

Abstract
Dimerization of G protein-coupled receptors has received much attention as a regulatory system of physiological function. Metabotropic glutamate receptors (mGluRs) are suitable models for studying the physiological significance of G protein-coupled receptor dimers because they form constitutive homodimers and function through dimeric rearrangement of their extracellular ligand binding domains. However, the molecular architecture of the transmembrane domains (TMDs) and their rearrangement upon agonist binding are still largely unknown. Here we show that the two helix Vs are arranged as the closest part in the dimeric TMDs and change their positions through synergistic control by the binding of two glutamates. The possibility that helix V is involved in an inter-protomer communication was first suggested by the finding that constitutively active mutation sites were identified on both sides of helix V. Then, comprehensive fluorescence resonance energy transfer (FRET) analysis using mGluRs whose cytoplasmic loops were labeled with donor and acceptor fluorescent proteins revealed that the third intracellular loop connecting helices V and VI of one protomer was in close proximity to the second and third intracellular loops of the other protomer and that all the intracellular loops became closer during the activation. Furthermore, FRET analysis of heterodimers in which only one protomer had ligand binding ability revealed the synergistic effect of the binding of two glutamates on the dimeric rearrangements of the TMD. Thus, the glutamate-dependent synergistic relocation of the helix Vs in the dimer is important for the signal flow from the extracellular ligand binding domain to the cytoplasmic surface of the mGluR.
Keywords: Fluorescence Resonance Energy Transfer (FRET), G Protein-coupled Receptors (GPCR), Glutamate Receptors Metabotropic, 7-Helix Receptor, Mutant, Protein Chemistry, Protein Structure, Protein-Protein Interactions, Rhodopsin, Signal Transduction
Introduction
G protein-coupled receptors (GPCRs)2 constitute the largest superfamily of signaling proteins with a seven-transmembrane domain (TMD). Recently, many reports about oligomerization of GPCRs have shown that not only the monomer but also homo- or heterodimers could work as a unit to regulate physiological functions and be used as a novel drug target (1–3). Metabotropic glutamate receptor (mGluR) is one of the typical GPCRs that function as a constitutive homodimer. Eight subtypes of mGluRs have been identified in mammalian genomes and are classified into three subgroups based on their sequence homology, signaling cascades, and pharmacology (4). Group I mGluRs (mGluR1 and mGluR5) couple with the Gq subtype of G protein, whereas group II (mGluR2 and mGluR3) and group III (mGluR4, -6, -7, and -8) mGluRs couple with Gi/Go subtypes.
All mGluR subtypes possess a large extracellular ligand binding domain (ECD) on the N-terminal side of the TMD. The x-ray crystallographic structures of ECDs revealed that the ligand-induced relocation of dimeric ECDs determines the resting and active states of mGluRs (5–7), and analyses of various mutants of mGluR5 demonstrated that the activation of mGluR is induced by an intersubunit rearrangement (8). However, the structural information available on TMDs that possess a dimeric arrangement is severely limited even though it is essential for the elucidation of the molecular mechanism of activation of mGluRs. In many GPCRs, the analysis of the mutants with high spontaneous activity (constitutive activity) has led to a better understanding of the structural changes of the receptor (9, 10). By taking such an approach in our previous studies, we identified several constitutively active mutation (CAM) sites in TMD and the intracellular loops of mGluR8 and suggested a model in which these residues participated in the intra-protomer interaction of the dimeric mGluR (11–13).
By identifying and analyzing additional CAM sites in the TMD of mGluR8, we found a unique region in helix V in the present study. That is, a cluster of CAM sites possibly located not only on the intra-protomer surface but also on an extra-protomer surface of helix V was identified. This finding suggested the possibility that helix V is involved in an inter-protomer communication via the CAM sites. Consequently, we performed comprehensive FRET analysis to obtain information about the dimeric arrangement of the TMD. We inserted GFP-derived fluorescent proteins Cerulean (14) and Venus (15) at the intracellular loops and the C-terminal region of mGluR8 and measured FRET efficiencies to compare the relative distance between two loop regions. Cerulean and Venus exhibit excitation maxima at 433 and 515 nm and emission maxima at 475 and 528 nm, respectively, so that we can observe a FRET signal from Cerulean to Venus by excitation of the labeled mGluR with 433-nm light if two fluorescent proteins are located close enough to each other. The results showed that the third intracellular loop (i3) of one protomer is adjacent to the second (i2) and third intracellular loops of the other protomer and that the two TMDs move closer to each other in accordance with the agonist-dependent conformational change of the ECDs. Furthermore, using a mutant of mGluR that exhibits deficient ligand binding in only one subunit, we demonstrated that the wild-type receptor that bound two glutamates exhibited an increase of FRET efficiency about four times higher than that of the mutant that bound only one glutamate. Thus, the bindings of two glutamates in the dimeric ECDs act synergistically to promote the dimeric rearrangement of the TMDs. Based on these findings, we will discuss the dimeric arrangement of mGluR and the mode of its rearrangement in the activation process.
EXPERIMENTAL PROCEDURES
Materials
[3H]LY341495 (1.28 TBq/mmol) and l-AP4 were purchased from Tocris Cookson, [35S]guanosine 5′-3-O-(thio)triphosphate (GTPγS) (37 TBq/mmol) from was PerkinElmer Life Sciences, and monoclonal anti-GFP, clone GFP-20 mouse ascites fluid were from Sigma.
Construction of mGluR Mutants
The mCerulean and Venus coding regions (Val-2—Leu-237) of mCerulean- and Venus-pCS2 were amplified by PCR and inserted into each intracellular loop region of mGluR1 (i1, Pro-621-Val-622; i2, Ile-685-Leu-686; i3, Pro-778-Ala-779; i4, just after Pro-881), mGluR3 (i1, Pro-605-Leu-606; i2, Ile-669-Phe-670; i3, Pro-762-Glu-763; i4, just after Leu-879), and mGluR8 (i1, Pro-612-Ile-613; i2, Ile-676-Phe-677; i3, Pro-774-Glu-775; i4, just after Ile-908) by In-Fusion PCR cloning (Clontech). To detect the expression of mGluRs by Western blotting analysis, the cDNAs of mGluRs were tagged with the epitope sequence of the anti-bovine rhodopsin monoclonal antibody Rho1D4 at the C terminus. The wild-type and mutant cDNAs of mGluRs were introduced into the mammalian expression vector pcDNA3.1 (Invitrogen) or pCAG-GS.
