

Abstract
The DNA helicase encoded by gene 4 of bacteriophage T7 forms a hexameric ring in the presence of dTTP, allowing it to bind DNA in its central core. The oligomerization also creates nucleotide-binding sites located at the interfaces of the subunits. DNA binding stimulates the hydrolysis of dTTP but the mechanism for this two-step control is not clear. We have identified a glutamate switch, analogous to the glutamate switch found in AAA+ enzymes that couples dTTP hydrolysis to DNA binding. A crystal structure of T7 helicase shows that a glutamate residue (Glu-343), located at the subunit interface, is positioned to catalyze a nucleophilic attack on the γ-phosphate of a bound nucleoside 5′-triphosphate. However, in the absence of a nucleotide, Glu-343 changes orientation, interacting with Arg-493 on the adjacent subunit. This interaction interrupts the interaction of Arg-493 with Asn-468 of the central β-hairpin, which in turn disrupts DNA binding. When Glu-343 is replaced with glutamine the altered helicase, unlike the wild-type helicase, binds DNA in the presence of dTDP. When both Arg-493 and Asn-468 are replaced with alanine, dTTP hydrolysis is no longer stimulated in the presence of DNA. Taken together, these results suggest that the orientation of Glu-343 plays a key role in coupling nucleotide hydrolysis to the binding of DNA.
Keywords: ATPases, DNA-binding Protein, DNA Enzymes, DNA Helicase, DNA-Protein Interaction, DNA Replication
Introduction
Helicases are molecular motors that translocate unidirectionally along single-stranded nucleic acids using energy derived from nucleotide hydrolysis (1–3). The gene 4 protein (gp4)2 encoded by bacteriophage T7 is a bifunctional protein that contains both a helicase and primase domain. The helicase domain, located in the C-terminal half of the protein, is tethered to the primase domain, located in the N-terminal half, by a flexible linker. In the presence of dTTP gp4 oligomerizes to form hexamers and heptamers, a process involving the helicase domain and linker region. Gp4 binds, as a hexamer, to single-stranded DNA (ssDNA) in the presence of dTTP and translocates in a 5′ to 3′ direction along the DNA strand using the energy derived from the hydrolysis of dTTP (4–6). The ssDNA passes through the center of the hexamer and binding of residues within the central core to the DNA stimulates nucleotide hydrolysis, a phenomenon also observed for other members of the SF IV group of helicases (3). The nucleotide-binding sites are located at the interfaces between the subunits with dTTP being the preferred nucleotide for unwinding of duplex DNA (7). Upon binding of dTTP, the helicase recruits ssDNA into its central core (8–11).
Like the SF IV group of helicases, AAA+ ATPases (ATPases associated with diverse cellular activities) also form oligomeric structures (often hexamers) that form a ring-shaped structure with a central pore. These proteins function as a molecular motor that couples ATP binding and hydrolysis to changes in conformational states. These conformational changes are propagated through the assembly to enable the protein to bind to its substrate, to translocate on it, and to remodel the substrate (12). These proteins are involved in a range of processes, including protein degradation, membrane fusion, microtubule severing, peroxisome biogenesis, signal transduction, and regulation of gene expression. AAA+ ATPases are characterized by the presence of 200–250 amino acid ATP-binding domains that contain Walker A and B motifs. Studies on AAA+ ATPases identified a conserved link between the ligand binding and ATPase sites (13, 14). This link regulates the ATPase activity in response to the binding of target ligands. During ATP hydrolysis, a conserved catalytic glutamate activates a water molecule to attack the γ-phosphate. Otherwise, the glutamate is close to a conserved basic or polar residue. Significantly, this second residue is located within a secondary structure implicated in binding of the target ligands. Thus, binding of the ligand has the potential to influence the ATPase activity of these proteins via the polar residue. Accordingly, this polar or basic residue has been termed the glutamate switch (13). Analysis of the crystal structures of several AAA+ proteins containing ATP or ADP with or without DNA enabled a comparison of the position of the glutamate in different states (13).
To identify the glutamate switch in T7 helicase, we have analyzed the crystal structure of the helicase domain of T7 gp4 reported by Singleton et al. (8). The nucleotide-binding sites in the structure reveals three different states of nucleotide occupancy designated as NTP-binding competent, NDP-binding competent, and empty state. In such a crystal structure model it became possible to locate the position of the catalytic glutamate in response to the presence or absence of nucleotide (AMPPNP) (Fig. 1A). In the NTP binding state, the catalytic glutamate (Glu-343) interacts with the γ-phosphate of AMPPNP (Fig. 1B). However, in the empty state Glu-343 interacts with Arg-493 of helicase motif IV from the adjacent subunit (Fig. 1C). Helicase motif IV forms a loop that projects into the central channel of the hexamer, where mutational analysis suggests it contacts ssDNA (15). An interaction between Asn-468 and Arg-493 is critical for a functional helicase and these residues can be swapped without losing helicase activity. Arg-493 can be replaced with a lysine; however, when substituted with a glutamine, the altered protein loses the ability to bind DNA. This study suggests that hydrogen bonding between Arg-493 and Asn-468 from adjacent subunits is critical for the DNA binding ability of the T7 helicase (16). In the NTP binding state Arg-493 is displaced from the Glu-343 by 15 Å, but is sufficiently close (∼3 Å) to interact with Asn-468. In the empty state, Arg-493 interacts with Glu-343 (∼2.9 Å), but is displaced from Asn-468 by 10 Å. Thus Arg-493 may play a crucial role in nucleotide hydrolysis by controlling the positioning of Glu-343 between active and inactive status.
