

Abstract
The expression of inducible nitric oxide synthase (iNOS) and the production of nitric oxide (NO) are important host defense mechanisms against pathogens in mononuclear phagocytes. The objectives of this study were to examine the roles of mitogen-activated protein kinases (MAPKs) and transcription factors (nuclear factor-κB [NF-κB] and activating protein 1 [AP-1]) in peptidoglycan (PGN)-induced iNOS expression and NO production in macrophages. PGN is a cell wall component of Gram-positive bacteria that stimulates inflammatory responses both ex vivo and in vivo. PGN stimulates the activation of all three classes of MAPKs, extracellular signal-related kinase (ERK), c-Jun N-terminal kinase (JNK), and p38mapk in macrophages, albeit with differential activation kinetics. Using a selective inhibitor of JNK (SP600125) and JNK1/2 small interfering RNA (siRNA) knocked-down macrophages, it was observed that PGN-induced iNOS and NO expression is significantly inhibited. This suggested that JNK MAPK plays an essential role in PGN-induced iNOS expression and NO production. In contrast, inhibition of the ERK pathway using PD98059 dose dependently enhanced PGN-induced iNOS expression and NO production. PGN-induced ERK activation was attenuated in ERK1/2 siRNA knocked-down macrophages; however, NO and iNOS expression were significantly enhanced. An electrophoretic mobility shift assay showed that SP600125 inhibited PGN-induced NF-κB and AP-1 activation, whereas inhibition of the ERK pathway enhanced NF-κB activation, but with no effect on AP-1. These results indicate that the JNK MAPK positively regulate PGN-induced iNOS and NO expression by activating NF-κB and AP-1 transcription factors, whereas the ERK pathway plays a negative regulatory role via affecting NF-κB activity.
INTRODUCTION
Sepsis represents a major challenge to the healthcare system, affecting about 751,000 people, causing ∼215,000 deaths, and costing nearly $17 billion annually in the United States (35). According to a recent report, the incidence of sepsis is rising at an astonishing annual rate of ∼8.7%, despite substantial prevention efforts and advancements in treatment (35, 49). Gram-positive bacteria have become the predominant organisms in sepsis cases since 1987 and accounted for >52% of all cases of sepsis in 2000 (35). Staphylococcus aureus is a leading cause of nosocomial pneumonia and wound infections and is one of the bacteria most commonly isolated from patients with sepsis (35, 49). Peptidoglycan (PGN), the main cell wall component of Gram-positive bacteria, activates host cells through the pattern recognition receptors Toll-like receptor 2 (TLR2) and CD14 and induces the expression of more than 120 genes in human monocytes (23, 43, 51). PGN has been reported to stimulate a variety of signal transduction elements such as MyD88, TRAF, protein tyrosine kinases, protein kinase Cδ, and mitogen-activated protein kinases (MAPKs) in macrophages and induces the production of inflammatory cytokines and nitric oxide (5, 8, 19, 48, 50, 51). However, the roles of MAPKs in PGN-induced activation of macrophages are still not well defined. The MAPKs constitute an important group of serine/threonine signaling kinases that modulate the phosphorylation and therefore the activation status of transcription factors, linking transmembrane signaling with gene induction in the nucleus. MAPKs are signaling molecules that play important roles in inflammatory processes. At least three MAPK cascades have been well described: extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK)/stress-activated protein kinase (15, 29, 42), and PGN has been reported to activate all three pathways (19). However, the physiological relevance of such MAPK signaling to macrophage function remains unclear.
Activated macrophages release oxygen and nitrogen radicals that are important bactericidal and cytostatic molecules (33, 38). However, massive production of these mediators can exert detrimental effects in the organism, as occurs during septic shock or persistent local inflammatory processes (7, 39). For this reason, the study of the mechanism of action of anti-inflammatory cytokines and drugs has constituted a subject of current interest (7, 27, 28, 31). It is well known that inducible nitric oxide synthase (iNOS) expression is regulated mainly at the transcription level due to the activation of several transcription factors that bind to the promoter region of the iNOS gene, such as nuclear factor-κB (NF-κB), activating protein 1 (AP-1), STAT1, and IRF-1 (32–34, 52, 54, 55). Several data point to NF-κB activation as a critical event in the expression of iNOS (16, 46, 55), and most studies focused on the analysis of anti-inflammatory mechanisms have suggested a prominent role for the inhibition of this transcription factor in their mode of action (2, 3).
The present investigations were undertaken to define the regulatory role of different MAPKs, in iNOS expression and nitric oxide (NO) production in mouse peritoneal macrophages activated with PGN from S. aureus in vitro. Our data support the hypothesis that while activation of all three subfamilies of MAPKs occurs in macrophages stimulated with PGN, the activation of JNK MAPK, which in turn activates NF-κB and AP-1, is necessary for iNOS and NO expression. Inhibition of ERK MAPK by PD98059 or small interfering RNA (siRNA) knockdown of ERK1/2 in macrophages results in an upregulation of PGN-induced iNOS expression, mainly through a mechanism that involves an enhanced activation of NF-κB versus macrophages treated with PGN alone.
MATERIALS AND METHODS
Mice.
Inbred strains of BALB/c mice of either sex at 8 to 10 weeks of age were used for obtaining peritoneal macrophages.
Cell culture and reagents.