Preparation of mGluR Wild-Type and Mutants
Expression of mGluRs in HEK293S cells was performed by the method previously reported (13). HEK293S cells were grown to ∼40% confluency in DMEM/F-12 supplemented with 10% fetal bovine serum and transfected with wild-type or mutant mGluR plasmid DNA (10 μg/100-mm dish) using the calcium phosphate method. The cells were collected by centrifugation 48 h after transfection. The cell membranes were homogenized in 50% sucrose in buffer A (50 mm HEPES (pH 6.5) and 140 mm NaCl) before centrifugation. The supernatant was diluted in two volumes of buffer A and re-centrifuged. The membrane pellets were washed 3 times for more than 2 h with low salt buffer (5 mm HEPES (pH 8.0) and 0.3 mm EDTA) to reduce autofluorescence derived from cell surface proteins. Washed membrane pellets were suspended in buffer B (50 mm HEPES (pH 8.0), 140 mm NaCl, and 3 mm MgCl2) for the ligand binding assay and the FRET assay.
Fluorescence Microscopy
To confirm the cellular expression of fluorescence protein-fused mGluR, the cells ∼30 h after transfection were observed in phosphate-buffered saline on a Nikon Eclipse TE300 inverted microscope with a TE-FM fluorescence attachment. A high pressure mercury lamp (HB 10103AF) with a 450–490-nm band pass filter was used for the excitation of fluorescence proteins, and fluorescence was detected with a 505-nm dichroic mirror and a 520-nm long-pass filter (Nikon B-2A filter set).
Ligand Binding Assay of mGluR8
The ligand binding assay was performed according to our previous report (13). [3H]LY341495 binding to membranes expressing mGluR8 was measured at room temperature. A mixture of membranes and 0∼400 nm [3H]LY341495 in buffer B was incubated for 30 min (final assay volume, 20 μl). After incubation, bound and free radioligands were separated by centrifugation. The membrane pellets were washed with buffer B and solubilized in 1 m NaOH. The amount of bound [3H]LY341495 in the solubilized membranes was quantified by assaying with a liquid scintillation counter. Nonspecific binding was determined with 1 mm l-AP4. Kd and Bmax values were calculated using the nonlinear one-site binding equation, y = Bmaxx/(Kd + x). Protein concentration in the membranes was determined by the Bradford method. Displacement by l-AP4 of [3H]LY341495 binding to the cell membranes expressing mGluR8 was also measured at room temperature. A mixture of membranes (30 μg of total protein), 100 nm [3H]LY341495, and 0∼100 μm l-AP4 in buffer B was incubated for 30 min (final assay volume, 20 μl). After incubation, bound and free radioligands were separated by centrifugation. The membrane pellets were washed with buffer B and solubilized in 1 m NaOH. The specific binding, defined using 1 mm l-AP4 as displacer, was ∼93% that of total binding estimated using 100 nm [3H]LY341495 and 30 μg of tissue protein. Competition binding curves were fitted to the one-site binding model, y = (Max-Basal)/(1 + x/IC50) + Basal.
GTPγS Binding Assay of mGluR8
The assay of G protein activation by mGluR8 was carried out according to our previous report (12). The mGluR8-containing membranes (final concentration, 2 nm) after sucrose flotation were suspended in 0.03% dodecylmaltoside in buffer C (50 mm HEPES (pH 7.2), 140 mm NaCl, and 3 mm MgCl2) and preincubated with Go-type G protein (final concentration, 200 nm) purified from pig cerebral cortex and the agonist l-AP4. After preincubation for 30 min at 10 °C, the GDP/GTPγS exchange reaction was started by adding GTPγS solution. The assay mixture (20 μl) consisted of 50 mm HEPES (pH 7.2), 140 mm NaCl, 5 mm MgCl2, 0.015% dodecylmaltoside, 0.03% sodium cholate, 0.8 mg/ml α-l-phosphatidylcholine, 0.1 μm GTPγS, and 3 μm GDP. After incubation for 3 min, the reaction was terminated by adding stop solution (200 μl: 20 mm Tris (pH 7.4), 100 mm NaCl, 25 mm MgCl2, 0.1 μm GTPγS, and 3 μm GDP) and immediately filtering the sample through a nitrocellulose membrane to trap [35S]GTPγS bound to G proteins.
Western Blotting
Wild-type and mGluR8 mutants were detected with the Rho1D4 monoclonal antibody against the tag sequence at the C terminus or anti-GFP monoclonal antibody against Cerulean and Venus. The cell membrane suspension in the buffer (62.5 mm Tris (pH 6.8), 4% SDS, 10% glycerol) with or without 2.5% β-mercaptoethanol was subjected to SDS-PAGE (5.5%), and then the electrophoresed proteins were transferred onto a polyvinylidene difluoride membrane and probed with the antibody. Immunoreactive proteins were detected by the ABC method and visualized using the horseradish peroxidase-diaminobenzidine reaction. Visualization was carried out according to a previous report (16).
FRET Measurement of mGluRs
Fluorescence spectra from Cerulean- and Venus-fused mGluRs were recorded at 20 °C with an RF-5300PC spectrofluorophotometer (Shimadzu). The membrane pellets prepared as described above were resuspended in buffer D (10 mm HEPES (pH 7.5) and 140 mm NaCl, 4 mm KOH, 1 mm MgCl2, and 1.5 mm CaCl2) and were excited at 433 nm for Cerulean or 500 nm for Venus. To block the scattering, cutoff filters (Y-45 for 433-nm excitation and Y-51 for 500-nm excitation) were placed in front of the detector. Autofluorescence spectra from the membranes containing wild-type mGluRs were also recorded in the same conditions as the base line. We estimated Eapp of each sample from the base-line-corrected fluorescence spectra by “spectra FRET” analysis as described in a previous report (17). The molar extinction coefficient of Venus at 433 nm (1100) was estimated from the excitation spectrum of Venus, referring to a previous report (15). Estimation of net FRET efficiency (E) from Eapp was performed as described under “Results.” Dose-response plots of E were fitted to the equation y = (Max-Basal)/(1 + (x/EC50)n) + Basal.
Estimation of the Amount of Cerulean- or Venus-fused mGluRs in Co-expressing Samples
The amount of Venus-fused mGluRs in co-expressing samples (Vexp) was quantified based on the maximum intensity of emission spectra excited at 500 nm (I500) according to the following equation,
 |
in which IV is the maximum intensity of emission spectra of a sample expressing only m8-V4 (82.5 ± 0.9), and AV is the absolute amount estimated by the ligand binding assay as described above (66.0 ± 0.4 pmol/mg of total protein). In the same manner, the amount of Cerulean-fused mGluRs in co-expressing samples (Cexp) was quantified based on the maximum intensity of emission spectra excited at 433 nm (I433) according to the equation,
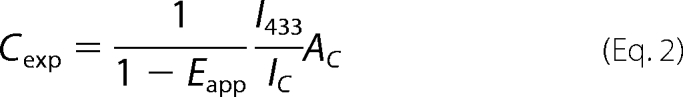 |
in which IC is the maximum intensity of emission spectra of a sample expressing only m8-C4 (615 ± 138), and AC is the absolute amount estimated by the ligand binding assay as described above (58.6 ± 8.6 pmol/mg of total protein). The decrease of Cerulean intensity by FRET in the co-expressing sample was corrected in Equation 2 based on the following equation:
 |
in which Inet is the net Cerulean intensity without FRET.