FIGURE 1.

Trans-interaction of Arg-493 with Glu-343 or Asn-468 in the nucleotide-binding site of T7 DNA helicase. A, the crystal structure of the helicase domain of T7 gp4 (Protein Data Bank accession code 1E0J) drawn in PyMOL. The structure reveals a ring-shaped molecule with a central core. The structure shows the location of a central β-hairpin (green), helicase motif Ia (cyan), and motif IV (blue) and their rearrangement in respect to the NTP bound or empty state of the nucleotide-binding pocket. B, a magnified view of the nucleotide bound state of the nucleotide-binding site shows the arrangement of the indicated amino acids in response to AMPPNP binding. C, a magnified view of the arrangement of the indicated amino acids in an empty site, devoid of a nucleotide in the nucleotide-binding site. Note that in the NTP bound state, Glu-343 interacts with the γ-phosphate. However, in the empty state Glu-343 interacts with Arg-493. Arg-493 is otherwise paired with Asn-468.
In the present study, we explore the possibility that Arg-493 acts as the glutamate switch in T7 DNA helicase. Toward this purpose we have altered Glu-343 (the catalytic glutamate) and Arg-493 (the proposed glutamate switch) in T7 DNA helicase. Biochemical characterization of the altered helicases along with the structural interpretation of the results provides evidence that Arg-493 acts as a glutamate switch to control ssDNA-dependent nucleotide hydrolysis, and nucleotide-dependent ssDNA binding.
EXPERIMENTAL PROCEDURES
Materials
Escherichia coli C600, E. coli HMS174(DE3), and pET11b were purchased from Novagen. Wild-type and gene 4-deficient T7 phage (T7Δ4-1) were from the laboratory collection. Oligonucleotides used in the assay to measure DNA binding (50-mer), to prepare helicase substrates (75-mer and 95-mer), and for mutagenesis of gene 4 were purchased from Integrated DNA Technologies. T4 polynucleotide kinase was purchased from New England Biolabs. The site-directed mutagenesis kit was purchased from Stratagene. All of the chemicals and reagents were from Sigma, unless otherwise mentioned.
Methods
Site-directed Mutagenesis, Protein Overproduction, and Protein Purification
Plasmid pET11gp4-63 was used for expression of gene 4 and overproduction of wild-type gp4 (17). This plasmid was also used to create point mutations using the QuikChange II mutagenesis kit (Stratagene) in accordance with the manufacturer's instructions. The sequence of primers used to construct various single and double mutations are available on request. Mutations were confirmed by DNA sequencing. Constructs harboring wild-type or genetically altered gene 4 were transferred to E. coli strain HMS-174(DE3) for overproduction of the corresponding proteins. Wild-type and altered gene 4 proteins were purified as described earlier (18). The purity was greater than 95% as judged by Coomassie staining of proteins after SDS-gel electrophoresis (supplemental Fig. S1).
Phage Complementation Assay
E. coli C600 containing a plasmid that expresses gene 4 under a T7 promoter was grown to an A600 of 1. Serially diluted T7 phage stocks were mixed with an aliquot of the E. coli culture in 0.7% soft agar and poured onto LB plates. After an incubation of 4–6 h at 37 °C, the number of plaques appearing on the plate were counted. The number of plaques formed by plasmids harboring wild-type gene 4 is normalized to 1. The relative efficiency of plating obtained with the mutated gene 4 constructs were determined by the number of plaques formed by the mutated gene 4 constructs divided by the plaque forming units of wild-type gene 4.
dTTP Hydrolysis Assay of Gp4
dTTP hydrolysis was carried out at 37 οC in a reaction mixture containing 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, 50 mm potassium glutamate, 100 nm wild-type or altered gp4, 5 nm M13 ssDNA and the indicated concentration of [α-32P]dTTP (18). After incubation for 30 min at 37 °C, EDTA was added to a final concentration of 250 mm to stop the reaction and then the reaction sample was kept on ice until spotting on TLC plate. [α-32P]dNDP, the product of hydrolysis, was separated from [α-32P]dNTP on TLC plates coated with polyethyleneimine in a solvent containing 0.5 m formic acid and 0.5 m lithium chloride. The TLC plates were scanned in a phosphorimager (Fuji) and the radiolabeled spots were measured by the help of ImageQuant software (FUJI). The data were further analyzed in GraphPad Prism software.
ssDNA Binding Assay
A nitrocellulose filter-binding assay was used to measure the ssDNA binding ability of wild-type or altered gp4. The reaction mixture (20 μl) containing 1 nm 5′-32P-labeled 50-mer oligonucleotide (5′-ATG ACC ATG ATT TCG ACG TTT TTT TTT TTG GGG ATC CTC TAA CCT GCG CA-3′), 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, 50 mm potassium glutamate, 1 mm β,γ-methylene dTTP or dTDP, and the indicated concentrations of wild-type or altered gp4 was incubated at 37 °C for 30 min. Then the reaction mixture was filtered through a nitrocellulose membrane laid above a ζ probe membrane in a dot-blot filtration apparatus. The quantity of protein-bound ssDNA and free ssDNA was measured by scanning the nitrocellulose and ζ probe membrane, respectively, in a phosphorimager.