Macrophage monolayers were generated as described previously (45). Macrophages were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, penicillin (100 U/ml), streptomycin (100 U/ml), and gentamicin (20 μg/ml) in a CO2 incubator (5% CO2). RPMI 1640 medium, PGN (Staphylococcus aureus, catalog no. 77140), TRI-reagent, lipopolysaccharide (LPS), gamma interferon, and most of the other reagents were obtained from Sigma-Aldrich Chemicals, St. Louis, MO. Fetal calf serum was purchased from Gibco, Invitrogen, NY. MEK1 inhibitor, PD98059 (catalog no. 513000); p38mapk inhibitor, SB202190 (catalog no. 559388); JNK inhibitor, SP600125 (catalog no. 420119) were purchased from Calbiochem, La Jolla, CA. Anti-NOS2 (sc-7271), anti-p-JNK (sc-12882), anti-p-ERK (sc-16982), anti-p-p38 (sc-17852), anti-p38 (sc-7972), anti-ERK2 (sc-153), anti-JNK2 (sc-7345), anti-actin (sc-1615), and all secondary antibodies and ECL reagent were purchased from Santa Cruz Biotechnology, Santa Cruz, CA. The one-step real-time reverse transcription-PCR (RT-PCR) kit was from Qiagen, Hilden, Germany, and the mouse primers for real-time PCR were purchased from Eurofins MWG Operon, Ebersberg, Germany. All of the reagents were endotoxin-free as determined by the Limulus lysate assay (sensitivity limit, 0.1 ng/ml).
Inhibitor studies.
Macrophage monolayers were cultured in serum-free medium in 24-well culture plates (106 cells/well) with SP600125 (10, 30, and 50 μM), PD98059 (10, 30, and 50 μM), or SB202190 (10 μM) or a vehicle control (dimethyl sulfoxide at a concentration of 0.1%) for 30 min. The monolayers were then washed twice with warm incomplete medium, followed by further culture in complete medium in the presence or absence of PGN in a CO2 incubator for 6 or 18 h. All inhibitors were used at the generally recommended concentrations (14). After inhibitor treatment, cell death was checked by using an MTT [3(4, 5)-dimethylthiazol-2, 5-diphenyltetrazolium bromide] assay (36).
Gene knockdown studies.
Macrophage monolayers were cultured overnight in complete medium and transfected with a JNK1/JNK2 or ERK1/ERK2 siRNA cocktail or with scrambled siRNA (Ambion, Austin, TX) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). siRNA Lipofectamine 2000 complexes were prepared according to the manufacturer's instructions using 25 μM siRNA and 1 μl of Lipofectamine 2000. The final siRNA concentration was 20 nM. Transfection was performed for 5 h. After 5 h, medium containing siRNA-Lipofectamine 2000 complex was removed and replaced by RPMI supplemented with 10% fetal calf serum. The medium was changed 24 h after transfection and, after an additional 48 h, the macrophages were stimulated with PGN for 30 min and 18 h, and the supernatant and cell lysate were used for NO detection and immunoblotting, respectively.
Real-time RT-PCR analysis.
Total RNA was isolated from the macrophages by using TRI-reagent according to supplier's instructions. Real-time RT-PCR was done by using a single-step real-time RT-PCR kit in Bio-Rad iQ5 real-time PCR machine (Bio-Rad Laboratories, Hercules, CA) according to the SYBR green detection protocol. The following gene specific primers were used to amplify genes: GAPDH, forward (TGA CCA CAG TCC ATG CCA TC) and reverse (GAC GGA CAC ATT GGG GGT AG); and iNOS, forward (ACA TCG ACC CGT CCA CAG TAT) and reverse (CAG AGG GGT AGG CTT GTC TC). The primers were designed using Beacon Designer software.
RT was performed for 30 min at 50°C, and then reverse transcriptase was inactivated at 95°C for 15 min. Amplification was performed with cycling conditions of 94°C for 15 s, 56°C for 30 s, and 72°C for 30 s for 35 cycles. After the amplification protocol was complete, the PCR product was subjected to melting-curve analysis using Bio-Rad iQ5 software. Comparative cycle threshold method (the ΔΔCT method) was used for relative quantitation of the gene expression (30). The CT values were calculated by software automatically after completion of a run. The ΔΔCT was calculated using the formula: ΔΔCT = (CT GOI − CT HG)experimental sample − (CT GOI − CT HG)control sample, where GOI is the gene of interest, and HG is the housekeeping gene.
The fold increase in gene expression was determined by using the formula: fold expression = 2−ΔΔCT.
Measurement of nitrite production.
The concentration of nitrite, the stable end product of NO, was determined on the basis of the Griess reaction (17). Nitrite content was quantified by extrapolation from the sodium nitrite standard curve in each experiment.
Electrophoretic mobility shift assay (EMSA).
Biotin-end-labeled single-stranded oligonucleotide probes (Metabion, Martinsried, Germany) were annealed by heating equimolar amounts of complementary strands to 95°C for 5 min in annealing buffer (10 mM Tris-HCl [pH 7.5], 0.1 M NaCl, 1 mM EDTA) and slowly cooling the reaction mixture to room temperature. Macrophages were pretreated with or without SP600125 or PD98059 for 30 min and then treated with PGN (10 μg/ml) for 1 h. Nuclear proteins were isolated by using an NE-PER nuclear and cytoplasmic extraction reagent kit (Thermo Scientific, Rockford, IL). For the binding reaction and detection, LightShift Chemiluminescent EMSA and a Chemiluminescent nucleic acid detection module (both from Thermo Scientific), respectively, were used. Briefly, 40 fmol of labeled probe was incubated with nuclear extract (∼5 μg of protein) in a total volume of 20 μl for 20 min at room temperature with 1× binding buffer. To prevent nonspecific binding of nuclear proteins, 100 ng of poly(dI-dC) was added, and the specificity of retarded bands was confirmed by including a 100× excess of unlabeled oligonucleotides. Protein-DNA complexes were separated from unbound DNA by using 6% (wt/vol) native PAGE and run in 0.5× Tris-borate-EDTA. The sequences of the probes were as follows: NF-κB probe, 5′-biotin-AGTTGAGGGGACTTTCCCAGGC-3′; and AP-1 probe, 5′-biotin-CGCTTGATGACTCAGCCGGAA-3′.
Densitometric analysis was carried out using GeneTools software from Syngene, a Division of Synoptics, Ltd., United Kingdom. The densities of each band are represented as raw volume.
