

Abstract
Arsenic inhibits DNA repair and enhances the genotoxicity of DNA-damaging agents such as benzo[a]pyrene and ultraviolet radiation. Arsenic interaction with DNA repair proteins containing functional zinc finger motifs is one proposed mechanism to account for these observations. Here, we report that arsenite binds to both CCHC DNA-binding zinc fingers of the DNA repair protein PARP-1 (poly(ADP-ribose) polymerase-1). Furthermore, trivalent arsenite coordinated with all three cysteine residues as demonstrated by MS/MS. MALDI-TOF-MS analysis of peptides harboring site-directed substitutions of cysteine with histidine residues within the PARP-1 zinc finger revealed that arsenite bound to peptides containing three or four cysteine residues, but not to peptides with two cysteines, demonstrating arsenite binding selectivity. This finding was not unique to PARP-1; arsenite did not bind to a peptide representing the CCHH zinc finger of the DNA repair protein aprataxin, but did bind to an aprataxin peptide mutated to a CCHC zinc finger. To investigate the impact of arsenite on PARP-1 zinc finger function, we measured the zinc content and DNA-binding capacity of PARP-1 immunoprecipitated from arsenite-exposed cells. PARP-1 zinc content and DNA binding were decreased by 76 and 80%, respectively, compared with protein isolated from untreated cells. We observed comparable decreases in zinc content for XPA (xeroderma pigmentosum group A) protein (CCCC zinc finger), but not SP-1 (specificity protein-1) or aprataxin (CCHH zinc finger). These findings demonstrate that PARP-1 is a direct molecular target of arsenite and that arsenite interacts selectively with zinc finger motifs containing three or more cysteine residues.
Keywords: ADP-ribosylation, DNA Repair, Mass Spectrometry (MS), Spectroscopy, Zinc Finger, PARP-1, Arsenite
Introduction
Environmental or occupational exposures to arsenic are associated with numerous acute and chronic health effects in humans (1–3), including increased risk of skin, lung, liver, and urinary tract cancers (2, 4–7). Although long recognized as a complete carcinogen with tumor-promoting and genotoxic actions, it is becoming more widely appreciated that low and non-cytotoxic concentrations of arsenic greatly enhance the carcinogenicity of other DNA-damaging agents (8–11). We and others have found that submicromolar and low micromolar arsenite (As(III)) concentrations amplify DNA damage (8-hydroxyl-2′-deoxyguanine, strand break, cyclobutane pyrimidine dimers, adducts) caused by UV radiation, H2O2, and benzo[a]pyrene (8, 12–17). Although there is strong evidence that arsenic inhibits DNA repair activities, the underlying mechanisms remain largely unknown. Our results and those of others suggest that DNA repair proteins with functional zinc finger motifs may be direct molecular targets of arsenic.
Zinc finger DNA repair proteins reported to interact directly with arsenic include PARP-1 (poly(ADP-ribose) polymerase-1, XPA (xeroderma pigmentosum group A), and bacterial formamidopyrimidine-DNA glycosylase (14, 18–22), and there is mixed evidence for arsenic inhibition of other DNA repair proteins such as DNA ligase III (23–25). PARP-1 is of particular interest because it is the only validated DNA repair zinc finger molecular target with demonstrated inhibition in arsenic-exposed cells in situ (14, 19, 26), and PARP-1 is highly sensitive to arsenic inhibition (detectable at 100–200 nm) (14, 19). PARP-1 is the founding member of the PARP protein family that catalyzes polymerization of ADP-ribose (poly(ADP-ribose)) units from donor NAD+ molecules onto target proteins and is responsible for >85% of total poly(ADP-ribose) synthesis stimulated by DNA damage (reviewed in Refs. 27–31). Because PARP-1 has numerous substrates in the base excision repair and nucleotide excision repair DNA damage repair networks (29–31), PARP-1 inhibition may broadly influence DNA repair processes.
We reported previously that arsenite can bind to a synthetic peptide representing the first zinc finger of PARP-1 at the expense of zinc and that addition of Zn(II) abolishes arsenite enhancement of UV radiation-stimulated 8-hydroxyl-2′-deoxyguanine formation and restores PARP-1 activity in cellular systems (14). These findings indicated functional interaction between arsenite and zinc within a specific target protein and suggested questions regarding determinants of arsenic target selectivity. Cysteine content and vicinal cysteine residues have been postulated as key factors for arsenic interaction with proteins (32, 33). One study using synthetic peptides based on the estrogen receptor demonstrated that cysteine is essential for arsenite binding and that trivalent arsenite binding to peptides favors those with two or more cysteines (32). Another report demonstrated that 10 μm arsenite can strongly disrupt the oxidative folding of riboflavin-binding protein (a protein with 18 cysteines) even in the presence of 5 mm reduced glutathione (a monothiol compound), indicating poor interaction of arsenite with single thiol groups (34). Collectively, the evidence suggests selectivity in arsenite binding to different cysteine-containing proteins.
In this study, we demonstrate that both zinc finger motifs in the DNA-binding domain of PARP-1 bind arsenite and that arsenite displays preferential binding to zinc fingers with three or four cysteine residues. Little binding was detected with zinc fingers containing a C2H2 motif as detected by MALDI-MS analysis. MS/MS revealed that all three cysteine residues covalently bound arsenite. Furthermore, cell exposure to arsenite led to decreased zinc content in PARP-1 isolated from exposed cells with a concomitant decrease in DNA binding, indicating disruption of zinc finger function. We observed comparable decreases in zinc content in the C4 zinc finger DNA repair protein XPA isolated from arsenite-exposed cells, but no change in zinc content in the prototypical C2H2 zinc finger proteins SP-1 (specificity protein-1) and APTX (aprataxin) or in C2H2 zinc finger mutants of PARP-1 expressed in cells. These findings strongly suggest that PARP-1 is a direct molecular target of arsenite and that arsenite displays selectivity for interaction with zinc finger targets based on the number of cysteine residues.