Double-stranded DNA (dsDNA) Unwinding by Gp4
The dsDNA unwinding activity of gp4 was measured using a DNA substrate consisting of a 5′-32P-labeled 75-mer oligonucleotide partially annealed to a 95-mer oligonucleotide (18). The helicase substrate was prepared by annealing a 5′-32P-end-labeled 75-mer oligo (5′-CGC CGG GTA CCG AGC TCG AAT TCA CTG GCC GTC GTT TTA CAA CGT CGT GAC ATG CCT19-3′) with an unlabeled 95-mer oligo (5′-T39 GGC ATG TCA CGA CGT TGT AAA ACG ACG GCC AGT GAA TTC GAG CTC GGT ACC CGG CG-3′). Reaction mixtures containing 100 nm labeled DNA substrate, 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, 50 mm potassium glutamate, 5 mm dTTP, and the indicated concentrations of wild-type or altered gp4 were incubated at 37 °C for 10 min. The reaction mixture was then mixed with 5× stop buffer to a final concentration of 0.4% (w/v) SDS, 40 mm EDTA, 8% (v/v) glycerol, and 0.1% (w/v) bromphenol blue. Unwound single-stranded oligonucleotides were separated from the helicase substrate in a 10% non-denaturing polyacrylamide gel. Subsequently, the gel was scanned in a phosphorimager and analyzed in ImageQuant and GraphPad Prism software.
Protein Oligomerization
Gp4s were examined for their ability to oligomerize in the presence of a non-hydrolyzable dTTP analog (β,γ-methylene dTTP) or dTDP. The reaction mixtures (15 μl) containing 2 μm (monomer) gp4, 40 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 10 mm DTT, 50 mm potassium glutamate, 0.01 to 1 mm β,γ-methylene dTTP or dTDP, and 1 μm 50-mer ssDNA were incubated for 20 min at 37 °C. The reactions were stopped by the addition of glutaraldehyde to 0.033% (v/v). The reaction sample was kept at 37 °C for another 5 min, and the reaction products were analyzed on a non-denaturing 10% polyacrylamide gel using a running buffer of 0.25× TBE. After staining with Coomassie Blue, the oligomerization status of gp4 was determined by gel analysis as described (19, 20). The mobility of hexamers and heptamers were distinguished as described earlier (20). The density of protein bands corresponding to monomers and higher order oligomers in each lane were measured using the software AlphaEase FC (AlphaImager 3400). The fraction of hexamers formed from monomers was determined by the density of hexamers and higher order oligomers divided by the total density of protein bands in the corresponding lane. The fraction of hexamers formed was plotted against the concentration of β,γ-methylene dTTP or dTDP, and the data were fit to the Hill equation (Equation 1) to obtain the apparent dissociation constant (Kd) for hexamer formation (22),
where YL is the fractional saturation, [L] is the concentration of β,γ-methylene dTTP or dTDP, Kd is the apparent dissociation constant for the hexamer, and n is the Hill coefficient. The apparent Kd values of β,γ-methylene dTTP or dTDP to convert monomers to hexamers are compared for each of the altered proteins with wild-type gp4.
RESULTS
To examine the role of Arg-493 of T7 gp4 in helicase function we altered gp4 at this position and examined its function both in vivo and in vitro. Because, as described under the Introduction, Arg-493 interacts with catalytic Glu-343 in the absence of nucleotide we also replaced this residue in gp4.
Both Glu-343 and Arg-493 of Gp4 Are Required for T7 Phage Growth
In the present study, the catalytic glutamate at position 343 was replaced with aspartate (gp4-E343D), glutamine (gp4-E343Q), or asparagine (gp4-E343N). Plasmids containing the altered gene 4 constructs were transferred to E. coli C600 and examined for their ability to complement gene 4 function in supporting T7Δ4 phage growth. T7Δ4 phage, lacking gene 4, can grow only in E. coli cells harboring a plasmid that produces a functional gp4 (17). Earlier studies have shown that plasmids encoding for gp4-R493Q and gp4-N468A are unable to support growth of T7Δ4 (11, 16). We have constructed a double mutant gp4-R493A/N468A, where both Arg-493 and Asn-468 are substituted with alanine. We find that gp4-E343Q, gp4-E343D, gp4-E343N, and, as expected, gp4-R493A/N468A do not complement for T7Δ4 growth, suggesting that both Glu-343 and Arg-493 are essential for phage growth (Table 1). Gp4-R493A also does not complement the growth of T7Δ4 (data not shown).
TABLE 1.
Plating efficiency of T7Δ4 on E. coli strains containing plasmids expressing wild-type or genetically altered gp4
The ability of plasmids encoding gp4 variants to support the growth of T7Δ4 in E. coli was examined. The number of plaques formed by plasmids harboring wild-type gp4 is normalized to 1. The relative efficiency of plating obtained with the altered gene 4 constructs was determined by the number of plaques formed by the mutated gene 4 constructs divided by the plaque forming units of wild-type gene 4. The efficiency of plating of ≤10−9 corresponds to the gene 4 construct unable to complement the T7Δ4 growths in the host bacteria.
| pET11b:gp4 construct | Efficiency of plating T7Δ4 |
|---|---|
| Wild-type | 1 |
| E343Q | ≤10−9 |
| E343D | ≤10−9 |
| E343N | ≤10−9 |
| N468A | ≤10−9 |
| R493Q | ≤10−9 |
| R493A/N468A | ≤10−9 |
Hydrolysis of dTTP
To examine the role of Arg-493 as a glutamate switch, we measured the rate of dTTP hydrolysis catalyzed by gp4 with substitutions for Arg-493, in the presence or absence of M13 ssDNA. The presence of ssDNA stimulates the rate of dTTP hydrolysis ∼100-fold in the case of wild-type gp4 (6). However, in the absence of ssDNA there is a measurable hydrolysis of dTTP, a reaction that occurs randomly among the six subunits (10).