Western blot analysis.
The macrophage monolayers were washed with ice-cold phosphate-buffered saline containing 1 mM Na3VO4, lysed in 15 μl/cm2 of lysis buffer (20 mM Tris-HCl [pH 8], 137 mM NaCl, 10% glycerol [vol/vol], 1% Triton X-100 [vol/vol], 1 mM Na3VO4, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 20 mM leupeptin, 0.15 U of aprotinin/ml) for 20 min at 4°C. The lysates were centrifuged at 15,000 × g for 20 min, and the supernatants were separated on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gels. The separated proteins (40 μg/lane) were transferred to a nitrocellulose membrane (1 h at 100 V) by using a Bio-Rad Mini Transblotter, and each membrane was blocked with 5% serum for 2 h at room temperature, followed by incubation with primary antibody for 1 h at room temperature and then with horseradish peroxidase-labeled secondary antibody for 1 h at room temperature. The blot was developed using ECL reagent. To monitor equal loading of protein, Western blot analysis with antibody directed against actin was performed.
Statistical analysis.
Results are expressed as means ± the standard deviations (SD) of triplicate determinations. The statistical significance was determined by using a Student t test (P < 0.05).
RESULTS
MAPKs are activated by PGN.
We have previously reported that activation of macrophages with PGN from S. aureus leads to iNOS expression at both the mRNA and the catalytic levels (5). To explore that the PGN-induced iNOS expression is mediated through MAPK activation, the role of three MAPKs in PGN-induced signal transduction was investigated by detecting their dually phosphorylated (Thr/Tyr) forms by Western blotting. The phosphorylation of ERK and p38mapk were detected early at 5 min and reached a maximum at 30 min of PGN (10 μg/ml) treatment, whereas JNK phosphorylation started at 15 min and reached a peak at 30 min. p38mapk and JNK returned to near basal levels at 2 h after adding PGN (Fig. 1). In contrast, the phosphorylation of ERK was sustained, although slight decreases in phospho-ERK levels were observed after 2 h with PGN stimulation (Fig. 1). These results demonstrate that PGN induces early phosphorylation of ERK and p38, whereas JNK phosphorylation occurs later.
Fig. 1.
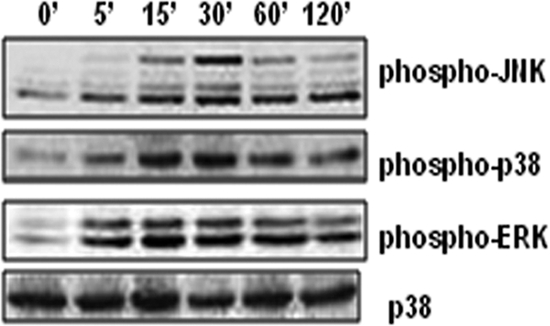
Activation (phosphorylation) of JNK, ERK, and p38 MAPKs on PGN treatment. Mouse peritoneal macrophages were treated with PGN (10 μg/ml) for 0, 5, 15, 30, 60, or 120 min. Macrophages were lysed, and the lysates were analyzed by immunoblotting with antibodies to phospho-JNK, phospho-ERK, and phospho-p38. The total p38 in each sample was used to ensure equal protein loading.
Activation of JNK MAPK is required for NO production and iNOS expression in response to PGN.
To further understand the mechanism of PGN-induced iNOS and NO expression, the role of JNK was investigated by using a selective inhibitor, SP600125 (4). Macrophages were pretreated with a different concentration of JNK inhibitor, SP600125, for 30 min and then incubated with PGN for 30 min, 6 h, and 18 h. After 30 min as shown in Fig. 2A, SP600125 dose dependently inhibited the PGN induced phosphorylation of c-Jun. SP600125 strongly inhibited PGN induced NO accumulation in a concentration-dependent fashion (Fig. 2B). At 10 μM SP600125, there was ca. 20% inhibition of NO accumulation, and with 30 and 50 μM SP600125 there was 58 and 89% inhibition, respectively, in macrophages treated with PGN for 18 h. Polymyxin B (10 μg/ml) markedly attenuated LPS (10 μg/ml) but not PGN-induced iNOS/NO production (see Fig. S1 in the supplemental material). It was further observed that there is an 18-fold increase in the expression of iNOS mRNA in macrophages activated with PGN for 6 h. However, macrophages pretreated with 10, 30, or 50 μM SP600125 and then incubated for 6 h with PGN showed 12.6-, 4.4-, and 2.78-fold-increased expression of iNOS mRNA, respectively (Fig. 2C). To determine whether this inhibition also occurred at the level of iNOS protein expression, macrophages were similarly pretreated with SP600125 before PGN treatment for 18 h, and whole-cell lysates were separated by SDS-PAGE and immunoblotted with iNOS polyclonal antibody. Significant inhibition in the expression of iNOS was observed at 30 μM SP600125, which was completely inhibited at 50 μM (Fig. 2D). No inhibition in PGN-induced NO production was observed in macrophages pretreated with different concentrations of the p38mapk inhibitor SB202190 (10, 30, or 50 μM) (see Fig. S2 in the supplemental material).
Fig. 2.

Inhibition of JNK MAPK by SP600125 inhibits PGN-induced iNOS expression and NO production. Mouse peritoneal macrophages were cultured with or without various concentrations of SP600125 (10, 30, or 50 μM) for 30 min, washed, and then treated with PGN (10 μg/ml) for 30 min, 6 h, or 18 h. (A) After 30 min, the phosphorylation of c-Jun was determined by immunoblotting in cell lysates with an antibody against phospho c-Jun or c-Jun. (B) After 18 h, NO production in culture supernatants of mouse peritoneal macrophages was evaluated by using the Griess reagent assay. Each bar represents the standard error of three independent experiments. *, P < 0.05 compared to PGN. (C) After 6 h, RNA was isolated and checked for iNOS transcripts by real-time RT-PCR. Each bar represents the fold expression relative to untreated macrophages. The data represent the means ± the SD of three independent experiments. #, P < 0.01 compared to control macrophages. (D) After 18 h, macrophages were lysed, and lysates were immunoblotted with a polyclonal anti-iNOS antibody. The same blot was reprobed with anti-actin antibody to demonstrate equal protein loading.