Molecular Modeling of TMD of mGluR8
A molecular model of TMD of mGluR8 was created using SWISS-MODEL (18, 19), the internet-based protein-modeling server. We selected the 2.2 Å crystal structure of bovine rhodopsin (Protein Data Bank code 1U19) (20) as a template for homology modeling. The alignment of amino acid sequence between mGluR and rhodopsin shown in Fig. 5a was according to previous reports (21–24) in which predictions of the binding pockets of allosteric modulators of family 3 GPCRs were successful using the molecular modeling based on the crystal structure of bovine rhodopsin. Especially, the alignment of helices III, V, VI, where the hot spots of CAM sites exist, is identical to the published report for calcium-sensing receptor (21). In the alignment mode of SWISS-MODEL, the energy-minimization based on the Gromos96 was performed to improve the stereochemistry of the model. The created model was depicted in Fig. 5 by using PyMol.
FIGURE 5.

Molecular model of TMD of mGluR8. a, alignment of amino acids sequences of mouse mGluR8 (MGR8) and bovine rhodopsin (OPSD). CAM sites of mGluR8 are indicated by bold letters. b, the whole image of the model as viewed from the lipid bilayer is shown. The seven helices are represented as ribbons colored using different colors (blue to red). The region shown in c is indicated by a dashed frame. c, an extended image of helices III, V, and VI as viewed from the intracellular side is shown. Representation of the models is the same as in b. The side chains of the CAM sites in the TMD of mGluR8 are shown as sticks in all figures.
RESULTS
Alanine Scanning Mutagenesis in Helix V of mGluR8
We previously found that Thr-764 in helix V of mGluR8 is a unique CAM site whose replacement with any amino acid caused elevation of the constitutive activity (Fig. 1a) (13). We also identified the residue (Tyr-663) in helix III that is the only residue suppressing the constitutive activity caused by Thr-764 replacement by comprehensive alanine scanning mutagenesis in the helices other than helix V (Fig. 1a). These results suggested that Tyr-663 is a candidate residue interacting with Thr-764 within the protomer of mGluR8, although it is possible that the apparent suppression by Tyr-663 replacement is due to a global conformational change throughout the receptor (13). Then we continued our research to identify CAM sites near Thr-764 in helix V and found an interesting region in which CAM sites are clustered (Fig. 1b). This region constitutes two helical turns containing 10 amino acids. The alanine mutagenesis of these residues showed that all mutants except for L759A were expressed at sufficiently high levels to measure the G protein activation ability by the GTPγS binding assay (Fig. 1d). Then we found that six of these amino acid residues including Thr-764 were the CAM sites (Figs. 1, b, c, and e). Because this region contains no helix breaker amino acids such as glycine or proline, it should form a normal helix with CAM sites distributed in both sides of the helix, as shown in Fig. 1c. Assuming that Thr-764 in this region would face toward the intra-protomer surface of the helix and interact with Tyr-663 in helix III, the CAM sites (Thr-762 and Tyr-766) in the side opposite to Thr-764 could face toward the extra-protomer surface of the helix and make some contribution to forming the dimeric interface (Fig. 1c), although there is another possibility, namely, that helix V is simply making multiple intra-protomer interactions. Thus, we performed a comprehensive FRET analysis to test the possibility that helix V makes some contribution to forming a dimer.
FIGURE 1.

Constitutively active mutations in helix V of mGluR8. a and b, shown is a two-dimensional diagram of mGluR8. The residues that were replaced by alanine (or serine) in the previous (13) (a) and present (b) alanine-scanning mutagenesis are indicated by either black or gray shading. Black shading indicates the residues whose substitution resulted in an increase of constitutive activity. c, shown is helical wheel projection of constitutively active mutation sites in helix V. Residues whose substitution resulted in an increase of basal activity are indicated by bold letters. d, ligand binding potency of alanine mutants in helix V is shown. [3H]LY341495 binding to wild-type and mutant-expressing HEK293 cell membranes was measured. Results are normalized to total specific binding of wild-type. e, constitutive activity of alanine mutants in helix V are shown. The asterisk indicates a significant increase of constitutive activity in the mutants (p < 0.05; Student's t test, two-tailed). All data were normalized to the activity of the mock-transfected membranes and are expressed as the mean ± S.D. of more than two independent experiments done in duplicate.
Construction and Expression of Fluorescent Protein-fused mGluRs
To obtain information about the relative position of each intracellular region of dimeric mGluRs, we introduced Cerulean or Venus into the intracellular regions of mGluR8 and co-transfected these mutants into HEK293 cells. In addition, Cerulean/Venus-fused mutants of mGluR1 and mGluR3, representatives of groups I and II, respectively, were also constructed to obtain insight into the common features of the arrangement of the intracellular regions among the three mGluR groups. We abbreviate these co-expressing samples as m(a)-C(b)V(c) (a, number of subtype; b and c, number of Cerulean- or Venus-inserted intracellular loop region, the C-terminal region is represented as “4”). The expression of the mGluR mutants was confirmed by fluorescence microscopy and immunoblotting. The expressed mGluR8 mutants were distributed not only in the subcellular organelles but also in the plasma membrane in the expression system (Fig. 2a). The immunoblotting analysis showed that the mutants of mGluR8 basically exist as dimers in the nonreducing condition like wild-type, and they turned into monomers when mercaptoethanol was added (Fig. 2b). Therefore, the insertion of fluorescence probes into the intracellular loop region caused little, if any, hindrance of dimerization.
FIGURE 2.