In the absence of ssDNA wild-type gp4 hydrolyzed dTTP at the rate of 0.22 μm s−1 (Fig. 2A, Table 2). Under similar reaction conditions, gp4 lacking the catalytic glutamate (gp4-E343D, gp4-E343Q, and gp4-E343N) does not hydrolyze dTTP, however, gp4-N468A, gp4-R493Q, and gp4-R493A/N468A hydrolyze dTTP, at rates comparable with wild-type gp4 (Fig. 2A and Table 2). Gp4-N468A hydrolyzes dTTP with a Vmax of 0.05 μm s−1, which is nearly 4-fold less than that of wild-type gp4 in the absence of ssDNA. Gp4-R493Q and gp4-R493A/N468A both hydrolyze dTTP in the absence of ssDNA with a maximal rate of 0.22 and 0.35 μm s−1, respectively (Table 2).
FIGURE 2.

dTTP hydrolysis activity in the absence and presence of ssDNA. dTTP hydrolysis assays were performed in 10-μl reactions containing the indicated concentrations of [α-32P]dTTP, 100 nm of either wild-type or altered gp4, and 1 nm circular M13 ssDNA where indicated. After incubation at 37 °C for 30 min, EDTA was added to stop the reaction. The reaction was spotted on TLC plates coated with polyethyleneimine and dTDP was separated from dTTP in a buffer containing 0.5 m LiCl2 and 0.5 m formic acid. The quantity of dTTP hydrolysis was calculated by measuring the intensities corresponding to [α-32P]dTDP and unhydrolyzed [α-32P]dTTP. Reaction kinetics was derived with the help of Michaelis-Menten equation in GraphPad Prism software and presented in Table 2. A, dTTP hydrolysis in the absence of ssDNA. B, dTTP hydrolysis in the presence of 1 nm M13 ssDNA. The values in the y axis are in log10 scale. Note that 1 nm M13 ssDNA could stimulate the rate of dTTP hydrolysis by wild-type gp4 up to 100-fold. C, effect of M13 ssDNA on dTTP hydrolysis catalyzed by gp4-R493A/N468A compared with wild-type gp4. The assays were carried out as described above, except that different concentrations of M13 ssDNA (0–5 nm) were added to the reactions containing 200 μm [α-32P]dTTP. The rate of dTTP hydrolysis by gp4-R493A/N468A (0.35 μm/s) and wild-type gp4 (0.22 μm/s) in the absence of M13 ssDNA were normalized to 1. The rates of dTTP hydrolysis in the presence of different concentrations of the M13 ssDNA were converted relative to the corresponding protein activity in the absence of DNA. Please note that in all cases less than 30% of the dTTP is hydrolyzed in 30 min for each of the concentrations examined.
TABLE 2.
dTTP hydrolysis activity of wild-type gp4 and gp4 variants
The rate of dTTP hydrolysis by wild-type gp4 and gp4 variants in the presence or absence of ssDNA are compared. dTTP hydrolysis reactions were performed as described under “Experimental Procedures.” Wild-type gp4 hydrolyzes dTTP approximately 90-fold faster in the presence of 1 nm circular M13 ssDNA as compared to the absence of DNA.
| Gp4-helicase | kcat, no DNA | Km | kcat/Km | kcat +M13 ssDNA | Km | kcat/Km |
|---|---|---|---|---|---|---|
| s−1 | mm | s−1mm1 | s−1 | mm | s−1mm1 | |
| Gp4-wild type | 0.22 ± 0.01 | 0.07 | 5.7 | 20 ± 0.9 | 2.6 | 7.7 |
| Gp4-E343Q | NDa | ND | ND | ND | ND | ND |
| Gp4-E343D | ND | ND | ND | ND | ND | ND |
| Gp4-E343N | ND | ND | ND | ND | ND | ND |
| Gp4-N468A | 0.05 ± 0.01 | ND | ND | 0.06 ± 0.02 | ND | ND |
| Gp4-R493Q | 0.22 ± 0.01 | 0.06 | 3.8 | 0.08 ± 0.008 | ND | ND |
| Gp4-R493A/N468A | 0.35 ± 0.01 | 0.07 | 5 | 0.05 ± 0.01 | ND | ND |
a ND, not determined.
Upon addition of ssDNA there is approximately a 100-fold stimulation of dTTP hydrolysis by wild-type gp4 (Fig. 2B, Table 2). Unlike wild-type gp4, the dTTP hydrolysis catalyzed by gp4-N468A, gp4-R493Q, and gp4-R493A/N468A is not stimulated by the addition of ssDNA (Fig. 2B and Table 2). The rate of DNA-dependent dTTP hydrolysis by gp4-R493A/N468A was also examined at different concentrations of ssDNA (0.5–5 nm) with a constant concentration of dTTP (200 μm). Increasing concentrations of ssDNA do not stimulate the rate of dTTP hydrolysis by gp4-R493A/N468A; instead the rate decreased severalfold, whereas the rate of dTTP hydrolysis by wild-type gp4 increased considerably (Fig. 2C). The results suggest that Arg-493 and Asn-468 are involved in the ssDNA-mediated stimulation of nucleotide hydrolysis.