EXPERIMENTAL PROCEDURES
Cell Culture and Exposures
The human keratinocyte cell line (HaCaT) was generously provided by Dr. Mitch Denning (Loyola University Medical Center, Maywood, IL). HaCaT cells were maintained as described previously (14). Normal neonatal human epidermal keratinocytes (HEKn) and DermaLife K culture medium were purchased from Lifeline Cell Technology (Oceanside, CA). 10 mm stock solutions of TPEN4 (Sigma), sodium arsenite and zinc chloride (Fluka Chemie, Buchs, Switzerland) with >99 and >99.99% purity, respectively, were prepared in double-distilled water and sterilized using a 0.22-μm syringe filter. Working solutions were prepared by diluting the stock with DMEM/F-12 containing 0.1% (w/v) bovine serum albumin. For experiments involving cell exposures, HaCaT cells were rinsed with PBS and placed into DMEM/F-12 with 0.1% bovine serum albumin, and HEKn cells were rinsed and placed in DermaLife K medium containing TPEN, arsenite, and/or zinc as indicated in the figure legends.
Site-directed Mutagenesis and Transfections
A PARP-1 expression plasmid was purchased from OriGene Technologies, Inc. (Rockville, MD), and site-directed mutagenesis of the first and second zinc fingers of PARP-1 was performed by Retrogen, Inc. (San Diego, CA). Mutant 1 was constructed by altering Cys-56 (zf1) and Cys-162 (zf2) to histidine, resulting in CCHH zinc fingers, and mutant 2 was constructed by altering Cys-21 (zf1) and Cys-125 (zf2) to histidine, resulting in HCHC zinc fingers. Mutations were verified by DNA sequencing.
HEKn cells were transfected with the wild-type and mutant PARP-1 expression vectors using Attractene transfection reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. Briefly, Attractene (1 μl/ml) was added to medium containing plasmid DNA (0.12 μg/ml), incubated at room temperature for 10 min, and added dropwise to cells. Cells were incubated for 48 h to allow protein expression before treatment with arsenite.
Spectroscopic Studies of Metal Binding to PARPzf
Cobalt chloride (Co(II)) and zinc chloride (Zn(II)) were obtained from Fluka Chemie with ≥99% purity. The solutions used in the metal binding experiments were prepared with metal-free reagents. Milli-Q purified water was further processed with Chelex resin (Sigma) to remove trace metals. Lyophilized peptide was suspended at a concentration of 2 mm in 10 mm Tris (pH 7.4) for UV-visible spectrometry. Stock solutions of 0.1 m zinc chloride and cobalt chloride were diluted to working concentrations in 10 mm Tris (pH 7.4). All solvents were degassed with argon prior to use. All solutions were prepared freshly before each experiment. An aliquot of Co(II) was added to 100 μm peptide solution, and an absorption spectrum was collected from 280 to 800 nm under argon at 25 °C on a Cary 50 spectrophotometer (Varian Australia Pty. Ltd.). The absorption spectrum of the apopeptide was subtracted from each spectrum. The typical UV-visible difference spectrum of CoPARPzf exhibits peaks at 309 and 665 nm with shoulders at 361 and 602 nm. The ability of Zn(II) to compete with Co(II) for the Cys-3 metal-binding sites of the PARP-1 zinc finger was assessed by titrating aliquots of the Zn(II) solution into the Co(II)·peptide complex along with a 3-fold molar excess of Co(II) relative to the amount of peptide present. An A665 value was used to indicate the complex of peptide with Co(II) (35, 36).
Mass Spectrometry Analysis
The N-terminally acetylated and C-terminally amidated peptides derived from the first zinc finger of human PARP-1 (apoPARPzf), aprataxin (apoAPTXzf), and specific site-directed mutations (see Fig. 3 and supplemental Table S1) were commercially synthesized by Genemed Synthesis Inc. (San Antonio, TX). Purity was assessed by HPLC to exceed 95%. Lyophilized peptide was suspended at a concentration of 1 mm in 20 mm Tris (pH 6.8) containing 1 mm dithiothreitol to allow the cysteine residues to remain in the reduced state. Stock solutions of metal ions were prepared at a concentration of 1 m in 20 mm Tris (pH 6.8). For experimental analysis, both metal ions and peptide were diluted to working concentrations in 20 mm Tris (pH 6.8). Aliquots of 100 μm apoPARPzf were incubated with 100 μm arsenite for 15 min at 25 °C. The samples were then diluted 100 times in 5 mg/ml α-cyano-4-hydroxycinnamic acid (Sigma) in a 1:1 (v/v) acetonitrile/water solution, and 1 μl of each sample was deposited in duplicate on the MALDI plate, allowed to dry at room temperature, and analyzed by MS. MALDI-TOF-MS analyses were performed on an Applied Biosystems 4700 Proteomics Analyzer (TOF/TOF) operating in MS reflector-positive ion mode. The total acceleration voltage was 20 kV. Desorption was performed using a neodymium/yttrium-aluminum-garnet laser (355 nm, 3-ns pulse width, and 200-Hz repetition rate). Mass spectra were acquired with a total of 1000 laser pulses over a mass range of m/z from 1000 to 4000 Da using a focus mass of 3450 Da. Final mass spectra were the summation of eight subspectra, each acquired with 125 laser pulses.
FIGURE 3.

Peptide substitution strategy to test arsenite binding selectivity. The schematic represents the native and mutant peptides tested in Figs. 4 and 5. Specific substitutions are indicated by underlined boldface characters. The full peptide sequences are presented in supplemental Table S1. P-like, PARP-1 like.