Effects of JNK1 and JNK2 siRNA on PGN-induced JNK activation and iNOS-NO expression in macrophages.
To further confirm the role of JNK MAPK in PGN-induced iNOS and NO expression in macrophages, JNK1 and JNK2 siRNA knocked-down macrophages were used. The molecular masses 46 and 54 kDa represent JNK1 and JNK2 isoforms (25). JNK1 and JNK2 siRNAs attenuated the expression of JNK1 (46-kDa) and JNK2 (54-kDa) protein in PGN-treated macrophages (Fig. 3A). Significant inhibition of PGN induced JNK and c-Jun phosphorylation were observed in JNK1/2 knocked-down macrophages compared to scramble siRNA (Scr) (Fig. 3A and B). PGN-induced iNOS and NO expression were further investigated in JNK1/2 knocked-down macrophages. Figure 3C and D shows that PGN-induced NO production and iNOS expression are significantly inhibited in JNK1/2 knocked-down macrophages compared to scrambled siRNA.
Fig. 3.

Knockdown of JNK1/2 by siRNA inhibits PGN-induced iNOS expression and NO production. Mouse peritoneal macrophages were transfected with JNK1 and JNK2 siRNA (JNK) or scrambled siRNA (Scr). (A) After 36 h of transfection, Macrophages were stimulated with PGN (10 μg/ml) for 30 min, and cell lysates were immunoblotted with antibodies against phospho-JNK (p-p46 and p-p54) and JNK2. The lower panel shows the densitometry analysis of phospho-JNK bands. (B) Macrophages were stimulated with PGN for 30 min, and cell lysates were immunoblotted with anti-phospho c-Jun and c-Jun antibody. (C) After 36 h of transfection, macrophages were stimulated with PGN (10 μg/ml) for 18 h, and the NO in culture supernatants was evaluated by using a Griess reagent assay. Each bar represents the standard error of three independent experiments. *, P < 0.05 compared to scramble siRNA. (D) Macrophages were similarly stimulated with PGN for 18 h, followed by immunoblotting of cell lysates with anti-iNOS polyclonal antibody. The same blot was reprobed with anti-actin antibody to demonstrate equal protein loading.
PGN-induced expression of iNOS and NO is inhibited by the ERK pathway.
To determine the role of the ERK pathway in the regulation of iNOS and NO expression, PD98059, a pharmacologic inhibitor of MEK1, the upstream kinase of p42 and p44 ERK, was used (18). Figure 4A shows that PD98059 dose dependently suppressed the PGN induced phosphorylation of ERK1/2. Macrophages were pretreated with different doses of PD98059 (10, 30, and 50 μM) for 30 min and then further incubated with PGN for 18 h. As demonstrated in Fig. 4B, Inhibition of the ERK pathway with PD98059 (at 30 or 50 μM) further augmented PGN-induced NO production in a dose-dependent manner. To determine whether PD98059 was inherently capable of inducing NO production, macrophages were pretreated with 50 μM PD98059 and then further activated with LPS (10 μg/ml) or IFN-γ (100 U/ml). No increase in NO production was observed in macrophages treated with IFN-γ, while LPS treatment resulted in enhanced iNOS expression and NO production (see Fig. S3 in the supplemental material).
Fig. 4.

Inhibition of MEK1-ERK by PD98059 augmented iNOS expression and NO production induced by PGN. Mouse peritoneal macrophages were treated with or without various concentrations of PD98059 (10, 30, or 50 μM) for 30 min, washed, and then further treated with PGN (10 μg/ml) for 30 min, 6 h, or 18 h. (A) After 30 min, the phosphorylation of ERK was determined by immunoblotting cell lysates with antibody specific for a phospho-ERK (p-p42 and p-p44). The bottom panel shows an immunoblot with an antibody to ERK2. (B) After 18 h, the NO production in culture supernatants of mouse peritoneal macrophages was evaluated by using a Griess reagent assay. Each bar represents the standard error of three independent experiments. *, P < 0.05 compared to PGN. (C) After 6 h, the RNA was isolated and checked for iNOS transcripts by real-time RT-PCR. Each bar represents the fold expression relative to untreated macrophages. The data represent the means ± the SD of three independent experiments. #, P < 0.01 compared to control macrophages. (D) After 18 h, the macrophages were lysed, and cell lysates were immunoblotted with a polyclonal anti-iNOS antibody. The same blot was reprobed with anti-actin antibody to demonstrate equal protein loading.
It was further observed that macrophages treated with PGN showed significantly increased transcription and translation of iNOS at 6 and 18 h, respectively. Macrophages treated with PGN showed an 18-fold increase in the expression of iNOS mRNA. In contrast, macrophages pretreated for 30 min with 10, 30, or 50 μM PD98059 and then activated with PGN showed 16.33-, 24.45-, and 42.42-fold-increased expression of iNOS mRNA, respectively (Fig. 4C). As shown in Fig. 4D, a similar increase in iNOS protein expression was observed with ERK pathway inhibition compared to that seen with PGN stimulation alone. Significant augmentations of iNOS protein was observed with 30 and 50 μM PD98059. These results demonstrate that the activation of ERK pathway negatively regulates PGN-induced iNOS expression and NO production. It was also observed in immunoblotting analyses that PD98059 does not inhibit PGN-induced phosphorylation of JNK and p38 (see Fig. S4 in the supplemental material).
