential therapeutic intervention. The current study has extended cellular modeling to include the ability of α-syn to induce the robust formation of filamentous tau aggregates with characteristics of NFTs. The ability of a small amount of preformed α-syn to induce large intracellular amyloid tau inclusions is consistent with the frequent coexistence of α-syn and tau inclusions in the diseased brain, and our study supports the notion that minute amounts of α-syn can be a physiological inducer of tau aggregation (Giasson et al., 2003). This ability of α-syn is also consistent with the mounting evidence that α-syn can be transported across cell membranes (Ahn et al., 2006; Lee et al., 2008; Desplats et al., 2009; Liu et al., 2009) and that amyloid diseases may spread by prion-like mechanisms (Lee et al., 2010; Goedert et al., 2010). Therefore, this research provides a likely mechanism to explain the frequent coexistence of hyperphosphorylated α-syn and tau pathology observed postmortem in disease. It is through the understanding of these factors that are associated with the formation of pathological aggregates and result in cross-seeding of pathological proteins that we may develop future therapeutics for disease.
Footnotes
This work was supported by National Institute on Aging Grant AG09215 and National Institute of Neurological Disorders and Stroke Grant NS053488. We thank the Biochemical Imaging Core Facility supported by Abramson Cancer Institute at the University of Pennsylvania for assistance in the electron and confocal microscopy studies.
The authors declare no competing financial interests.
References
- Ahn KJ, Paik SR, Chung KC, Kim J. Amino acid sequence motifs and mechanistic features of the membrane translocation of alpha-synuclein. J Neurochem. 2006;97:265–279. doi: 10.1111/j.1471-4159.2006.03731.x. [DOI] [PubMed] [Google Scholar]
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Perez M, Armas-Portela R, Avila J. Polymerization of tau peptides into fibrillar structures. The effect of FTDP-17 mutations. FEBS Lett. 1999;446:199–202. doi: 10.1016/s0014-5793(99)00210-0. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- Biernat J, Mandelkow EM, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323:577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekhoorn K, Terwel D, Biemans B, Borghgraef P, Wiegert O, Ramakers GJA, de Vos K, Krugers H, Tomiyama T, Mori H, Joels M, van Leuven F, Lucassen PJ. Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci. 2006;26:3514–3523. doi: 10.1523/JNEUROSCI.5425-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Congdon EE, Kim S, Bonchak J, Songrug T, Matzavinos A, Kuret J. Nucleation-dependent tau filament formation: the importance of dimerization and an estimation of elementary rate constants. J Biol Chem. 2008;283:13806–13816. doi: 10.1074/jbc.M800247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Olesen OF, Jakes R, Goedert M. The microtubule binding repeats of tau protein assemble into filaments like those found in Alzheimer's disease. FEBS Lett. 1992;309:199–202. doi: 10.1016/0014-5793(92)81094-3. [DOI] [PubMed] [Google Scholar]
- Crowther T, Goedert M, Wischik CM. The repeat region of microtubule-associated protein tau forms part of the core of the paired helical filament of Alzheimer's disease. Ann Med. 1989;21:127–132. doi: 10.3109/07853898909149199. [DOI] [PubMed] [Google Scholar]
- Crystal AS, Giasson BI, Crowe A, Kung MP, Zhuang ZP, Trojanowski JQ, Lee VM. A comparison of amyloid fibrillogenesis using the novel fluorescent compound K114. J Neurochem. 2003;86:1359–1368. doi: 10.1046/j.1471-4159.2003.01949.x. [DOI] [PubMed] [Google Scholar]
- Dayanandan R, Van Slegtenhorst M, Mack TG, Ko L, Yen SH, Leroy K, Brion JP, Anderton BH, Hutton M, Lovestone S. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett. 1999;446:228–232. doi: 10.1016/s0014-5793(99)00222-7. [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, Meyer HE, Mandelkow EM, Mandelkow E. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270:7679–7688. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Duda JE, Lee VMY, Trojanowski JQ. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61:121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Miller DC, Golbe LI, Lee VMY, Trojanoswki JQ. Concurrence of α-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol (Berl) 2002;104:7–11. doi: 10.1007/s00401-002-0563-3. [DOI] [PubMed] [Google Scholar]