Isolation of Zinc Finger Proteins and Zinc Content Measurement
Zinc finger proteins were isolated by immunoprecipitation. Cells were cultured as described above and treated as described in the figure legends. Cells were no more than 75% confluent at the time of collection. Cells were harvested in lysis buffer (20 mm Tris (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm sodium vanadate, 1 μg/ml leupeptin, and 1 mm PMSF), sonicated, and centrifuged at 10,000 rpm for 5 min at 4 °C to remove cellular debris. Protein (500 μg in 500 μl) was incubated with 5 μl of rabbit polyclonal antibody (PARP-1, Cell Signaling 9542; XPA, Abcam ab85914; SP-1, Cell Signaling 5931; or aprataxin, Abcam 31841) or mouse monoclonal antibody (DDK, OriGene TA50011) for at least 1 h at 4 °C, protein A beads (Invitrogen) were added in a 1:1 slurry, and samples were incubated for an additional 1–2 h at 4 °C. The beads were recovered by centrifugation at 10,000 rpm for 5 min at 4 °C and washed five times with 1 ml of lysis buffer.
To elute protein, the pellets were incubated with 100 μl of 0.1 m citric acid (pH 3.0) for 30 min, followed by centrifugation at 10,000 rpm for 5 min at 4 °C. The resultant supernatant was adjusted to pH 7 with 10 n sodium hydroxide and then incubated with 10 mm hydrogen peroxide for a minimum of 3 h at 4 °C to release zinc from protein. Zinc content was measured by adding 10 μl of 1 mm 4-(2-pyridylazo)resorcinol to 100 μl of protein sample and scanning the spectra at 350–550 nm on a Beckman Coulter DU 800 spectrophotometer. The resorcinol indicator absorbance shifts from 411 to 493 nm in the presence of zinc, and the 493 nm peak is recorded and compared with a standard curve for zinc content (35, 36). The relative zinc content was normalized to protein concentration. The activity of PARP-1 proteins expressing mutant zinc finger domains was assessed using an automodification assay as described previously (37). Briefly, immunoprecipitated protein (5 μg) was incubated with activated DNA (Trevigen, Gaithersburg, MD) on ice for 20 min, and the reaction was initiated with NAD+ (5 mm; Trevigen), followed by further incubation for 2 min at room temperature. The reaction was terminated by the addition of 3× SDS buffer (187.5 mm Tris-HCl (pH 6.8), 6% (w/v) SDS, 30% (v/v) glycerol, 150 mm DTT, and 0.03% (w/v) bromphenol blue), samples were resolved by SDS-PAGE and transferred to nitrocellulose membrane, and PARP-1 was detected by immunoblotting. PARP-1 activation was detected by a shift in PARP-1 electrophoretic mobility due to automodification (37).
Measurement of PARP-1 Binding to DNA
PARP-1 protein was isolated by immunoprecipitation as described above, and EMSA was performed to measure DNA binding (38). The synthetic 42-nucleotide double-stranded DNA polynucleotide (Integrated DNA Technologies) probe had an upper strand nucleotide sequence of 5′-GAGTGTTGCATTCCTCTCTGGGCGCCGGGCAGGTACCTGCTG-3′ (38). PARP-1 binding to double-stranded DNA was measured using an EMSA kit (Molecular Probes E33075) according to the manufacturer's instructions. DNA·PARP-1 complexes were resolved on a nondenaturing 6% polyacrylamide gel, and the gels were stained with SYBR Green EMSA nucleic acid gel stain (Molecular Probes). The stained nucleic acids were visualized using 254 nm UV epi-illumination (Kodak IS4000MM Image Station). The excitation filter was 465 nm, and the emission filter was 535 nm. The relative intensity was obtained using the density of the PARP-1 complex divided by the density of the control (probe only).
Statistical Analysis
Data were analyzed using Student's t test. Differences between means were regarded as significant if p < 0.05, and significant differences are indicated by an asterisk in the figures.
RESULTS
Arsenite Interacts with All Three Cysteine Residues of the PARP-1 CCHC Zinc Finger Motif
We reported previously that cell exposure to arsenite enhances UV radiation-stimulated 8-hydroxyl-2′-deoxyguanine generation largely through inhibition of PARP-1 activity (14). In addition, preincubating cells with zinc reverses the arsenite-dependent inhibition of PARP-1 activity and abolishes the arsenite augmentation of UV radiation-induced 8-hydroxyl-2′-deoxyguanine formation (14) and strand breaks (16) in a zinc concentration-dependent manner. Because arsenite binding to the zinc finger motifs of PARP-1 may be the critical event leading to inhibition of PARP-1 activity, we used synthetic peptides representing each PARP-1 zinc finger (PARPzf) sequence of the DNA-binding domain to study the molecular mechanisms of arsenite interaction. Incubation of arsenite with the synthetic PARP-1 apopeptide of zf1 or zf2 led to the formation of AsPARPzf at monoisotopic m/z [M + H]+ = 3525.7 (zf1) and m/z [M + H]+ = 3502.6 (zf2) as detected by MALDI-TOF-MS (Fig. 1, A and B). The +72 m/z shift corresponds to the covalent binding of arsenite to ApoPARPzf after the release of three hydrogens, suggesting arsenite binding with three cysteines.
FIGURE 1.

Arsenite binds to both zinc finger peptides derived from the PARP-1 DNA-binding domain. Arsenite binding to PARP-1 zinc finger apopeptides was analyzed by MALDI-TOF as described under “Experimental Procedures.” A, arsenite (As) binding to the PARPzf1 peptide was detected by MALDI-TOF-MS at m/z = 3526, which reflects a +72 m/z shift against apoPARPzf1 (m/z = 3454). B, arsenite binding to the PARPzf2 peptide (m/z = 3502) also displays a +72 m/z shift relative to apoPARPzf2 (m/z = 3430).