Effects of ERK1 and ERK2 siRNA on PGN-induced ERK activation and iNOS-NO production in macrophages.
To further confirm the negative regulatory role of ERK in iNOS expression and NO production, ERK1 and ERK2 knocked-down macrophages were used. The molecular masses of 44 and 42 kDa represent the ERK1 and ERK2 isoforms (6). The PGN-induced expression of both ERK1/2 and phosphorylated ERK1/2 is attenuated in ERK1/2 knocked-down macrophages compared to scrambled siRNA (Fig. 5A). As shown in Fig. 5B and C, ERK1/2 knocked-down macrophages showed enhanced PGN-induced NO production and iNOS protein expression compared to scramble siRNA.
Fig. 5.

Knockdown of ERK1/2 augmented PGN-induced iNOS expression and NO production. Macrophages were transfected with ERK1 and ERK2 siRNA (ERK) or scrambled siRNA (Scr). After 36 h of transfection, macrophages were stimulated with PGN (10 μg/ml) for 30 min. (A) Phosphorylated ERK and total ERK protein were detected by immunoblotting with antibody specific for phospho-ERK (p-p42 and p-p44) or ERK2. The lower panel shows the densitometry analysis of phospho-ERK bands. (B) After 36 h of transfection, macrophages were stimulated with PGN (10 μg/ml) for 18 h, and the NO in the culture supernatant was evaluated by Griess reagent assay. Each bar represents the standard error of three independent experiments. *, P < 0.05 compared to scramble siRNA. (C) After 18 h, PGN-treated macrophages were lysed, and the cell lysates were immunoblotted with anti-iNOS polyclonal antibody. The same blot was reprobed with anti-actin antibody to demonstrate equal protein loading.
Role of JNK and ERK MAPKs in PGN-induced NF-κB and AP-1 activation.
The iNOS gene contains DNA-protein interaction sites for the transcription factors AP-1 and NF-κB (34). To determine whether JNK and ERK MAPKs are involved in the PGN-mediated activation of these transcription factors in mouse peritoneal macrophages, EMSAs were performed. Nuclear extracts were isolated from macrophages pretreated with PD98059 (50 μM) and SP600125 (50 μM) for 30 min and further incubated with PGN for 1 h. Using NF-κB and AP-1 consensus sequence oligonucleotides, we observed that PGN induces increased NF-κB and AP-1 binding activity. Pretreatment with the JNK inhibitor SP600125 (50 μM) reduced NF-κB activation (compare lanes 2 and 3 in Fig. 6 A) and AP-1 activation (compare lane 2 and 3 in Fig. 6B) at 1 h, whereas PD98059 (50 μM) further enhanced NF-κB activation (compare lanes 2 and 5 in Fig. 6A) but had no effect on the AP-1 DNA-binding ability (compare lanes 2 and 4 in Fig. 6B). These experiments thus demonstrate that both NF-κB and AP-1 transcription factors are activated in PGN-treated murine macrophages and that SP600125 suppresses NF-κB and AP-1 activation. In contrast, PD98059 enhances PGN-induced NF-κB activity.
Fig. 6.

Inhibition of JNK MAPK abolished the activation of NF-κB and AP-1, whereas ERK pathway inhibition enhanced PGN-induced activation of NF-κB. An EMSA was performed with nuclear proteins isolated from murine macrophages pretreated for 30 min with or without specific inhibitors of MAPKs ERK (PD98059; 50 μM) and JNK (SP600125; 50 μM) and then further stimulated with PGN for 1 h. (A) Biotin-labeled NF-κB oligonucleotides probes were incubated in the presence of nuclear extract. (B) Biotin-labeled AP-1 oligonucleotides probes were incubated in the presence of nuclear extract. The data are representative of three independent experiments. The lower panel shows the densitometry analysis of the EMSA bands.
DISCUSSION
The signaling mechanisms and trans-acting factors that mediate the induction of iNOS expression during stimulation with PGN have not been very well documented. We have previously reported the role of protein tyrosine kinase, PKCδ and NF-κB in PGN (S. aureus)-induced iNOS expression and NO production in macrophages (5). In the present study, the role of MAPKs and transactivating molecules (NF-κB and AP-1) in PGN-induced iNOS expression and NO production have been investigated. This is probably the first time that evidence for the involvement of the JNK MAPK pathway in iNOS and NO expression in PGN-treated macrophages, independent of p38mapk, has been reported, whereas the ERK pathway plays a negative regulatory role. Utilization of particular signaling pathways in regulating cellular function or inducing gene expression appears to be dependent on, among other factors, the type of stimulus and cell examined. In this regard, augmenting, inhibitory, and neutral roles for the MAPKs have been reported in signaling gene expression, including that for iNOS. For example, JNK MAPK was shown to be necessary for iNOS induction by interleukin-1β (IL-1β) in rat glomerular mesangial cells (22), by tumor necrosis factor alpha (TNF-α) in macrophages (10), and by IFN-γ plus TNF-α in mouse macrophages (11). In contrast, JNK has been shown to play a neutral role in iNOS induction by TNF-α plus IL-1α in astrocytes (13) and by IFN-γ plus LPS in glioma cells (40).