Binding to ssDNA
We measured the ability of each protein to bind a 50-mer ssDNA in the presence of the non-hydrolyzable dTTP analog β,γ-methylene dTTP using a nitrocellulose DNA binding assay (Fig. 3A). The dissociation constants of wild-type and altered gp4 for the 50-mer oligonucleotide are shown in Table 3. Wild-type gp4 binds the oligonucleotide with a dissociation constant of 16 nm, similar to the binding of gp4-E343D (Kd of 18 nm) (Fig. 3A). Gp4-E343Q binds the ssDNA 7-fold less tightly (Kd of 111 nm) but still binds nearly 85% of the substrate at higher protein concentrations (250 nm). Gp4-E343N binds only 50% of the ssDNA even at the higher protein concentrations with a Kd of 140 nm (Table 3). Gp4-R493A/N468A binds ssDNA so poorly that we could not determine dissociation constant. This result was expected, because neither gp4-R493Q nor gp4-N468A bind ssDNA (11, 16).
FIGURE 3.

Effect of nucleotides on the binding of gp4 to ssDNA. Binding of T7 gp4 to ssDNA was measured in a nitrocellulose filter assay. Reactions contained 1 nm 5′-32P-labeled 50-mer oligonucleotide, 1 mm β,γ-methylene dTTP, 10 mm MgCl2, 40 mm Tris-HCl (pH 7.5), 10 mm DTT, 50 mm potassium glutamate, and the indicated concentrations of either wild-type or altered gp4. After incubation at 37 °C for 30 min, the reaction mixture was filtered through a nitrocellulose membrane atop a charged ζ probe membrane. Protein-bound DNA on the nitrocellulose membrane and unbound free DNA on the ζ probe membrane were measured by radioactive intensity in a PhosphorImager. The percentage of oligonucleotide bound by each of the proteins is plotted against the corresponding protein concentration. The dissociation constant (KD) calculated for each of the proteins using GraphPad Prism software is presented in Table 3. A, the graph shows the ability of wild-type and variants of gp4 to bind the 50-mer ssDNA in the presence of β,γ-methylene dTTP. B, the graph shows the ability of wild-type and variants of gp4 to bind 1 nm 50-mer ssDNA in the presence of dTDP. Note that wild-type gp4 and gp4-R493A/N468A are unable to bind ssDNA in this reaction. The error bars represent the S.D. obtained from three independent experiments.
TABLE 3.
ssDNA binding by gp4 variants
The dissociation constants for wild-type or altered gp4 were determined in a nitrocellulose DNA-binding assay. The reactions were carried out in a 10-μl volume containing a range of concentrations of gp4, 1 nm 5′-32P labeled 50-mer ssDNA, 1 mm β,γ−methylene dTTP or dTDP and incubated for 30 min at 37 °C as described under “Experimental Procedures.” The reaction mixture was filtered through a nitrocellulose membrane laid above a ζ probe membrane in a dot-blot filtration apparatus. The quantity of protein bound ssDNA and free ssDNA was measured by scanning the nitrocellulose and ζ probe membrane, respectively, in a PhosphorImager. The binding by the gp4 variants to the oligonucleotide in the presence of β,γ−methylene dTTP or dTDP were compared with the wild type activity.
| Gp4-helicase | Kd (β,λ−methylene dTTP) | Maximal binding | Kd (dTDP) | Maximal binding |
|---|---|---|---|---|
| nm | % | nm | % | |
| Gp4-Wild-type | 16 ± 3 | 100 | NDa | ≤2 |
| Gp4-E343Q | 111 ± 34 | 85 | 44 ± 10 | 90 |
| Gp4-E343D | 18 ± 4 | 85 | 108 ± 37 | 50 |
| Gp4-E343N | 140 ± 49 | 50 | 142 ± 55 | 50 |
| Gp4-R493A/N468A | ND | ≤2 | ND | ≤2 |
a ND, not determined.
Subsequently, we measured the ssDNA binding ability of wild-type and altered gp4 in the presence of dTDP. Glu-343 is involved in interacting with the γ-phosphate of the bound nucleoside triphosphate through a water molecule. Therefore, we measured the effect of dTDP on the DNA binding activity of gp4 with substitutions for Glu-343. In the presence of dTDP, wild-type gp4 does not bind ssDNA (Fig. 3B). Unlike wild-type protein, gp4-E343Q binds ssDNA considerably more tightly with a Kd of 44 nm. Similarly gp4-E343D and gp4-E343N also bind the oligonucleotide but with dissociation constants of 108 and 142 nm, respectively (Table 3). Gp4-E343D and gp4-E343N could bind only 50% of the ssDNA in these experiments; 2-fold less than gp4-E343Q (Fig. 3B). We did not observe ssDNA binding of gp4-R493A/N468A under any of the conditions in our study.
Taken together, these results demonstrate that gp4-E343D binds to ssDNA similar to wild-type gp4 in the presence of β,γ-methylene dTTP. The binding affinity of these proteins for ssDNA decreases in the presence of dTDP. Gp4-E343Q binds to ssDNA in the presence of dTDP or β,γ-methylene dTTP. Gp4-E43N binds poorly to ssDNA in the presence of either of the nucleotides.