Both of the DNA-binding zinc fingers of PARP-1 are CCHC motifs, so we investigated whether arsenite is capable of forming covalent bonds with all three cysteine residues using MS/MS. The MS2 spectrum of apoPARPzf (precursor ion m/z = 3453.9) is shown in Fig. 2A, in which b and y ions are detected. In the MS2 spectrum of AsPARPzf (precursor ion m/z = 3525.8) (Fig. 2B), we detected the arsenic-bound b and y ions (b14/b15 and y12/y14), which have a +72 m/z shift to the relevant apo-ions. In the same spectrum, apo-b and apo-y fragment ions produced by AsPARPzf are detected in the MS2 spectrum, which can be confirmed by the MS2 spectrum of the apoPARPzf peptide. The matches of fragment ions are summarized in supplemental Table S2. The detection of arsenite binding to each of the cysteine-containing fragment ions strongly suggests that arsenite binds to all three cysteine residues.
FIGURE 2.
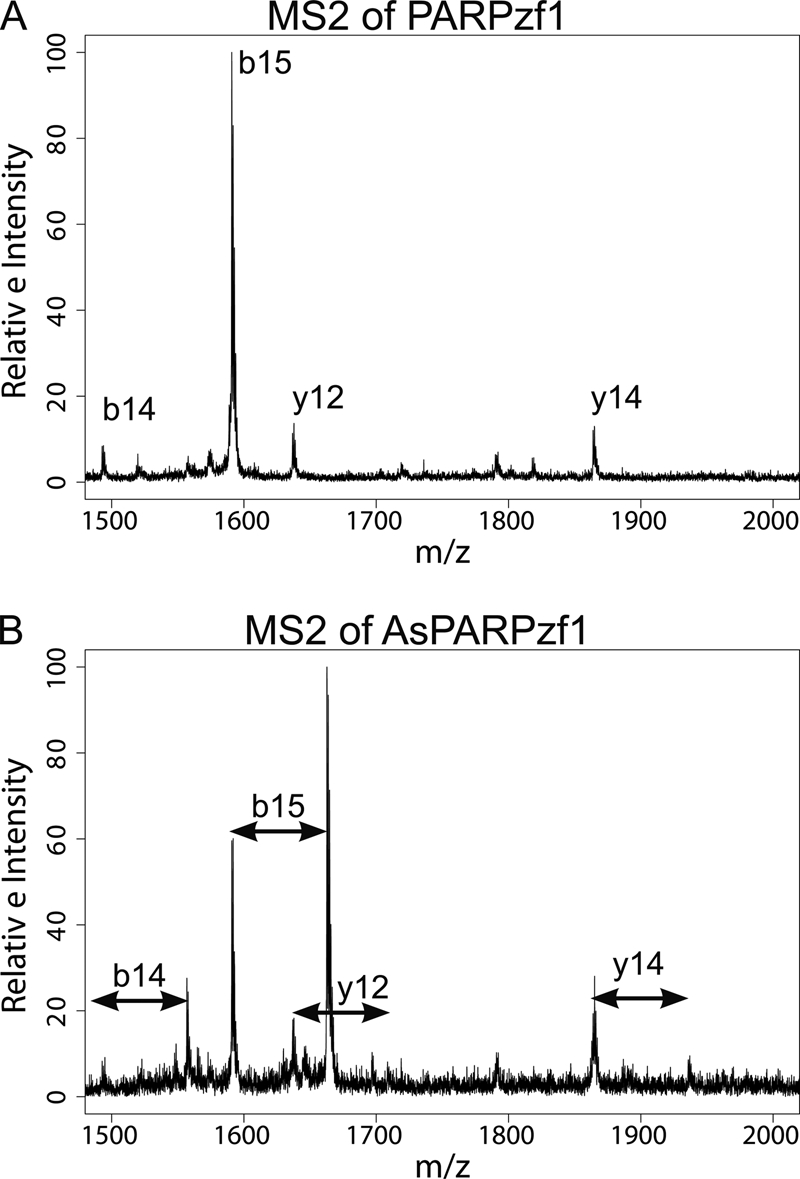
Arsenite forms bonds with all three cysteine residues of the PARP-1 zinc finger. Shown are the results from MS/MS analysis of arsenite-bound PARPzf1. A, apoPARPzf1 was analyzed by MS/MS, and apo-b and apo-y ions are shown. B, arsenite-bound b and y ions were detected by MS/MS. Additional information regarding the bound ions is provided in supplemental Table S2.
Selectivity of Arsenite Binding to PARP-1-derived Zinc Finger Peptides
On the basis of the evidence that arsenite interacts with all three cysteine residues of the PARP-1 zinc finger peptide, we next probed whether all three cysteines are required for binding. Specific substitutions were introduced in the PARP-1 zinc finger peptide to modify the total number or spacing of cysteine residues (Fig. 3 and supplemental Table S1). We evaluated arsenite binding to the PARP-1 apopeptides by MALDI-TOF-MS. Two mutant PARP-1 C2H2 zinc finger peptides were synthesized, one without vicinal cysteine residues (CHHC) (Fig. 4A) and one retaining the vicinal cysteine configuration (CCHH) (Fig. 4B). No arsenite-bonded peptide product was detected for either peptide (Fig. 4, A and B), suggesting that two cysteines, even if vicinal, did not confer arsenite binding to the PARP-1 zinc finger peptide. Extending the distance between the two vicinal cysteines of the PARP-1 CCCH peptide or mutation of the peptide from CCHC to CCCC did not disrupt arsenite interaction (Fig. 4, C and D). To confirm that these native and mutant peptides were capable of binding with zinc, we conducted experiments using cobalt spectrophotometry, which has been used extensively to probe zinc sites in nucleic acid-binding and gene regulatory proteins (35, 36). The method is based on the principle that complexes of paramagnetic Co(II) are highly colored and that the addition of Co(II) to the peptide yields absorption bands in the visible/near-IR region and near-UV region. The former is typical of d-d electronic transitions of Co(II) ion in a tetrahedral coordination, and the latter is characteristic of S → Co(II) ligand-to-metal charge transfer transitions. Fig. 4E shows that addition of Co(II) to PARPzf-CCHC resulted in a Co(II) concentration-dependent increase in both absorption bands. Adding Zn(II) to preformed CoPARPzf-CCHC (Fig. 4F) or CoPARPzf-CCHH (Fig. 4G) caused the absorption bands to decrease in a Zn(II) concentration-dependent manner. These findings show that although arsenic does not bind to the PARP-1 CCHH zinc finger (Fig. 4G) or all other PARP-1-derived peptides depicted in Fig. 3 (data not shown), zinc is capable of binding to all tested zinc finger peptides, including C2H2. Together, these results demonstrate selectivity of arsenite binding to various zinc finger peptides.