- Engel T, Lucas JJ, Gómez-Ramos P, Moran MA, Avila J, Hernández F. Cooexpression of FTDP-17 tau and GSK-3beta in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol Aging. 2006;27:1258–1268. doi: 10.1016/j.neurobiolaging.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Uryu K, Trojanowski JQ, Lee VMY. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Jakes R, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VMY. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson's disease. J Neurosci Res. 2000;59:528–533. doi: 10.1002/(SICI)1097-4547(20000215)59:4<528::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Murray IVJ, Trojanowski JQ, Lee VMY. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanoswki JQ, Lee VMY. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Golbe LI, Graves CL, Kotzbauer PT, Trojanoswki JQ, Lee VMY. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Ann Neurol. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- Godemann R, Biernat J, Mandelkow E, Mandelkow EM. Phosphorylation of tau protein by recombinant GSK-3[beta]: pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS Lett. 1999;454:157–164. doi: 10.1016/s0014-5793(99)00741-3. [DOI] [PubMed] [Google Scholar]
- Goedert M. Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and alpha-synucleinopathies. Philos Trans R Soc Lond B Biol Sci. 1999;354:1101–1118. doi: 10.1098/rstb.1999.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Hasegawa M, Smith MJ, Spillantini MG. Intraneuronal filamentous tau protein and alpha-synuclein deposits in neurodegenerative diseases. Biochem Soc Trans. 1998;26:463–471. doi: 10.1042/bst0260463. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999;450:306–311. doi: 10.1016/s0014-5793(99)00508-6. [DOI] [PubMed] [Google Scholar]
- Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010;33:317–325. doi: 10.1016/j.tins.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Goode BL, Feinstein SC. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol. 1994;124:769–782. doi: 10.1083/jcb.124.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem. 2005;280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- Han D, Paudel HK. FTDP-17 missense mutations site-specifically inhibit as well as promote dephosphorylation of microtubule-associated protein tau by protein phosphatases of HEK-293 cell extract. Neurochem Int. 2009;54:14–27. doi: 10.1016/j.neuint.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, Seereeram A, Reynolds CH, Ward MA, Anderton BH. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. 2007;282:23645–23654. doi: 10.1074/jbc.M703269200. [DOI] [PubMed] [Google Scholar]
- Hansen L, Salmon D, Galasko D, Masliah E, Katzman R, DeTeresa R, Thal L, Pay MM, Hofstetter R, Klauber M, Rice V, Butters N, Alford M. The Lewy body variant of Alzheimer's disease: a clinical and pathologic entity. Neurology. 1990;40:1–8. doi: 10.1212/wnl.40.1.1. [DOI] [PubMed] [Google Scholar]
- He X, Saint-Jeannet JP, Woodgett J, Varmus H, Dawid I. Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature. 1995;374:617–622. doi: 10.1038/374617a0. [DOI] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Wisniewski HM, Grundke-Iqbal I, Korthals JK, Terry RD. Chemical pathology of neurofibrils. Neurofibrillary tangles of Alzheimer's presenile-senile dementia. J Histochem Cytochem. 1975;23:563–569. doi: 10.1177/23.7.1141687. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. Glycogen synthase kinase 3[beta] is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325:167–172. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–32076. doi: 10.1074/jbc.M805300200. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Spooren W, Fuss B, Mallon B, Macklin WB, Fujiwara H, Hasegawa M, Iwatsubo T, Kretzschmar HA, Haass C. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 2002;3:583–588. doi: 10.1093/embo-reports/kvf109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JB, Loy CT, Hamilton G, Lau E, Hallupp M, Williams J, Owen MJ, Broe GA, Tang N, Lam L, Powell JF, Lovestone S, Schofield PR. Glycogen synthase kinase-3beta and tau genes interact in Alzheimer's disease. Ann Neurol. 2008;64:446–454. doi: 10.1002/ana.21476. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of nonprion protein aggregates. Nat Rev Neurol. 2010;6:702–706. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanoswki JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lee VM, Giasson BI, Trojanowski JQ. More than just two peas in a pod: common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci. 2004;27:129–134. doi: 10.1016/j.tins.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul MM, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2003;32:1091–1105. doi: 10.1023/B:NEUR.0000021904.61387.95. [DOI] [PubMed] [Google Scholar]