Unwinding of DNA
A relatively short duplex DNA with a preformed fork (see inset to Fig. 4) at one end was used to measure the DNA unwinding activity of the proteins. To initiate unwinding of DNA gp4 requires a 5′-ssDNA tail onto which it assembles and a 3′-ssDNA tail to exclude that strand from the center of the helicase (7, 22). The latter strand is labeled with 32P at its 5′ terminus to detect the displaced strand on a polyacrylamide gel. As expected, wild-type gp4 unwinds the duplex (Fig. 4). Gp4-E343Q, gp4-E343D, gp4-E343N, all containing substitutions for the catalytic glutamate do not unwind the DNA. No unwinding activity was observed with gp4-R493A/N468A. Gp4-R493Q and gp4-N468A have been shown previously to be devoid of unwinding activity (11, 16). These results are not surprising because the Glu-343 altered proteins do not hydrolyze dTTP and the dTTP hydrolysis activity of gp4-R493A/N468A is not stimulated by ssDNA. These altered helicases do not support leading strand synthesis mediated by T7 DNA polymerase and helicase (data not shown).
FIGURE 4.
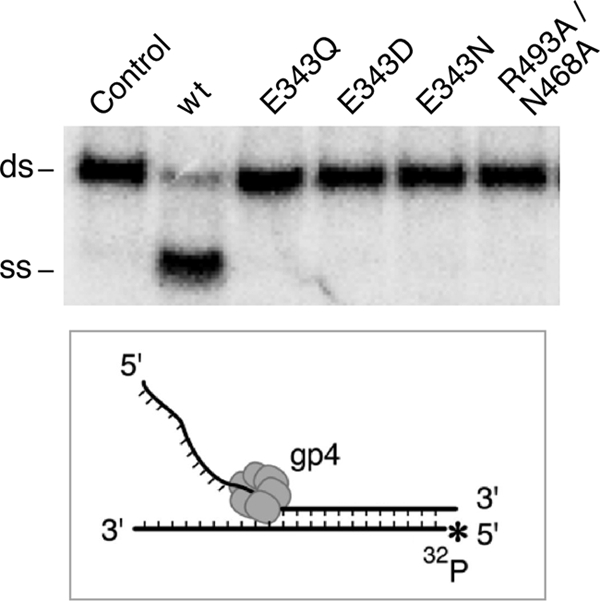
Unwinding of duplex DNA by T7 DNA helicase. The DNA used to measure the unwinding activity of gp4 was a short duplex DNA with a 5′- and 3′-ssDNA tail at one end of the molecule. The latter strand has a 32P label at its 5′ terminus. The DNA substrate was prepared by annealing a radioactive labeled 75-mer with an unlabeled 95-mer, generating a duplex region of 56 bp. Unwinding assays were carried out in a reaction containing 100 nm DNA substrate, 100 nm gp4, and 5 mm dTTP as described under “Experimental Procedures.” After incubation at 37 °C the reaction was stopped after 10 min. The unwound ssDNA was separated from the dsDNA substrate in a 10% non-denaturing polyacrylamide gel. The identity of each of the gp4 variants is indicated. The control reaction does not contain gp4. The separation of unwound ssDNA from the dsDNA substrate can be seen from the gel picture.
Oligomerization of Gp4
In the presence of dTTP, wild-type gp4 forms both hexamers and heptamers. Only hexamers are found to bind ssDNA (20). Nucleotides play an essential role in the formation of stable hexamers. Wild-type gp4 monomers or dimers convert to the hexamer species in the presence of dTTP (21). At saturated concentrations of β,λ-methylene dTTP and ssDNA, wild-type gp4 and altered gp4s do not show any significant difference in forming hexamers even at low concentrations of protein (supplemental Fig. S2). Furthermore, we have compared the ability of Glu-343 and Arg-493 altered helicases to form hexamers in the presence of different concentrations of β,λ-methylene dTTP or dTDP and saturated ssDNA (Fig. 5, A and B). We have plotted the fraction of hexamers and higher order oligomers formed for each of the helicases at various nucleotide concentrations in the presence of a fixed concentration of ssDNA (1 μm of 50-mer). The data were fit to the Hill equation and an apparent Kd was determined for each of the altered proteins in the presence of β,λ-methylene dTTP or dTDP (Fig. 5C). Wild-type gp4 predominantly forms hexamers and dodecamers in the presence of β,λ-methylene dTTP with an apparent Kd of 1.2 μm. The Kd value remains the same when dTDP is added to the reaction instead of β,λ-methylene dTTP. Gp4-E343Q exhibits a relatively higher Kd (6.8 μm β,γ-methylene dTTP and 4.9 μm dTDP) for hexamer formation. However, other altered helicases exhibit Kd values comparable with wild-type proteins for oligomerization (Fig. 5C). Wild-type gp4 predominately forms heptamers in the presence of dTDP (Fig. 5B). Unlike wild-type gp4, the gp4-E343D-altered proteins form hexamers to bind ssDNA in the presence of dTDP. Gp4-E343Q and gp4-E343N also form hexamers at relatively higher concentrations of dTDP and also bind ssDNA in the presence of 1 mm dTDP (Figs. 3B and 5B). Gp4-R493A/N468A forms both hexamers and heptamers in the presence of dTDP and forms predominately hexamers in the presence of β,λ-methylene dTTP. In the absence of nucleotides, all the proteins, including wild-type gp4, form a range of oligomeric forms from monomers to heptamers (data not shown).
FIGURE 5.