We report here a simultaneous activation of ERK, p38, and JNK MAPKs by PGN. The PGN-induced activation of ERK can be observed as early as 5 min of treatment, with sustained, though slightly decreased, activation for up to 2 h thereafter. In contrast, p38 or JNK MAPK rapidly attenuated after the initial peak at 30 min. Inhibition of the ERK or JNK signaling pathway by specific inhibitors or siRNAs had distinct effects on PGN-induced iNOS expression and NO production in mouse peritoneal macrophages. The present study showed that SP600125 dose dependently inhibited PGN-induced c-Jun phosphorylation, iNOS expression, and NO production in mouse peritoneal macrophages. This observation was confirmed by using JNK1/2 siRNA knocked-down macrophages in which PGN-induced iNOS expression and NO production was significantly inhibited. These results indicate that a positive signaling pathway for iNOS expression induced by PGN in mouse macrophages is mediated through JNK MAPK. These data are in concord with the findings of others who have reported that the treatment of Sertoli epithelial cells with SP600125 completely inhibited IL-1β-induced iNOS expression and NO production in a dose-dependent manner (26). In addition, JNK MAPK also appears to play a positive regulatory role in transducing the LPS-mediated induction of iNOS in murine macrophages and murine skeletal muscle cells (21, 47). To better understand the PGN signaling, MAPK involvement in activation of NF-κB and AP-1 transcription factors that regulate the expression of the iNOS gene was investigated. JNK and ERK MAPKs are able to independently and synergistically activate or increase the expression of a number of transcription factors, including c-Fos, c-Jun, NF-κB, activating transcription factor 2, the Ets family, serum response factor, and CREB (41). Our data on PGN-induced activation of NF-κB and AP-1 is in accordance with the previous reports (24, 50). Sinke et al. and Chen et al. (12, 44) reported that the JNK inhibitor SP600125 inhibited induced iNOS expression, with concomitant inhibition in NF-κB and AP-1 activation in astrocytes and murine macrophages, respectively. The observations of these researchers are consistent with our data here suggesting that JNK MAPK plays a key role in the PGN-stimulated induction of iNOS in macrophages through activation of NF-κB and AP-1 binding to the iNOS promoter.
Several studies have shown that different MAPKs may have opposing effects on gene expression. More specifically, activation of the p38 and JNK MAPKs with simultaneous inhibition of the ERK MAPK was found to be critical for apoptosis in PC-12 pheochromocytoma cells stimulated with nerve growth factor (53). In another study, the p38 MAPK was shown to enhance the accumulation of IL-12 mRNA, whereas ERK MAPK inhibited IL-12 transcription in murine macrophages stimulated with LPS (20). We observed here that inhibition of MEK1-ERK pathway with PD98059 suppresses ERK1/2 activation and increases PGN-induced iNOS expression and NO production in mouse peritoneal macrophages. PD98059 also enhanced LPS-induced NO production, whereas it does not have any effect on IFN-γ-induced NO production. To reinforce these observations, ERK1/2 knocked-down macrophages was used. PGN-induced iNOS expression and NO production was significantly enhanced in knocked-down macrophages. LPS induces enhanced NO production in murine BV-2 microglial cells compared to RAW 264.7 cells but is unable to activate ERK (52). Aga et al. reported that the cotreatment of LPS with nucleotide receptor agonist enhances NO production and attenuates ERK activation (1). These reports support our hypothesis that ERK activation is involved in the negative regulation of iNOS/NO production. The constitutively active MEK→ERK pathway is known to negatively regulate NF-κB-dependent gene expression (9). It was also observed that inhibition of the ERK pathway increased the transcriptional activity of NF-κB by ∼2-fold (Fig. 6A, lanes 2 and 5). It is possible that this enhanced activation of NF-κB might contribute to favor the signaling by autocrine factors released in response to PGN challenge (TNF-α and several proinflammatory interleukins), and it might lead to an augmentation in iNOS expression. Inhibition of the MEK/ERK pathway-enhanced cisplatin-induced NF-κB activation via a pathway different from conventional IKK/IκBα/NF-κB signaling has also been reported (56). The data presented here demonstrate that the ERK pathway regulates PGN-induced iNOS expression negatively through NF-κB.
These observations show that the PGN-induced JNK MAPK is required for the enhanced NF-κB and AP-1 activation observed in mouse peritoneal macrophages. NF-κB and AP-1 activation then relates to increased expression of the iNOS gene and the subsequent generation of NO. Finally, the activation of ERK pathway and the suppression of NF-κB activity and NO production may represent the microbe's strategies to avoid or delay activation of the immune response, providing a window of opportunity for the bacteria to establish infection. This might contribute to our understanding of the mechanism(s) of action of the anti-inflammatory cytokines (IL-13 and IL-4) that activate ERK MAPK in the course of their intracellular signaling (37). These findings suggest that therapeutic inhibition of ERK activation during bacterial infection may promote NO responses and inhibit microbe replication.
Supplementary Material
ACKNOWLEDGMENTS
This study has been supported by a research grant to Ajit Sodhi from the Department of Biotechnology, Government of India, New Delhi, India. K.H.B. is an SRF of the Council of Industrial and Scientific Research, Government of India, New Delhi, India, and Rituparna Chakraborty is a JRF of the University Grant Commission, New Delhi, India.
Footnotes
Supplemental material for this article may be found at http://cvi.asm.org/.
Published ahead of print on 30 March 2011.