- Liu F, Li B, Tung EJ, Grundke-Iqbal I, Iqbal K, Gong CX. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci. 2007;26:3429–3436. doi: 10.1111/j.1460-9568.2007.05955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang JP, Shi M, Quinn T, Bradner J, Beyer R, Chen S, Zhang J. Rab11a and HSP90 regulate recycling of extracellular alpha-synuclein. J Neurosci. 2009;29:1480–1485. doi: 10.1523/JNEUROSCI.6202-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Mishizen AJ, Giasson BI, Lynch DR, Thomas SA, Nakashima A, Nagatsu T, Ota A, Ischiropoulos H. Cytosolic catechols inhibit alpha-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J Neurosci. 2006;26:10068–10078. doi: 10.1523/JNEUROSCI.0896-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, Yen S-H. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999;447:195–199. doi: 10.1016/s0014-5793(99)00294-x. [DOI] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Muller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishie M, Mori F, Fujiwara H, Hasegawa M, Yoshimoto M, Iwatsubo T, Takahashi H, Wakabayashi K. Accumulation of phosphorylated alpha-synuclein in the brain and peripheral ganglia of patients with multiple system atrophy. Acta Neuropathol (Berl) 2004;107:292–298. doi: 10.1007/s00401-003-0811-1. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of alpa-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Paxinou E, Chen Q, Weisse M, Giasson BI, Norris EH, Rueter SM, Trojanowski JQ, Lee VM, Ischiropoulos H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci. 2001;21:8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Rankin CA, Sun Q, Gamblin TC. Tau phosphorylation by GSK-3beta promotes tangle-like filament morphology. Mol Neurodegener. 2007;2:12. doi: 10.1186/1750-1326-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Tatebayashi Y, Akagi T, Chui DH, Murayama M, Miyasaka T, Planel E, Tanemura K, Sun X, Hashikawa T, Yoshioka K, Ishiguro K, Takashima A. Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J Biol Chem. 2002;277:42060–42065. doi: 10.1074/jbc.M202241200. [DOI] [PubMed] [Google Scholar]
- Schneider A, Biernat J, von BM, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GVW, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, Lee VMY. Detection of phosphorylated Ser in fetal tau, adult tau, and paired helical filament tau. J Biol Chem. 1995;270:18917–18922. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- Sun Q, Gamblin TC. Pseudohyperphosphorylation causing AD-like changes in tau has significant effects on its polymerization. Biochemistry. 2009;48:6002–6011. doi: 10.1021/bi900602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teclemariam-Mesbah R, Wortel J, Romijn HJ, Buijs RM. A simple silver-gold intensification procedure for double DAB labeling studies in electron microscopy. J Histochem Cytochem. 1997;45:619–621. doi: 10.1177/002215549704500414. [DOI] [PubMed] [Google Scholar]
- Terry RD, Gonatas NK, Weiss M. Ultrastructural studies in Alzheimer's presenile dementia. Am J Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, Zhang B, Hong M, Trojanowski JQ, Lee VM. Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell. 2000;11:4093–4104. doi: 10.1091/mbc.11.12.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J Neuropathol Exp Neurol. 2008;67:402–416. doi: 10.1097/NEN.0b013e3186fc995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI. A novel, high-efficiency cellular model of fibrillar alpha-synuclein inclusions and the examination of mutations that inhibit amyloid formation. J Neurochem. 2010;113:374–388. doi: 10.1111/j.1471-4159.2010.06592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Covy JP, Bukh I, Li X, Dawson TM, Giasson BI. Leucine-rich repeat kinase 2 expression leads to aggresome formation that is not associated with alpha-synuclein inclusions. J Neuropathol Exp Neurol. 2009;68:785–796. doi: 10.1097/NEN.0b013e3181aaf4fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem. 1999;274:19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]