Oligomerization of gp4. A, oligomerization reactions contained 2 μm wild-type or altered proteins, 0.01–1 mm β,γ-methylene dTTP, and 1 μm 50-mer oligonucleotide and incubated at 37 °C for 20 min as described under “Experimental Procedures.” Glutaralderhyde was added to 0.033% to stabilize the oligomeric forms. The proteins were analyzed by electrophoresis on a 10% non-denaturing polyacrylamide gel. The state of the oligomerization of the proteins was determined after staining the gel with Coomassie Blue. The formation of hexamers, higher order oligomers, and lower order oligomers are shown in the gel picture. The identity of the protein is also indicated in the gel picture. B, the gel picture shows the oligomerization ability of wild-type and altered proteins in the presence of different concentrations of dTDP (0.01–1 mm). The experiments were carried out as described in A except that β,γ-methylene dTTP was replaced with dTDP. C, fraction of hexamers and higher order oligomers formed by each of the proteins in the presence of different concentrations of β,γ-methylene dTTP or dTDP are quantitated by the software AlphaEase FC (AlphaImager 3400). The data were fit to the Hill equation and provided an apparent Kd for β,γ-methylene dTTP or dTDP-dependent hexamer formation. The error bars represent the S.D. of the result from three independent experiments.
DISCUSSION
In the present study, we address the molecular mechanism for nucleotide-dependent ssDNA binding by the helicase domain of T7 gp4. Gp4 binds nucleotides in the nucleotide-binding pockets located at the subunit interfaces. The nucleotide is stabilized by several amino acids located in the active site. These residues reside in either of two subunits that form the nucleotide-binding site. Several amino acids including Lys-318 and Ser-319 (Walker motif A), Glu-343 of helicase motif Ia (catalytic glutamate), Asp-424 (Walker motif B), His-465 of helicase motif III (phosphate sensor), and Arg-522 from the adjacent subunit (arginine finger) are important players in the hydrolysis of dTTP. Walker motifs A and B are involved in coordinating a magnesium ion that promotes catalysis (23). The arginine finger plays a role in polarizing the γ-phosphate of dTTP to facilitate hydrolysis (8, 24). His-465 acts as a phosphate sensor and plays an important role in coupling nucleotide hydrolysis to DNA binding (11, 20). The catalytic glutamate polarizes a water molecule for in-line attack of the γ-phosphate of dTTP to initiate hydrolysis (8). Replacement of Glu-343 with glutamine eliminates dTTP hydrolysis but does not alter the affinity of the protein for ssDNA (10).
Gp4 forms heptamers and hexamers in the presence of dTTP. However, only hexamers are observed in the presence of ssDNA and the non-hydrolyzable β,γ-methylene dTTP. dTDP favors the presence of heptamers but does not promote ssDNA binding. The inability of dTDP to promote DNA binding suggests that the γ-phosphate of dTTP plays a major role in maintaining a conformation that binds DNA. In addition to the catalytic glutamate, the arginine finger and phosphate sensor also interact with the γ-phosphate of the bound nucleotide. In the crystal structure of T7 DNA helicase Glu-343 interacts with Arg-493 of helicase motif IV when there is no nucleotide in the nucleotide-binding site (empty state). Structural and biochemical evidence, in turn, suggest that Arg-493 interacts with Asn-468 of the central β-hairpin on the adjacent subunit and that this interaction is critical for DNA binding in the central core of the hexamer (16). These interactions led us to hypothesize that Glu-343 plays a role in controlling the DNA binding activity of the helicase in response to nucleotide binding in the nucleotide-binding pocket. When dTTP is present in the nucleotide-binding site (NTP bound state), Glu-343 interacts with the γ-phosphate of dTTP through a water molecule. As a result Arg-493 of the adjacent subunit is displaced from the nucleotide-binding site and positioned in the center of the ring. In the central core of the hexameric gp4, Arg-493 interacts with Asn-468 and facilitates ssDNA binding by the helicase (Fig. 1). In the presence of dTDP, the lack of γ-phosphate in the nucleotide probably facilitates the interaction of Glu-343 with Arg-493 of the adjacent subunit as it does in the empty state, thus interrupting the interaction of Arg-493 with Asn-468, which in turn disrupts DNA binding.
Our results also convincingly show that gp4-E343Q binds ssDNA in the presence of dTDP. As shown in the model (Fig. 6) a glutamine in the place of Glu-343 cannot make an interaction with the γ-phosphate of the bound nucleotide. The altered amino acid is also unable to make a hydrogen bridge with Arg-493 from the adjacent subunit in the empty state. As a result Arg-493 makes an interaction with Asn-468 in the center of the ring, which facilitates the altered helicase, gp4-E343Q in ssDNA binding. Thus in the presence of β,γ-methylene dTTP or dTDP gp4-E343Q binds ssDNA. Like a glutamate, an aspartate residue at position 343 can interact with the γ-phosphate of the bound nucleotide. Consequently, gp4-E343D retains wild-type levels of ssDNA binding in the presence of β,γ-methylene dTTP, but unlike wild-type gp4 the altered gp4 is unable to hydrolyze dTTP. This inability to hydrolyze dTTP may arise from a defect in activation of a water molecule for an inline attack to the γ-phosphate of dTTP. Unlike wild-type gp4, gp4-E343D exhibits ssDNA binding in the presence of dTDP. This binding in the presence of dTDP could arise from the ability of the aspartate at position 343 to make a weak interaction with Arg-493 at the nucleotide-binding site (Fig. 6). Furthermore, an asparagine at position 343 can only interact with Arg-493, but not with the γ-phosphate of dTTP. Thus, we see a significant loss of ssDNA binding by gp4-E343N in the presence of β,γ-methylene dTTP or dTDP. These results suggest that the interaction of Glu-343 with the γ-phosphate of dTTP (active) or Arg-493 (inactive) is a decisive factor in nucleotide-dependent DNA binding by the helicase (Fig. 6).
FIGURE 6.