Spectra FRET analysis of m8-C3V3 sample. a, shown are fluorescence and difference interference contrast images of HEK293 cells expressing m8-C3 (i and ii), m8-V3 (iii and iv), m8-C3V3 samples (v and vi). The fluorescence images (i, iii, and v) were colored cyan, yellow, and green, respectively. b, immunoblotting analysis of wild-type and fluorescence protein fused-mGluR8 is shown. The same amounts of membrane expressing wild-type and mutants of mGluR8 were electrophoresed with (+Me) or without (−Me) β-mercaptoethanol and probed with Rho 1D4 (upper) and anti-GFP antibody (lower). Because the epitope for 1D4 antibody at the C terminus was replaced by fluorescent proteins in m8-C4V4, no band was observed in the upper panel. The lack of fluorescent proteins in wild-type (WT) caused the lack of a detectable band in the lower panel. In the upper panel, an upward shift of bands in mutants compared with WT was observed, reflecting the fusion of fluorescence proteins. c, emission spectra of m8-C3V3 samples excited at 433 nm in 0, 1, 4, 10, 40, 100, and 400 μm, 1 mm, and 10 mm l-glutamate are shown. Bleed-through shows the emission spectrum of Cerulean. All the spectra were normalized by the fluorescence intensity at 480 nm. d, shown are difference spectra between the emission spectra of m8-C3V3 sample and bleed-through in c. Cross-talk shows the emission spectrum of Venus in the m8-C3V3 sample directly excited at 433 nm, which can be calculated from the emission spectrum of the m8-C3V3 sample excited at 500 nm referring to the emission spectra of Venus excited at 433 and 500 nm. All the difference spectra were normalized at the peak value of cross-talk. e, are shown dose-response plots of E values of m8-C3V3 with (open circles) or without (closed circles) 10 μm LY341495. Because LY341495 was solubilized in ethanol, ethanol (5% final concentration) was added instead of LY341495 as a control. Data are expressed as mean ± S.D. of more than three experiments.
We prepared the cell membranes of HEK293 cells containing mGluRs using sucrose flotation and washed them three times with low salt buffer to remove cell surface proteins and riboflavin and to suppress autofluorescence. We quantified the amount of expression of Cerulean- or Venus-fused mGluRs in co-expressing samples based on the fluorescence intensity of Cerulean or Venus, referring to the ratio between the fluorescence intensity of the sample containing only m8-C4 or -V4 and its absolute amount estimated from the ligand binding assay (see “Experimental Procedures”). Using the above methods, we were able to obtain the active proteins of all the mutants except for m1-C3 and m1-V3, but the expression levels varied depending on the positions of the inserted fluorescent protein (Table 1 and 2). The total expression levels of mGluR8 mutants quantified by fluorescence intensity were consistent with those seen by immunoblotting analysis (Fig. 2b).
TABLE 1.
Amount of expression of Cerulean- or Venus-fused mGluRs in co-expressing “homo” samples
ND, not determined.
| mGluR mutants | Cerulean |
Venus |
||||
|---|---|---|---|---|---|---|
| mGluR1 | mGluR3 | mGluR8 | mGluR1 | mGluR3 | mGluR8 | |
| C1V1a | 22.1 ± 3.00b | 7.34 ± 0.33 | 5.62 ± 0.91 | 21.4 ± 3.8 | 9.80 ± 0.46 | 4.88 ± 1.00 |
| C2V2 | 37.2 ± 1.47 | 28.6 ± 5.66 | 27.6 ± 2.00 | 22.3 ± 1.29 | 26.9 ± 4.76 | 16.8 ± 1.34 |
| C3V3 | ND | 27.2 ± 3.96 | 34.4 ± 3.16 | ND | 27.5 ± 3.22 | 25.7 ± 1.75 |
| C4V4 | 28.9 ± 2.05 | 35.3 ± 1.71 | 44.5 ± 1.18 | 22.4 ± 1.68 | 29.6 ± 7.7 | 42.2 ± 5.02 |
a C1V1∼C4V4 indicate the loop region of Cerulean or Venus insertion.
b The amount of expression (pmol/mg of total protein) of Cerulean- or Venus-fused mGluRs in co-expressing samples was estimated from emission spectra of each sample, calculated by referring to the ratio between the emission peak intensity of only m8-C4- or -V4-expressing sample, and its absolute amount was estimated using the ligand binding assay (see “Experimental Procedures”), in which the amount of expression of wild-type mGluR8 was 63 pmol/mg of total protein. Each amount of expression is expressed as the mean ± S.D. of three experiments.
Spectra FRET Analysis in m8-C3V3
The spectral change of the fluorescence emission of m8-C3V3 was observed when this protein was excited with 433-nm light in the presence of various concentrations of glutamate (Fig. 2c). Fig. 2d shows a series of the difference emission spectra between m8-C3V3 and bleed-through. These spectra include the enhanced emission that resulted from FRET (ΔR) and the cross-talk signal derived from direct excitation of Venus (R0). R0 was estimated from the emission spectra of m8-C3V3 irradiated at 500 nm to excite only Venus, referring to the emission spectra of Venus excited at 433 and 500 nm. According to a previous report (17), apparent FRET efficiency (Eapp) can be calculated as,
 |
in which ϵC and ϵV are the molar extinction coefficients of Cerulean (43000) and Venus (1100) at 433 nm, respectively. R0 is an index of the total amount of Venus in m8-C3V3, which includes the heterodimer composed of m8-C3 and m8-V3 (CV) and the homodimer of m8-V3 (VV). Thus, Eapp is underestimated in comparison with net FRET efficiency (E). E can be calculated according to the equation,
 |
in which cv and vv are the molar ratio of CV and VV, respectively, in the samples co-expressing m8-C3 and m8-V3. We estimated E from Eapp based on the hypothesis that random dimerization without discrimination between Cerulean- and Venus-fused mGluRs occurs in the co-transfected cells. In that case, E can be calculated as,
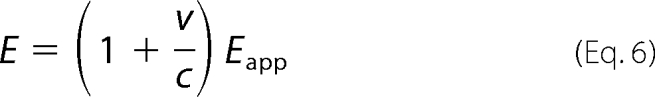 |
in which v/c is the molar ratio between m8-V3 and m8-C3 in the co-expressing sample (Table 1). Fig. 2e shows the profile of E of m8-C3V3 measured at various concentrations of glutamate with or without LY341495, a competitive antagonist of mGluRs in groups II and III. In the absence of LY341495, E was elevated in a glutamate-dependent manner (EC50, 26 ± 3.7 μm). In contrast, the glutamate-induced increase of E was suppressed by the addition of LY341495 (EC50, >10 mm). Because E depends on the inverse sixth power of the distance between donor and acceptor (25), these results demonstrated that the third intracellular loops in the mGluR8 dimer move closer to each other upon agonist binding.