FIGURE 4.

Arsenite binding selectivity for PARP-1-derived peptides. A–D, arsenite (As) binding to native and substituted PARPzf1 peptides was assessed by MALDI-TOF-MS analysis as described under “Experimental Procedures.” A, lack of arsenite binding to a PARPzf1-CHHC peptide mutant. B, lack of arsenite binding to a PARPzf1 peptide with a CCHH configuration that retains vicinal cysteine residues. C, arsenite binding to a PARPzf1-CCHC peptide with an insertion between the first two vicinal cysteine residues (CCHC-far) was detected at m/z = 3526. D, arsenite binding to a PARPzf1-CCCC peptide mutant was detected at m/z = 3491. E–G, cobalt spectrometry analysis of zinc binding to native and substituted PARPzf1 peptides. E, 100 μm native PARPzf1 peptide in 5 mm Tris (pH 7.4) was titrated with Co(II) over a range of 10–100 μm in increments of 10 μm. F, 100 μm native PARPzf1 peptide was saturated with 300 μm Co(II) and back-titrated with Zn(II) over a range of 10–50 μm in increments of 10 μm under argon. G, 100 μm mutant PARPzf1 peptide (CCHH) was saturated with 300 μm Co(II) and back-titrated with Zn(II) over a range of 10–40 μm in increments of 10 μm in each addition under argon. Similar results were obtained with each of the PARP-1 peptides shown in Fig. 3 (data not shown).
To verify the binding preference of arsenite for a zinc finger peptide with three or more cysteine residues, we tested arsenite interaction with native (CCHH) and mutant (CCHC) peptides derived from the zinc finger motif of the DNA repair protein APTX (Fig. 3). Arsenite did not bind to the native zinc finger peptide (Fig. 5A), but did bind to a peptide containing a single histidine-to-cysteine substitution rendering a CCHC zinc finger (Fig. 5B). Zinc binding to both peptides was confirmed by cobalt spectrophotometry (Fig. 5, C and D). Collectively, these findings (Figs. 4 and 5) illustrate that arsenite displays binding selectivity based on the number of cysteine residues and that this selectivity is not unique to the PARP-1 zinc finger peptide.
FIGURE 5.

Arsenite binding to the aprataxin zinc finger peptide is conferred upon the addition of a cysteine residue. Arsenite (As) binding to native (CCHH) and mutant (CCHC) APTX zinc finger peptides was assessed by MALDI-TOF-MS as described under “Experimental Procedures.” A, arsenite did not bind to the APTX zinc finger peptide (CCHH configuration). B, arsenite binding to the CCHC mutant of the APTX zinc finger peptide was detected by MALDI-TOF-MS at m/z = 3356. C and D, 100 μm native (CCHH; C) or mutant (CCHC; D) APTX zinc finger peptide was saturated with 300 μm Co(II) and back-titrated with Zn(II) over a range of 10–50 μm in increments of 10 μm under argon.
Functional Evidence of Arsenite-induced Zinc Finger Disruption
Arsenite binding to the zinc finger peptides of PARP-1 (Fig. 1), coupled with functional data that zinc reverses arsenite-dependent inhibition of PARP-1 activity and enhancement of DNA damage (14), suggests that the PARP-1 zinc finger domain is a molecular target for arsenic. To test the biological impact of arsenite binding to zinc finger motifs, we incubated cells with arsenite or the cell-permeable zinc chelator TPEN and isolated PARP-1 protein by immunoprecipitation. PARP-1 protein isolated from exposed cells was tested for zinc content and DNA binding. The zinc content of PARP-1 protein was reduced in an arsenite concentration-dependent manner (Fig. 6A). The decrease was comparable with that of PARP-1 prepared from cells treated with TPEN. The decrease in zinc associated with PARP-1 corresponded to decreased PARP-1 DNA binding as detected by EMSA analysis (Fig. 6, B and C). These findings suggest that the effect of arsenite on PARP-1 function is intrinsic to the protein and that reduced PARP-1 DNA-binding capacity is associated with the loss of zinc from PARP-1. We next wanted to determine whether the arsenite binding selectivity for zinc finger peptides (Figs. 4 and 5) predicts zinc content in four zinc finger DNA-binding proteins isolated from cells pretreated with arsenite or TPEN (Fig. 7A). As expected, cell treatment with the zinc chelator TPEN reduced the zinc content of all four proteins. XPA is a DNA repair protein containing C4 zinc fingers and is an established arsenic target (20). As shown in Fig. 7A, arsenite exposure greatly diminished XPA zinc content comparable with that found for PARP-1. In contrast, the CCHH zinc finger transcription factor SP-1 retained zinc following cell exposure to arsenite. As predicted by the lack of arsenite binding to the native C2H2 APTX peptide (Fig. 5), arsenite exposure did not cause loss of zinc from APTX isolated from cells. Similarly, arsenite exposure decreased zinc content in native PARP-1 expressed in keratinocytes, but zinc was retained in expressed PARP-1 harboring zinc fingers mutated to CCHH (mutant 1) or HCHC (mutant 2) motifs (Fig. 7B). These findings are consistent with the biochemical studies and indicate that, within the cellular environment, arsenite displays target selectivity based on zinc fingers with three or more cysteine residues.