REFERENCES
- 1. Aga M., et al. 2004. Evidence for nucleotide receptor modulation of cross talk between MAP kinase and NF-κB signaling pathways in murine RAW 264.7 macrophages. Am. J. Physiol. Cell Physiol. 286:C923–C930 [DOI] [PubMed] [Google Scholar]
- 2. Auphan N., DiDonato J. A., Rosette C., Helmberg A., Karin M. 1995. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science 270:286–289 [DOI] [PubMed] [Google Scholar]
- 3. Barnes P. J., Karin M. 1997. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336:1066–1071 [DOI] [PubMed] [Google Scholar]
- 4. Bennett B. L., et al. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U. S. A. 98:13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhatt K. H., Pandey R. K., Dahiya Y., Sodhi A. 2010. Protein kinase Cδ and protein tyrosine kinase regulate peptidoglycan-induced nuclear factor-κB activation and inducible nitric oxide synthase expression in mouse peritoneal macrophages in vitro. Mol. Immunol. 47:861–870 [DOI] [PubMed] [Google Scholar]
- 6. Boulton T. G., et al. 1991. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65:663–675 [DOI] [PubMed] [Google Scholar]
- 7. Brennan F. M., Feldmann M. 1996. Cytokines in autoimmunity. Curr. Opin. Immunol. 8:872–877 [DOI] [PubMed] [Google Scholar]
- 8. Carl V. S., Brown-Steinke K., Nicklin M. J., Smith M. F., Jr 2002. Toll-like receptor 2 and 4 (TLR2 and TLR4) agonists differentially regulate secretory interleukin-1 receptor antagonist gene expression in macrophages. J. Biol. Chem. 277:17448–17456 [DOI] [PubMed] [Google Scholar]
- 9. Carter A. B., Hunninghake G. W. 2000. A constitutive active MEK→ERK pathway negatively regulates NF-κB-dependent gene expression by modulating TATA-binding protein phosphorylation. J. Biol. Chem. 275:27858–27864 [DOI] [PubMed] [Google Scholar]
- 10. Chan E. D., Riches D. W. H. 1998. Potential role of the JNK/SAPK signal transduction pathway in the induction of iNOS by TNFα. Biochem. Biophys. Res. Commun. 253:790–796 [DOI] [PubMed] [Google Scholar]
- 11. Chan E. D., et al. 1999. Evaluation of the role of mitogen-activated protein kinases in the expression of inducible nitric oxide synthase by IFN-γ and TNFα in mouse macrophages. J. Immunol. 162:415–422 [PubMed] [Google Scholar]
- 12. Chen C. C., Tsai P. C., Wei B. L., Chiou W. F. 2008. 8-Prenylkaempferol suppresses inducible nitric oxide synthase expression through interfering with JNK-mediated AP-1 pathway in murine macrophages. Eur. J. Pharmacol. 590:430–436 [DOI] [PubMed] [Google Scholar]
- 13. Da Silva J., Pierrat B., Mary J. L., Lesslauer W. 1997. Blockade of p38 mitogen-activated protein kinase pathway inhibits inducible nitric-oxide synthase expression in mouse astrocytes. J. Biol. Chem. 272:28373–28380 [DOI] [PubMed] [Google Scholar]
- 14. Davies S. P., Reddy H., Caivano M., Cohen P. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351:95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Derijard B., et al. 1995. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267:682–685 [DOI] [PubMed] [Google Scholar]
- 16. Diaz-Guerra M. J., Velasco M. M., Martin-Sanz P., Bosca L. 1996. Evidence for common mechanisms in the transcriptional control of type II nitric oxide synthase in isolated hepatocytes: requirement of NF-κB activation after stimulation with bacterial cell wall products and phorbol esters. J. Biol. Chem. 271:30114–30120 [DOI] [PubMed] [Google Scholar]
- 17. Ding A. H., Nathan C. F., Stuehr D. J. 1988. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages: comparison of activating cytokines and evidence for independent production. J. Immunol. 141:2407–2412 [PubMed] [Google Scholar]
- 18. Dudley D. T., Pang L., Decker S. J., Bridges A. J., Saltiel A. R. 1995. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U. S. A. 92:7686–7689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dziarski R., Jin Y. P., Gupta D. 1996. Differential activation of extracellular signal-regulated kinase (ERK) 1, ERK2, p38, and c-Jun NH2 terminal kinase mitogen-activated protein kinases by bacterial peptidoglycan. J. Infect. Dis. 174:777–785 [DOI] [PubMed] [Google Scholar]
- 20. Feng G. J., et al. 1999. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. J. Immunol. 163:6403–6412 [PubMed] [Google Scholar]
- 21. Frost R. A., Nystrom G. J., Lang H. C. 2004. Lipopolysaccharide stimulates nitric oxide synthase-2 expression in murine skeletal muscle and C2C12 myoblasts via Toll-like receptor-4 and c-Jun NH2-terminal kinase pathways. Am. J. Physiol. 287:C1605–C1615 [DOI] [PubMed] [Google Scholar]
- 22. Guan Z., Buckman S. Y., Springer L. D., Morrison A. R. 1999. Both p38α(MAPK) and JNK/SAPK pathways are important for induction of nitric-oxide synthase by interleukin-1β in rat glomerular mesangial cells. J. Biol. Chem. 273:12901–12908 [DOI] [PubMed] [Google Scholar]
- 23. Gupta D., Kirkland T. N., Viriyakosol S., Dziarski R. 1996. CD14 is a cell-activating receptor for bacterial peptidoglycan. J. Biol. Chem. 271:23310–23316 [DOI] [PubMed] [Google Scholar]
- 24. Gupta D., Wang Q., Vinson C., Dziarski R. 1999. Bacterial peptidoglycan induces CD14-dependent activation of transcription factors CREB/ATF and AP-1. J. Biol. Chem. 274:14012–14020 [DOI] [PubMed] [Google Scholar]
- 25. Ip Y. T., Davis R. J. 1998. Signal transduction by the c-Jun N-terminal kinase (JNK): from inflammation to development. Curr. Opin. Cell Biol. 10:205–219 [DOI] [PubMed] [Google Scholar]
- 26. Ishikawa T., Morris P. L. 2006. Interleukin-1β signals through a c-Jun N-terminal kinase-dependent inducible nitric oxide synthase and nitric oxide production pathway in Sertoli epithelial cells. J. Androl. 147:5424–5430 [DOI] [PubMed] [Google Scholar]
- 27. Jiang C., Ting A. T., Seed B. 1998. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 391:82–86 [DOI] [PubMed] [Google Scholar]