Comparative FRET Analysis of mGluR
We also prepared samples co-expressing two mGluR mutants that had Cerulean or Venus in the corresponding intracellular region (C1V1, C2V2, C3V3, and C4V4) and compared the alteration of their FRET efficiencies among the three mGluR subtypes (Fig. 3a). Interestingly, we observed that the relative relationship of E of the co-expressed samples was similar among the three mGluR subtypes and that all the samples except for m1-C4V4 and m8-C4V4 showed a significant elevation of E in a glutamate-dependent manner (p < 0.03; Student's t test, two-tailed). These results suggested that the three mGluR subtypes share a similar arrangement of the two TMDs of the mGluR dimer and that the two TMDs move closer to each other after the dimeric conformational change of ECDs. Although E of m1-C3V3 could not be determined because of the low expression, E of the m3- and m8-C3V3 samples were significantly larger than those of the C1V1, C2V2, and C4V4 samples (p < 0.01; Student's t test, two-tailed). These results demonstrated that the relative distance between the two i3s in mGluRs is shorter than that between other respective loop regions.
FIGURE 3.

Comparative spectra FRET analysis of mGluRs. a, shown is a summary of glutamate-induced changes in FRET efficiency of C1V1 (blue curve), C2V2 (green curve), C3V3 (yellow curve), and C4V4 (red curve) samples. E values of mGluR1, mGluR3, and mGluR8 samples are shown using squares, triangles, and circles, respectively. b, shown is a comparison of the dose-response plot between m8-C1V2 (blue closed circles), m8-C1V3 (green closed squares), and m8-C2V3 (brown closed triangles). c, shown is a comparison of the dose-response plot between m8-C2V1 (blue open circles), m8-C3V1 (green open squares), and m8-C3V2 (brown open triangles).
Next, we prepared a series of the samples co-expressing other pairs of mGluR8 mutants that had Cerulean or Venus in the intracellular region (m8-C1V2, -C2V1, -C1V3, -C3V1, -C2V3, and -C3V2) to examine the dimeric arrangement of mGluR in more detail. The E of m8-C2V3 was larger than those of m8-C1V2 and -C1V3 (Fig. 3b) (p < 0.01; Student's t test, two-tailed). When the fluorescent proteins were swapped in these pairs (m8-C2V1, -C3V1, and -C3V2), the E of the swapped pairs were not completely identical with those of the original ones (Fig. 3c). However, m8-C3V2 exhibited higher E compared with those of m8-C2V1 and -C3V1 (p < 0.03; Student's t test, two-tailed). Moreover, the dose-dependent changes in E of the mGluR8 samples generally showed glutamate-induced elevation of FRET efficiencies, which supports the model of the agonist-dependent approach of the two TMDs. Moreover, the values of E of m8-C2V3 and -C3V2 were followed by that of m8-C3V3, the largest one among the mGluR8 samples shown in Fig. 3a (p < 0.03; Student's t test, two-tailed). This observation strongly suggests that i3 of one subunit is adjacent to the i2 of the other subunit of the mGluR8 dimer.
FRET Analysis of mGluR8 R75A Mutants
Next, to analyze the effect of defects in ligand binding on the conformational changes of the TMD in the dimer, we introduced the R75A mutation into m8-C3V3, which had the largest change in E among m8-C1V1, -C2V2, -C3V3, and -C4V4 (Fig. 3a). The arginine residue of mGluR1 at the corresponding position is located in the ligand binding pocket in the crystal structure of ECD (5), and substitution by alanine at this position in mGluR4 severely impairs the ligand binding ability (26). In mGluR8, the R75A mutation also caused loss of ligand binding and G protein activation abilities (Fig. 4, a and b, respectively). The R75A mutation into m8-C3V3 did not affect the subcellular distribution (Fig. 4c) or dimerization (Fig. 4d) of the receptor. The FRET analysis for this mutant clearly showed that the R75A mutation caused severe suppression of the glutamate-induced increase of the E value (R75A homo in Fig. 4g). The effect of this mutation was similar to the antagonizing effects of LY341495 (Fig. 2e).
FIGURE 4.

Spectra FRET analysis of m8-C3V3 R75A mutants. a, ligand binding potency of R75A mutant of mGluR8 is shown. [3H]LY341495 binding to wild-type and R75A mutant-expressing HEK293 cell membranes was measured. Results are presented as the percentage of total specific binding of wild-type. b, shown is the initial rate of the G protein activation ability of the R75A mutant of mGluR8. [35S]GTPγS binding to G protein was measured in the presence of 1 mm l-AP4. Results are presented as the percentage of total specific binding of wild-type. c, shown are fluorescence and difference interference contrast images of HEK293 cells expressing R75A-C3 (i and ii), R75A-V3 (iii and iv), and R75A-C3V3 samples (v and vi). The fluorescence images (i, iii, and v) are shown in gray scale. d, immunoblotting analysis of samples coexpressing WT-C3 (V3) and R75A-V3 (C3). The same amounts of membranes co-expressing WT-C3 (V3) and R75A-V3 (C3) were electrophoresed with (+Me) or without (−Me) β-mercaptoethanol and probed with Anti-GFP antibody. The amount of cDNA in the co-transfection is indicated under the each lane (μg/100-mm dish). e, shown is the amount of expression (pmol/mg of total protein) of Cerulean- or Venus-fused mGluR8 in coexpressing samples estimated from emission spectra of each sample. The amounts of Cerulean, Venus, and total expression are indicated by white, gray, and black bars, respectively. The amount of cDNA in the co-transfection is indicated under each bar (μg/100-mm dish). f, plots of Eapp versus molar ratio between Cerulean and Venus (c/v ratio) in WT+R75A hetero samples with (closed squares) or without (open circles) 10 mm l-glutamate. The c/v ratio was estimated from the values shown in e. The curves were fitted to Equation 8. The inset shows the reciprocal plots fitted to Equation 9. g, comparison of the dose-response plot of FRET efficiency among WT-homo (open circles), WT+R75A hetero (closed triangles), and R75A-homo (open squares) is shown. All the data are expressed as mean ± S.D. of more than three experiments.
We also analyzed WT+R75A heterodimer samples, in which only one subunit of mGluR dimer had the mutation in the ECD. When the same amounts of the plasmid DNA of WT-C3 (or -V3) and R75A-V3 (or -C3) were co-transfected into HEK293 cells, the expression of R75A-V3 (or -C3) was too severely suppressed for the measurement of FRET (data not shown). Therefore, we reduced the amount of the plasmid DNA of WT-C3 (or -V3) to 1/4 or 1/10 compared with that of R75A-V3 (or -C3). In these conditions, the dimerization of the receptors was intact (Fig. 4d), and the c/v ratio was varied depending on the amount of cDNA transfected (Fig. 4e). The mechanism of the suppression of the expression yield of R75A mutant by co-expression with wild-type is not clear at present. Because it is questionable whether one can assume the random dimerization of WT-C3 (V3) and R75A-V3 (C3), we examined it by analyzing the correlation between Eapp and v/c ratio. The following equation is a generalized form of Equation 6 considering that the probability of formation of VV is n times higher than that of CV when the collision rate of all protomers is equally possible.
 |
Namely, n should be one if the assumption of random dimerization holds true for WT+R75A heterodimer. Fig. 4f shows the relationship between Eapp and the c/v ratio of these samples. The curves were well fitted with the following equations, which are variants of Equation 7 (see Fig. 4f and the inset).