FIGURE 6.

Arsenite decreases the zinc content and DNA binding of PARP-1 isolated from cells. A–C, cells were treated with arsenite (As) or TPEN, and the indicated proteins were isolated by immunoprecipitation as described under “Experimental Procedures.” A, cells were treated with the indicated concentrations of arsenite for 40 h or with 5 μm TPEN for 24 h, PARP-1 was immunoprecipitated, and zinc content was measured as described under “Experimental Procedures.” Values are normalized to the untreated control and represent the mean ± S.D. of three independent experiments. *, p < 0.05. B, cells were treated as described for A, and PARP-1 DNA binding was measured by EMSA as described under “Experimental Procedures.” C, shown is the quantification of data in B based on densitometry. The concentration of zinc in the culture medium was 1.5 μm, so at the highest arsenite concentration, the molar ratio of arsenite to zinc in the tissue culture medium was 1.33:1.0. Values represent the mean ± S.D. of three independent experiments. *, p < 0.05.
FIGURE 7.

Arsenite-induced zinc release is selective for zinc fingers containing three or more cysteine residues. A, HaCaT cells were cultured to ∼60% confluence in 15-cm plates and then treated with 2 μm arsenite (As) for 48 h or with 5 μm TPEN for 24 h. PARP-1, XPA, SP-1, and APTX were immunoprecipitated from cell lysates, and zinc content was measured as described under “Experimental Procedures.” Values represent the mean ± S.D. of three independent experiments. *, p < 0.05 (significantly different from similarly treated samples). B, HEKn cells were cultured to ∼50% confluence in 15-cm plates. Cells were then transfected with plasmids containing WT PARP-1, zinc finger mutant 1 (M1; CCHH), or zinc finger mutant 2 (M2; HCHC) as described under “Experimental Procedures” before exposure to arsenite (1 μm) for 24 h. Cell lysates were prepared, and expressed WT or mutant PARP-1 protein was isolated by immunoprecipitation using antibody directed against the DDK epitope tag. The zinc release assay was performed as described for A. The zinc content of PARP-1 mutants was not statistically different from that of the WT protein. The lower panels show the activity of the WT and zinc finger-mutated PARP-1 proteins based on DNA-dependent automodification. Modified PARP-1 (upper bands) was normalized to total PARP-1 for each construct to generate relative activity values. The results represent the findings from three independent experiments. Values are normalized to untreated (A) or untransfected (B) control samples (data not shown) and represent the mean ± S.D. of three independent experiments. *, p < 0.05 (significantly different from the no arsenite-matched sample).
DISCUSSION
Emerging evidence by our laboratory and others suggests that inhibition of DNA repair is an important mechanism for arsenic carcinogenesis. A limited number of zinc finger DNA repair proteins such as PARP-1 and XPA have been demonstrated to be arsenite targets. PARP-1 is an important target because inhibition by arsenite is evident at environmentally relevant submicromolar concentrations (14, 19), and it is the only validated DNA repair zinc finger molecular target with demonstrated inhibition in arsenic-exposed cells in situ (14, 19, 26). PARP-1 activation is involved in cellular response to genotoxic insult, and lethality and carcinogenesis in response to DNA-damaging agents are greatly enhanced in a PARP-1 null background (28). PARP-1 recognizes and binds to damaged DNA, rapidly catalyzing the covalent attachment of poly(ADP-ribose) units to acceptor proteins (31). PARP-1 substrates in the base excision repair and nucleotide excision repair DNA damage repair networks include (but are not limited to) XPA, XRCC-1, DNA ligase III, DNA polymerase, the DNA-dependent protein kinase catalytic subunit, Ku70, proliferating cell nuclear antigen, histones, and topoisomerases required for chromatin remodeling, in addition to other nuclear proteins (29–31). Thus, PARP-1 inhibition is predicted to affect numerous aspects of DNA repair; however, it is not clear how arsenic inhibits PARP-1 activity and why PARP-1 is such a sensitive target for arsenic.
A recent study demonstrated that the zf1 and zf2 domains of the PARP-1 DNA-binding domain perform different functions (39). The zf2 domain displays high DNA-binding affinity, whereas the zf1 domain is required for DNA-dependent PARP-1 activity. In this study, we have demonstrated that arsenite can bind to both PARP-1 zinc finger motifs (Fig. 1) and that cell exposure to arsenite causes loss of PARP-1 activity (14), depletion of zinc from the protein (Fig. 7), and greatly diminished DNA-binding capacity (Fig. 6). These findings suggest that the exquisite sensitivity of PARP-1 to arsenite may be due to zinc loss from both zinc fingers, leading to impairment of DNA binding and catalytic activities governed by these domains.
Furthermore, we have demonstrated arsenite binding selectivity for zinc finger peptides containing a C3H1 or C4 configuration. C2H2 peptides derived from PARP-1 or APTX did not bind arsenite, whereas site-directed substitutions of histidine for cysteine conferred arsenite binding (Figs. 4 and 5). An earlier study reported that trivalent arsenicals are able to release zinc from an XPA-derived zinc finger peptide (20). The peptide results were confirmed by zinc loss in C4 and CCHC (but not C2H2) proteins that were isolated from cells exposed to arsenite (Fig. 7). These findings suggest that PARP-1 and XPA are direct molecular targets for arsenite within living cells. Arsenite-dependent loss of zinc leading to disruption of protein function may be similar to zinc deficiency, which has been shown to contribute to DNA single- and double-strand breaks and oxidative DNA damage that increase risk for cancer development (40). Our previous report demonstrated that additional zinc could oppose the impact of arsenite on PARP activity and DNA repair (14), which is consistent with the notion that zinc and arsenite compete for the same binding site in certain target proteins.