- 28. Laskin D. L., Pendino K. J. 1995. Macrophages and inflammatory mediators in tissue injury. Annu. Rev. Pharmacol. Toxicol. 35:655–677 [DOI] [PubMed] [Google Scholar]
- 29. Lin A., et al. 1995. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science 268:286–290 [DOI] [PubMed] [Google Scholar]
- 30. Livak K. J., Schmittgen T. D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 31. Lopez-Collazo E., Hortelano S., Rojas A., Bosca L. 1998. Triggering of peritoneal macrophages with IFN-α/β attenuates the expression of inducible nitric oxide synthase through a decrease in NF-κB activation. J. Immunol. 160:2889–2895 [PubMed] [Google Scholar]
- 32. Lowenstein C. J., et al. 1993. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon-γ and lipopolysaccharide. Proc. Natl. Acad. Sci. U. S. A. 90:9730–9734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MacMicking J., Xie Q. W., Nathan C. 1997. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15:323–350 [DOI] [PubMed] [Google Scholar]
- 34. Marks-Konczalik J., Chu S. C., Moss J. 1998. Cytokine-mediated transcriptional induction of the human inducible nitric oxide synthase gene requires both activator protein 1 and nuclear factor κB-binding sites. J. Biol. Chem. 273:22201–22208 [DOI] [PubMed] [Google Scholar]
- 35. Martin G. S., Mannino D. M., Eaton S., Moss M. 2003. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 348:1546–1554 [DOI] [PubMed] [Google Scholar]
- 36. Mizel S. B. 1982. Interleukin 1 and T cell activation. Immunol. Rev. 63:51–72 [DOI] [PubMed] [Google Scholar]
- 37. Moore P. E., Church T. L., Chism D. D., Panettieri R. A., Jr., Shore S. A. 2002. IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: a role for ERK. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L847–L853 [DOI] [PubMed] [Google Scholar]
- 38. Nathan C. 1992. Nitric oxide as a secretory product of mammalian cells. FASEB J. 6:3051–3064 [PubMed] [Google Scholar]
- 39. Nathan C. 1997. Inducible nitric oxide synthase: what difference does it make? J. Clin. Invest. 100:2417–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishiya T., et al. 1997. Activation of Stat1 and subsequent transcription of inducible nitric oxide synthase gene in C6 glioma cells is (sic) independent of interferon-γ-induced MAPK activation that is mediated by p21ras. FEBS Lett. 408:33–38 [DOI] [PubMed] [Google Scholar]
- 41. Rose D. M., et al. 1997. Fcg receptor cross-linking activates p42, p38, and JNK/SAPK mitogen-activated protein kinases in murine macrophages. J. Immunol. 158:3433–3438 [PubMed] [Google Scholar]
- 42. Sanchez I., et al. 1994. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature 372:794–798 [DOI] [PubMed] [Google Scholar]
- 43. Schwandner R., Dziarski R., Wesche H., Rothe M., Kirschning C. J. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J. Biol. Chem. 274:17406–17409 [DOI] [PubMed] [Google Scholar]
- 44. Sinke A. P., et al. 2008. NF-κB in the mechanism of ammonia-induced astrocyte swelling in culture. J. Neurochem. 106:2302–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sodhi A., Singh S. M. 1986. Release of cytolytic factor(s) by murine macrophages in vitro on treatment with cisplatin. Int. J. Immunopharmacol. 8:701–707 [DOI] [PubMed] [Google Scholar]
- 46. Spink J., Cohen J., Evans T. J. 1995. The cytokine responsive vascular smooth muscle cell enhancer of inducible nitric oxide synthase: activation by nuclear factor-κB. J. Biol. Chem. 270:29541–29547 [DOI] [PubMed] [Google Scholar]
- 47. Uto T., Fujii M., Hou D. X. 2005. 6-(Methylsulfinyl)hexyl isothiocyanate suppresses inducible nitric oxide synthase expression through the inhibition of Janus kinase 2-mediated JNK pathway in lipopolysaccharide-activated murine macrophages. Biochem. Pharmacol. 70:1211–1221 [DOI] [PubMed] [Google Scholar]
- 48. Wang J. E., et al. 2000. Peptidoglycan and lipoteichoic acid from Staphylococcus aureus induce tumor necrosis factor alpha, interleukin 6 (IL-6), and IL-10 production in both T cells and monocytes in a human whole blood model. Infect. Immun. 68:3965–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang J. E., et al. 2004. Peptidoglycan of Staphylococcus aureus causes inflammation and organ injury in the rat. Crit. Care Med. 32:546–552 [DOI] [PubMed] [Google Scholar]
- 50. Wang Q., Dziarski R., Kirschning C. J., Muzio M., Gupta D. 2001. Micrococci and peptidoglycan activate TLR2→MyD88→IRAK→TRAF→NIK→IKK→NF-κB signal transduction pathway that induces transcription of interleukin-8. Infect. Immun. 69:2270–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Z.-M., Liu C., Dziarski R. 2000. Chemokines are the main pro-inflammatory mediators in human monocytes activated by Staphylococcus aureus, peptidoglycan, and endotoxin. J. Biol. Chem. 275:20260–20267 [DOI] [PubMed] [Google Scholar]
- 52. Watters J. J., et al. 2002. A differential role for the mitogen-activated protein kinases in lipopolysaccharide signaling: the MEK/ERK pathway is not essential for nitric oxide and interleukin 1β production. J. Biol. Chem. 277:9077–9087 [DOI] [PubMed] [Google Scholar]
- 53. Xia Z., Dickens M., Raingeaud J., Davis R. J., Greenberg M. E. 1995. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326–1331 [DOI] [PubMed] [Google Scholar]
- 54. Xie Q. W., Whisnant R., Nathan C. 1993. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon-γ and bacterial lipopolysaccharide. J. Exp. Med. 177:1779–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xie Q. W., Kashiwabar Y., Nathan C. 1994. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J. Biol. Chem. 269:4705–4708 [PubMed] [Google Scholar]
- 56. Yeh P. Y., Yeh K. H., Chuang S. E., Song Y. C., Cheng A. L. 2004. Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-κB activation by protein phosphatase 4-mediated NF-κB p65 Thr dephosphorylation. J. Biol. Chem. 279:26143–26148 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.