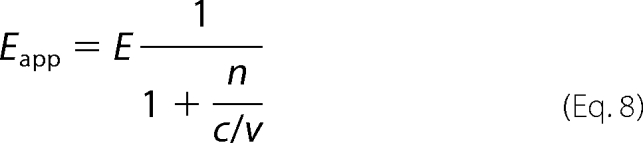 |
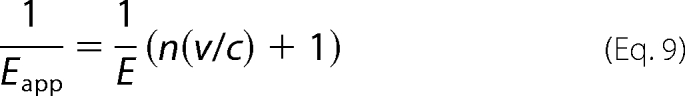 |
In the fitting curves, the values of n were almost 1 (1.06 ± 0.03 for 0 mm glutamate and 1.01 ± 0.07 for 10 mm glutamate) suggesting that the assumption of random dimerization is true for WT+R75A heterodimer.
Fig. 4g shows the E of WT+R75A heterodimer at various concentrations of glutamate that were estimated from the experiments shown above. When there was a defect of ligand binding ability in one protomer, a significant increase of E in a glutamate-dependent manner was observed in the WT+R75A heterodimer (p < 0.001; Student's t test, two-tailed), but the extent of the increase was about 4 times smaller than that for the WT homodimer. Although E theoretically has an inverse sixth power relationship with distance, it can be approximated that there is a linear relationship between E and the distance in this range (25). Thus, the change of mean distance in the WT+R75A heterodimer was also four times smaller than that in the WT homodimer. If the bindings of glutamates to two sites in the dimeric ECDs occur independently and each binding promotes the dimeric rearrangement of TMD at constant magnitude, about half of the increase in E observed in the WT homodimer should be observed in WT+R75A heterodimer. Thus, these results strongly suggested that two glutamate bindings acted synergistically on the conformational change in the dimeric TMDs. It is notable that the values of E were almost the same among WT homodimer, WT+R75A heterodimer, and R75A homodimer in the absence of glutamate, suggesting that R75A mutation did not alter the dimeric conformation of TMDs in the resting state. In addition, the heterodimer exhibited an EC50 (29 ± 7 μm) about half that of the WT (61 ± 11 μm), indicating weak negative cooperativity of glutamate binding to the dimeric ECD (27).
DISCUSSION
Distribution of CAM Sites in the TMD of mGluR8
In the present and previous studies (13), we identified 14 CAM sites in the TMD of mGluR8 from the comprehensive alanine scanning mutagenesis, and among them six sites were clustered in helix V (Fig. 1). To assist with the interpretation of the positional relationship of the cluster of CAM sites, we constructed a three-dimensional model of TMD of mGluR8, which is based on the crystal structure of bovine rhodopsin (Protein Data Bank code: 1U19) by using SWISS-MODEL (18, 19) as described under “Experimental Procedures.” It should be noted that the alignment between rhodopsin and family 3 GPCRs for construction of the present model (Fig. 5a) was identical to that in previous reports (21–24), in which the models created were validated by site-directed mutagenesis of presumed binding sites of allosteric ligands. Although our model has low accuracy for discussion of the amino acid interaction in detail, which is partly because of the low homology between rhodopsin and family 3 GPCRs, it would be useful for speculation about the helical arrangements in each protomer. In the created model, a hot spot of CAM sites was predicted in the space among helices III, V, and VI (Fig. 5c). In this region, Thr-764 in helix V is oriented to helix III, consistently with our previous observation (13), and other CAM sites in helix V are distributed on two different surfaces (Fig. 5, b and c). Namely, three CAM sites (Ile-757, Met-760, and Val-761) are located one or two helical turns above the Thr-764 oriented toward helices III or VI, but the other two (Thr-762 and Tyr-766) are located on the opposite surface of Thr-764 oriented to the lipid bilayer membrane. These results suggested that the CAM sites oriented toward the lipid bilayer should be involved in the switching between the resting and active states of the receptor through a direct or indirect interaction with the other protomer in the mGluR dimer.
The Dimeric Arrangement of mGluR in the Resting and Active States
Subsequently, the comprehensive FRET analysis of mGluR8 revealed some details of the dimeric arrangement of TMDs. In contrast with the previous FRET analysis on mGluR1, in which there was no information about i3 (28), here we successfully obtained active proteins of all mGluR8 mutants having the fluorescent protein in each intracellular loop region (Tables 1 and 2). The most straightforward interpretation of the data shown in Fig. 3 is that the distance between i2 and i3 (i2-i3) is the shortest, and the next shortest is that between i3-i3. If the helical arrangement in one subunit of mGluR dimer is similar to that of bovine rhodopsin (as shown in Fig. 5) and if two TMDs form a symmetric dimer, the dimeric arrangement can be speculated to be that as depicted in Fig. 6. Because i3 of one subunit is adjacent to i2 and i3 of the other protomer, helix V should be the closest part in TMD dimer. Furthermore, comparative FRET analyses of the three mGluR subtypes showed that the arrangement of the TMDs in the mGluR dimer is basically common to all of the mGluR subtypes (Fig. 3a).
TABLE 2.
Amount of expression of Cerulean- or Venus-fused mGluR8 in co-expressing samples
| mGluR mutants | Cerulean |
Venus |
||||
|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C1 | C2 | C3 | |
| V1a | 5.62 ± 0.91b | 16.2 ± 1.95 | 27.1 ± 2.97 | 4.88 ± 1.00 | 3.58 ± 0.26 | 3.36 ± 0.11 |
| V2 | 3.45 ± 0.70 | 27.6 ± 2.00 | 26.1 ± 5.45 | 5.37 ± 0.78 | 16.8 ± 1.34 | 11.2 ± 1.56 |
| V3 | 4.79 ± 0.59 | 18.8 ± 3.05 | 34.4 ± 3.16 | 11.8 ± 0.49 | 18.5 ± 2.00 | 25.7 ± 1.75 |
a C1∼C4 and V1∼V4 indicate the loop region of Cerulean or Venus insertion.
b The amount of expression (pmol/mg total protein) of Cerulean- or Venus-fused mGluR8 in co-expressing samples was estimated from emission spectra of each sample, calculated as described in Table I. Each amount of expression is expressed as the mean ± S.D. of three experiments.