Binding of trivalent arsenicals to cysteine-rich proteins such as hemoglobin, tubulin, and metallothionein has long been recognized (reviewed in Refs. 41 and 42), and there has been considerable focus on arsenic interaction with cysteine-containing peptides, including those derived from zinc finger domains of DNA repair proteins. Our observation of preferential arsenite interaction based on the number of cysteine residues within a zinc finger is significant because the majority of zinc finger proteins in the human genome are of the C2H2 variety, suggesting that arsenite may have distinct molecular zinc finger targets in cells. Our studies support the idea that trivalent arsenite binds directly to sulfur in each of the three cysteines of the PARP-1 zinc fingers based on a mass shift of +72 m/z for the AsPARPzf product, which corresponds to the covalent binding of arsenic (75 mass units) to apoPARPzf after the release of three hydrogens (Fig. 1) and detection of arsenite binding to each of the cysteine-containing fragments as detected in the tandem mass spectrum (Fig. 2). Our findings are consistent with the arsenite-peptide interaction model reported by Kitchin and Wallace (43).
Although it has been suggested that vicinal cysteine residues are required for high affinity arsenite binding (33), our current findings indicate that vicinal cysteines are not necessarily sufficient for arsenite binding to peptides (Figs. 4 and 5) or disruption of zinc binding in cells (Figs. 6 and 7). Our observation of arsenite binding selectivity for peptides and cellular target proteins with three or more cysteine residues is consistent with chemical studies demonstrating substantial differences in the Kd values for radioactive arsenite binding to model peptides based on the number of free cysteines (32). Another important consideration is demonstrated by the elegant work of Kitchin and Wallace (43). Using rapid vacuum filtration assays and radioactive arsenite, dramatic differences in the chemical kinetics of arsenite-peptide interactions were revealed. The half-life of the arsenite·trithiol peptide complex is >2 orders of magnitude greater than that of the corresponding arsenite·dithiol peptide complex (155 versus 1.29 min), suggesting that an arsenite-bonded C3 or C4 complex is much more stable than a C2 complex. Our studies demonstrate arsenite binding selectivity in both peptide and protein with a link between interaction and functional consequences for protein activity. Although arsenite may be capable of binding to a C2H2 zinc finger (32, 33, 43), the final product may not persist for a sufficient length of time to be detected by MALDI-TOF-MS. This difference in chemical kinetics could have major impact in cells, where a more stable interaction of arsenite with a C3 or C4 zinc finger protein may be necessary to sustain biological impact. This idea is supported by findings from the cellular studies demonstrating that arsenite exposure led to loss of zinc from C3 or C4 (PARP-1 and XPA), but not C2 (SP-1, APTX, and PARP-1 zinc finger mutants), zinc finger proteins (Fig. 7).
In summary, the findings presented here demonstrate the preferential interaction of arsenite with zinc finger peptides containing three or more cysteine residues. We have also provided strong evidence that arsenite decreases the zinc content of zinc finger protein targets harboring C3H1 (but not C2H2) motifs, thereby demonstrating target selectivity within the cellular environment. Thus, the ability of arsenite to disrupt zinc finger function in living systems may be restricted to a subset of potential zinc finger targets, and this may inform future studies to identify additional direct cellular targets for arsenic.
Supplementary Material
Acknowledgment
We thank Dr. Charlotte Mobarak for assistance with the mass spectrometry studies.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 ES15826 and R01 ES15826-03S1. This work was also supported by University of New Mexico Cancer Research and Treatment Center Grant NIH P30 CA118100 and the University of New Mexico Proteomics/Mass Spectrometry Facility.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2.
- TPEN
- N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine
- zf
- zinc finger.
REFERENCES
- 1. Agency for Toxic Substances and Disease Registry (2007) Toxicological Profile for Arsenic, United States Department of Health and Human Services, Public Health Service, Atlanta, GA [Google Scholar]
- 2. Schuhmacher-Wolz U., Dieter H. H., Klein D., Schneider K. (2009) Crit. Rev. Toxicol. 39, 271–298 [DOI] [PubMed] [Google Scholar]
- 3. Smith A. H., Steinmaus C. M. (2009) Annu. Rev. Public Health 30, 107–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rahman M., Vahter M., Wahed M. A., Sohel N., Yunus M., Streatfield P. K., El Arifeen S., Bhuiya A., Zaman K., Chowdhury A. M., Ekström E. C., Persson L. A. (2006) J. Epidemiol. Community Health 60, 242–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schoen A., Beck B., Sharma R., Dubé E. (2004) Toxicol. Appl. Pharmacol. 198, 253–267 [DOI] [PubMed] [Google Scholar]