FIGURE 6.

Hypothetical model of arrangement of TMD dimer and its rearrangement in the activation process. The model is viewed from the intracellular side. The helical arrangement within one subunit of mGluR dimer is based on the x-ray structure of bovine rhodopsin. The two TMDs are arranged based on the relative relationships of the distances between each loop region estimated from the rank of FRET efficiencies.
The overall observations of the glutamate-induced elevation in FRET efficiency demonstrated that the two TMDs moved closer to each other after the dimeric conformational change of the ECDs (Fig. 3). This agrees with the observation that the distance between the C-terminal ends of the dimeric ECDs in the active conformation is shorter than that in the resting conformation in the crystal structures (5, 6). The comparison of the values of E of the mGluR8 samples before and after agonist stimulation showed that the relative relationship of the values of E is conserved between the resting and active states (Fig. 3). This suggests that the dimeric arrangement is not changed significantly during the activation process. Thus, a local relocation of helix V appears to be sufficient to regulate the activation of mGluRs.
Taken together, the present results suggest that helix V is involved in the process of activation of mGluRs via an interprotomer interaction of TMDs. It is an open question whether or not a direct contact between TMDs exists. However, if one can assume that there is a direct contact at least in the active state, helix V is the most likely candidate for the dimeric interface. The elucidation of the role of CAM sites at positions 762 and 766 in the active conformation of mGluR8 will help to clarify the possible dimeric interface of the TMDs in the future.
The previous evolutionary trace analysis on ∼700 TMDs of GPCRs predicted that the functionally important residues are clustered in the external faces of helices V and VI in each family of GPCRs (1). In the case of family 1 GPCRs, helices IV and V were proposed to form a dimeric interface in rhodopsin based on analyses using atomic force microscopy (29), FRET and cross-linking studies (30). Moreover, helices I, IV, and V were identified in serotonin 5HT2c receptor by cross-linking analysis, and the interface composed of helices IV and V was shown to be sensitive to the activation state of the receptor (31). Recently, the crystal structure of chemokine CXCR4 demonstrated that the helices V and VI form a dimeric interface (32). The present results are generally compatible with those reports and do not rule out the possibility that helix IV or VI also participates in the dimeric interface together with helix V. Especially, helix VI might participate in the dimeric interface, as a cluster of CAM sites was also identified previously at the middle of helix VI (Figs. 1a and 5).
Differences between One and Two Glutamate Binding States in the Conformation and Function of mGluR8
According to the crystal structures of dimeric ECDs, a model was proposed in which glutamate bindings modulate the dynamic equilibrium between resting and active conformations, which were defined by the dimeric interface (7). Thus, it can be presumed that the dimeric TMDs are also in equilibrium between resting and active states. However, it is less certain how the ratio of the two states is changed by the first and second bindings of glutamates. We analyzed the effect of the R75A mutation on the dimeric rearrangement of mGluR8 to elucidate the functional coupling of the conformations of the ECDs and TMDs. R75A+WT heterodimer showed about a 4-fold lower glutamate-induced increase of FRET efficiency in comparison with WT homo-dimer (Fig. 4g), suggesting the synergistic effect of the binding of two glutamates on the conformational change of the dimeric TMDs. Namely, the first binding of glutamate induces only 25% elevation of the population of active conformations, and the second binding results in 100% elevation. These results are in accord with a previous report, demonstrating that agonist binding to one ECD of a dimer induces partial activation and that binding to both ECDs is required for full activity (33). In other words, the relative population of active states defined by the dimeric arrangement in the mGluR dimer is well correlated with the ability to activate G protein. Taken together with the negative cooperativity of glutamate binding to the dimeric ECDs (27), the synergy of the two glutamate bindings should accentuate the contrast between the effects of low and high concentrations of glutamate.
A Possible Model for the Mechanism of Activation of mGluR
It is an open question how the dimeric rearrangement of the TMD of mGluR is related to G protein activation. Several lines of evidence obtained from structural, spectroscopic, and biochemical analyses of bovine rhodopsin have clearly shown that light-induced helical movements within the monomer cause exposure of the site that interacts with G protein (34). Although intra-protomer helical movement in mGluR1 dimer was not detected in a previous FRET analysis (28), our previous studies indicated that rearrangement of the cytoplasmic regions within one subunit of mGluR dimer is essential for the G protein activation (12). In addition, ECD-deleted mGluR5 can function as a rhodopsin-like GPCR when exposed to allosteric modulators that directly bind within the TMD of each subunit (35). These observations suggest that intra-protomer helical movement in the TMD of mGluR is important for efficient G protein activation, just as it is for rhodopsin-like GPCRs.
Taking all these facts together, the mechanism of activation of mGluR can be summarized as follows. Glutamate binding to the ECDs stabilizes the active conformations of the ECDs in the dimer, which triggers dimeric rearrangement of the TMD and structural changes within the TMD of each protomer. These two events should be closely correlated to each other through the reorganization of the dimeric interface possibly composed of helix V (Fig. 6). The relocation of the dimeric arrangement would cause modulations of intra-protomer interactions. The changes of interaction between Thr-764 in helix V and Tyr-663 in helix III is one of the possible candidates of such modulations. The exposure of interaction sites for G protein would follow after these events.
In conclusion, comprehensive alanine-scanning mutagenesis and FRET analysis provided novel insights into the mechanism of activation of mGluRs. That is, helix V could be involved in the inter-protomer communication in the TMD of mGluRs, and the binding of two glutamates to the dimeric ECDs synergistically promotes the dimeric rearrangement of TMD. Further combinations of FRET and mutational analyses will clarify the structure-function relationships of mGluRs.
Acknowledgments
We thank Prof. J. Nathans for providing the HEK293S cell line, Prof. R. S. Molday for the generous gift of the Rho 1D4-producing hybridoma, Prof. H. Niwa for the pCAG-GS vector, Prof. D. W. Piston for the clone of mCerulean, and Prof. A. Miyawaki for the clone of Venus. We are also grateful to Dr. Y. Imamoto for very helpful discussions and Dr. E. Nakajima for critical reading of our manuscript and invaluable comments.
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology (MEXT, Japan) (Grants-in-aid for Scientific Research 20227002 and 21026019 and the Global Center of Excellence Program “Formation of a Strategic Base for Biodiversity and Evolutionary Research; from Genome to Ecosystem” (Program A06)) and by Japan Society for the Promotion of Science Research Fellowship for Young Scientists Grant 08J01259.
- GPCR
- G protein-coupled receptor
- mGluR
- metabotropic glutamate receptor
- TMD
- transmembrane domain
- ECD
- extracellular domain
- i1
- the first intracellular loop
- i2
- the second intracellular loop
- i3
- the third intracellular loop
- i4
- C-terminal region
- CAM
- constitutively active mutation
- L-AP4
- L-(+)-2-amino-4-phosphonobutyric acid
- LY341495
- (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate.
REFERENCES
- 1. Dean M. K., Higgs C., Smith R. E., Bywater R. P., Snell C. R., Scott P. D., Upton G. J., Howe T. J., Reynolds C. A. (2001) J. Med. Chem. 44, 4595–4614 [DOI] [PubMed] [Google Scholar]
- 2. George S. R., O'Dowd B. F., Lee S. P. (2002) Nat. Rev. Drug Discov. 1, 808–820 [DOI] [PubMed] [Google Scholar]
- 3. Szidonya L., Cserzo M., Hunyady L. (2008) J. Endocrinol. 196, 435–453 [DOI] [PubMed] [Google Scholar]
- 4. Ferraguti F., Shigemoto R. (2006) Cell Tissue Res. 326, 483–504 [DOI] [PubMed] [Google Scholar]
- 5. Kunishima N., Shimada Y., Tsuji Y., Sato T., Yamamoto M., Kumasaka T., Nakanishi S., Jingami H., Morikawa K. (2000) Nature 407, 971–977 [DOI] [PubMed] [Google Scholar]
- 6. Muto T., Tsuchiya D., Morikawa K., Jingami H. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 3759–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsuchiya D., Kunishima N., Kamiya N., Jingami H., Morikawa K. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 2660–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brock C., Oueslati N., Soler S., Boudier L., Rondard P., Pin J. P. (2007) J. Biol. Chem. 282, 33000–33008 [DOI] [PubMed] [Google Scholar]
- 9. Seifert R., Wenzel-Seifert K. (2002) Naunyn Schmiedebergs Arch. Pharmacol. 366, 381–416 [DOI] [PubMed] [Google Scholar]
- 10. Smit M. J., Vischer H. F., Bakker R. A., Jongejan A., Timmerman H., Pardo L., Leurs R. (2007) Annu. Rev. Pharmacol. Toxicol. 47, 53–87 [DOI] [PubMed] [Google Scholar]
- 11. Yamashita T., Kai T., Terakita A., Shichida Y. (2004) J. Neurochem. 91, 484–492 [DOI] [PubMed] [Google Scholar]
- 12. Yamashita T., Terakita A., Kai T., Shichida Y. (2008) J. Neurochem. 106, 850–859 [DOI] [PubMed] [Google Scholar]
- 13. Yanagawa M., Yamashita T., Shichida Y. (2009) Mol. Pharmacol. 76, 201–207 [DOI] [PubMed] [Google Scholar]
- 14. Rizzo M. A., Springer G. H., Granada B., Piston D. W. (2004) Nat. Biotechnol. 22, 445–449 [DOI] [PubMed] [Google Scholar]
- 15. Nagai T., Ibata K., Park E. S., Kubota M., Mikoshiba K., Miyawaki A. (2002) Nat. Biotechnol. 20, 87–90 [DOI] [PubMed] [Google Scholar]
- 16. Terakita A., Takahama H., Tamotsu S., Suzuki T., Hariyama T., Tsukahara Y. (1996) Vis. Neurosci. 13, 539–547 [DOI] [PubMed] [Google Scholar]
- 17. Takanishi C. L., Bykova E. A., Cheng W., Zheng J. (2006) Brain Res. 1091, 132–139 [DOI] [PubMed] [Google Scholar]
- 18. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 19. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) Nucleic Acids Res. 37, D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Okada T., Sugihara M., Bondar A. N., Elstner M., Entel P., Buss V. (2004) J. Mol. Biol. 342, 571–583 [DOI] [PubMed] [Google Scholar]
- 21. Petrel C., Kessler A., Maslah F., Dauban P., Dodd R. H., Rognan D., Ruat M. (2003) J. Biol. Chem. 278, 49487–49494 [DOI] [PubMed] [Google Scholar]
- 22. Malherbe P., Kratochwil N., Knoflach F., Zenner M. T., Kew J. N., Kratzeisen C., Maerki H. P., Adam G., Mutel V. (2003) J. Biol. Chem. 278, 8340–8347 [DOI] [PubMed] [Google Scholar]
- 23. Malherbe P., Kratochwil N., Zenner M. T., Piussi J., Diener C., Kratzeisen C., Fischer C., Porter R. H. (2003) Mol. Pharmacol. 64, 823–832 [DOI] [PubMed] [Google Scholar]
- 24. Pin J. P., Galvez T., Prézeau L. (2003) Pharmacol. Ther. 98, 325–354 [DOI] [PubMed] [Google Scholar]
- 25. Stryer L., Haugland R. P. (1967) Proc. Natl. Acad. Sci. U.S.A. 58, 719–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hampson D. R., Huang X. P., Pekhletski R., Peltekova V., Hornby G., Thomsen C., Thøgersen H. (1999) J. Biol. Chem. 274, 33488–33495 [DOI] [PubMed] [Google Scholar]
- 27. Suzuki Y., Moriyoshi E., Tsuchiya D., Jingami H. (2004) J. Biol. Chem. 279, 35526–35534 [DOI] [PubMed] [Google Scholar]
- 28. Tateyama M., Abe H., Nakata H., Saito O., Kubo Y. (2004) Nat. Struct. Mol. Biol. 11, 637–642 [DOI] [PubMed] [Google Scholar]
- 29. Liang Y., Fotiadis D., Filipek S., Saperstein D. A., Palczewski K., Engel A. (2003) J. Biol. Chem. 278, 21655–21662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kota P., Reeves P. J., Rajbhandary U. L., Khorana H. G. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 3054–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mancia F., Assur Z., Herman A. G., Siegel R., Hendrickson W. A. (2008) EMBO. Rep. 9, 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kniazeff J., Bessis A. S., Maurel D., Ansanay H., Prézeau L., Pin J. P. (2004) Nat. Struct. Mol. Biol. 11, 706–713 [DOI] [PubMed] [Google Scholar]
- 34. Hofmann K. P., Scheerer P., Hildebrand P. W., Choe H. W., Park J. H., Heck M., Ernst O. P. (2009) Trends Biochem. Sci. 34, 540–552 [DOI] [PubMed] [Google Scholar]
- 35. Goudet C., Gaven F., Kniazeff J., Vol C., Liu J., Cohen-Gonsaud M., Acher F., Prézeau L., Pin J. P. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]