- 6. Yoshida T., Yamauchi H., Fan Sun G. (2004) Toxicol. Appl. Pharmacol. 198, 243–252 [DOI] [PubMed] [Google Scholar]
- 7. Yu H. S., Liao W. T., Chai C. Y. (2006) J. Biomed. Sci. 13, 657–666 [DOI] [PubMed] [Google Scholar]
- 8. Germolec D. R., Spalding J., Yu H. S., Chen G. S., Simeonova P. P., Humble M. C., Bruccoleri A., Boorman G. A., Foley J. F., Yoshida T., Luster M. I. (1998) Am. J. Pathol. 153, 1775–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maier A., Schumann B. L., Chang X., Talaska G., Puga A. (2002) Mutat. Res. 517, 101–111 [DOI] [PubMed] [Google Scholar]
- 10. Rossman T. G., Uddin A. N., Burns F. J. (2004) Toxicol. Appl. Pharmacol. 198, 394–404 [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto S., Konishi Y., Matsuda T., Murai T., Shibata M. A., Matsui-Yuasa I., Otani S., Kuroda K., Endo G., Fukushima S. (1995) Cancer Res. 55, 1271–1276 [PubMed] [Google Scholar]
- 12. Ding W., Hudson L. G., Liu K. J. (2005) Mol. Cell. Biochem. 279, 105–112 [DOI] [PubMed] [Google Scholar]
- 13. Ding W., Hudson L. G., Sun X., Feng C., Liu K. J. (2008) Free Radic. Biol. Me.d 45, 1065–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding W., Liu W., Cooper K. L., Qin X. J., de Souza Bergo P. L., Hudson L. G., Liu K. J. (2009) J. Biol. Chem. 284, 6809–6817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qin X. J., Hudson L. G., Liu W., Ding W., Cooper K. L., Liu K. J. (2008) Chem. Res. Toxicol. 21, 1806–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin X. J., Hudson L. G., Liu W., Timmins G. S., Liu K. J. (2008) Toxicol. Appl. Pharmacol. 232, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi H., Hudson L. G., Ding W., Wang S., Cooper K. L., Liu S., Chen Y., Shi X., Liu K. J. (2004) Chem. Res. Toxicol. 17, 871–878 [DOI] [PubMed] [Google Scholar]
- 18. Asmuss M., Mullenders L. H., Eker A., Hartwig A. (2000) Carcinogenesis 21, 2097–2104 [DOI] [PubMed] [Google Scholar]
- 19. Hartwig A., Blessing H., Schwerdtle T., Walter I. (2003) Toxicology 193, 161–169 [DOI] [PubMed] [Google Scholar]
- 20. Hartwig A., Pelzer A., Asmuss M., Bürkle A. (2003) Int. J. Cancer 104, 1–6 [DOI] [PubMed] [Google Scholar]
- 21. Piatek K., Schwerdtle T., Hartwig A., Bal W. (2008) Chem. Res. Toxicol. 21, 600–606 [DOI] [PubMed] [Google Scholar]
- 22. Schwerdtle T., Walter I., Hartwig A. (2003) DNA Repair 2, 1449–1463 [DOI] [PubMed] [Google Scholar]
- 23. Hu Y., Su L., Snow E. T. (1998) Mutat. Res. 408, 203–218 [DOI] [PubMed] [Google Scholar]
- 24. Lynn S., Lai H. T., Gurr J. R., Jan K. Y. (1997) Mutagenesis 12, 353–358 [DOI] [PubMed] [Google Scholar]
- 25. Snow E. T., Sykora P., Durham T. R., Klein C. B. (2005) Toxicol. Appl. Pharmacol. 207, 557–564 [DOI] [PubMed] [Google Scholar]
- 26. Walter I., Schwerdtle T., Thuy C., Parsons J. L., Dianov G. L., Hartwig A. (2007) DNA Repair 6, 61–70 [DOI] [PubMed] [Google Scholar]
- 27. Hassa P. O., Haenni S. S., Elser M., Hottiger M. O. (2006) Microbiol. Mol. Biol. Rev. 70, 789–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malanga M., Althaus F. R. (2005) Biochem. Cell Biol. 83, 354–364 [DOI] [PubMed] [Google Scholar]
- 29. Rouleau M., Patel A., Hendzel M. J., Kaufmann S. H., Poirier G. G. (2010) Nat. Rev. Cancer 10, 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schreiber V., Dantzer F., Ame J. C., de Murcia G. (2006) Nat. Rev. Mol. Cell Biol. 7, 517–528 [DOI] [PubMed] [Google Scholar]
- 31. Woodhouse B. C., Dianov G. L. (2008) DNA Repair 7, 1077–1086 [DOI] [PubMed] [Google Scholar]
- 32. Kitchin K. T., Wallace K. (2005) Toxicol. Appl. Pharmacol. 206, 66–72 [DOI] [PubMed] [Google Scholar]
- 33. Kitchin K. T., Wallace K. (2006) J. Biochem. Mol. Toxicol. 20, 35–38 [DOI] [PubMed] [Google Scholar]
- 34. Ramadan D., Rancy P. C., Nagarkar R. P., Schneider J. P., Thorpe C. (2009) Biochemistry 48, 424–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kopera E., Schwerdtle T., Hartwig A., Bal W. (2004) Chem. Res. Toxicol. 17, 1452–1458 [DOI] [PubMed] [Google Scholar]
- 36. Payne J. C., Rous B. W., Tenderholt A. L., Godwin H. A. (2003) Biochemistry 42, 14214–14224 [DOI] [PubMed] [Google Scholar]
- 37. Langelier M. F., Servent K. M., Rogers E. E., Pascal J. M. (2008) J. Biol. Chem. 283, 4105–4114 [DOI] [PubMed] [Google Scholar]
- 38. Huang K., Tidyman W. E., Le K. U., Kirsten E., Kun E., Ordahl C. P. (2004) Biochemistry 43, 217–223 [DOI] [PubMed] [Google Scholar]
- 39. Langelier M. F., Planck J. L., Roy S., Pascal J. M. (2011) J. Biol. Chem. 286, 10690–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ho E. (2004) J. Nutr. Biochem. 15, 572–578 [DOI] [PubMed] [Google Scholar]
- 41. Aposhian H. V., Aposhian M. M. (2006) Chem. Res. Toxicol. 19, 1–15 [DOI] [PubMed] [Google Scholar]
- 42. Kitchin K. T., Wallace K. (2008) J. Inorg. Biochem. 102, 532–539 [DOI] [PubMed] [Google Scholar]
- 43. Kitchin K. T., Wallace K. (2006) J. Biochem. Mol. Toxicol. 20, 48–56 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.