Role of glutamate switch in the NTP-dependent ssDNA binding by T7 DNA helicase. Interaction of Arg-493 (glutamate switch) with either Glu-343 (catalytic glutamate) or Asn-468 (central β-hairpin) from the adjacent subunit depends on the nature of nucleotide in the nucleotide-binding site. The model is derived from our experimental results and the crystal structures of the helicase domain of T7 gp4 (PDB code 1E0J as presented in Fig. 1). Arrangement of the three indicated amino acid residues in response to the presence of either dTTP or dTDP in the nucleotide-binding site are depicted for wild-type gp4, gp4-E343Q, gp4-E343D, and gp4-E343N. Note that when Arg-493 interacts with Asn-468 of the neighboring subunit, the helicase binds ssDNA in its central core. In wild-type gp4, Glu-343 interacts with the γ-phosphate of dTTP. Arg-493 interacts with Asn-468 and the helicase binds ssDNA. In the presence of dTDP, Arg-493 interacts with Glu-343. An interruption in the Arg-493 and Asn-468 interaction makes the wild-type gp4 defective in ssDNA binding. In gp4-E343Q, Gln-343 neither interacts with the γ-phosphate of dTTP nor Arg-493. Consequently Arg-493 interacts with Asn-468 at the central core and facilitates the altered helicase, gp4-E343Q to bind ssDNA irrespective of the dTTP or dTDP present in the nucleotide-binding site. In gp4-E343D, Asp-343 can interact with the γ-phosphate of dTTP and Arg-493 interacts with Asn-468 at the central core of the helicase ring and facilitates ssDNA binding, but in the presence of dTDP, Arg-493 can interact with either Asp-343 or Asn-468. The choice of Arg-493 to interact with Asp-343 compromises the interaction of Arg-493 with Asn-468 in the central core leads to moderate ssDNA binding by gp4-E343D. In gp4-E343N, Asn-343 cannot bind the γ-phosphate of dTTP but can interact with Arg-493. Thus irrespective of the NTP present in the nucleotide-binding site, Arg-493 can interact with either Asn-343 or Asn-468 leading to the observed DNA binding.
In a hexameric structure of gp4, each subunit hydrolyzes dTTP independently in the absence of ssDNA. Our results show that gp4-N468A exhibits 4-fold slower dTTP hydrolysis activity compared with wild-type gp4. On the basis of our model (Fig. 6) we speculate that in the absence of ssDNA, interaction of Arg-493 with Glu-343 plays a crucial role in controlling dTTP hydrolysis activity in wild-type gp4. Therefore substitution of Asn-468 with Ala leads to a favorable interaction of Arg-493 with Glu-343 and thus reduces dTTP hydrolysis. However, substitution of Arg-493 would lead to a faster hydrolysis of dTTP because there would be no control on Glu-343 in the dTTP hydrolysis activity. In a previous study, it was shown that in T7 gp4, Arg-493 can be substituted with a lysine or a histidine with no significant loss in dTTP hydrolysis activity, ssDNA binding activity, dsDNA unwinding activity, and T7 DNA polymerase (gp5/trx)-mediated strand displacement DNA synthesis. Surprisingly, gp4-R493H is unable to complement gp4 function in vivo for supporting the growth of T7Δ4 phage in E. coli. In a similar case gp4-R493K could support the growth of T7Δ4 in E. coli. Additionally, substitution of Arg-493 with an asparagine, asparatate, or glutamate also leads to a severe defect in all the helicase-associated activities of gp4 both in vitro and in vivo. On the basis of our model (Fig. 6) we believe that a lysine or a histidine perhaps interacts with either Glu-343 or Asn-468, such as the Arg-493, and thus leads to no significant reduction in the observed activities in the corresponding altered proteins. Gp4-R493Q maintained only the basal level of dTTPase activity but did not exhibit activation in the dTTP hydrolysis activity in the presence of ssDNA.
During the unidirectional translocation of T7 DNA helicase on ssDNA the DNA strand is transferred from one subunit to an adjacent subunit in the central core of gp4 (8, 10). The transfer of DNA from one subunit to another is coupled to a sequential pattern of dTTP hydrolysis in the nucleotide-binding sites around the hexameric ring (8, 10). Because gp4-R493A/N468A binds poorly to ssDNA the mechanism by which this altered protein inhibits dTTP hydrolysis is not clear. However, a low affinity of gp4-R493A/N468A for ssDNA cannot be ruled out from our experiments. Results suggest that Arg-493 and Asn-468 play an important role in binding ssDNA. Thus, alteration of these residues abolishes the ssDNA-dependent stimulation of dTTPase activity. These data, in combination with the crystal structure of the helicase support the identity of Arg-493 as the glutamate switch. This glutamate switch not only controls ssDNA-dependent stimulation of dTTPase activity, but also plays a role in the nucleotide-dependent DNA binding.
Supplementary Material
Acknowledgments
We thank Steven Moskowitz (Advanced Medical Graphics) and Joseph Lee for illustrations. We give special thanks to Dr. Jaya Singh for valuable suggestions on the manuscript. We greatly appreciate the gift of gp4-R493Q from Seung Joo Lee. We are grateful to all the members of the Richardson lab for helpful discussions and constructive comments.
This work was supported, in whole or in part, by National Institutes of Health Grant GM54397 (to C. C. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- gp4
- gene 4 protein
- ssDNA
- single-stranded DNA
- dsDNA
- double-stranded DNA
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate.
REFERENCES
- 1. Lohman T. M., Bjornson K. P. (1996) Annu. Rev. Biochem. 65, 169–214 [DOI] [PubMed] [Google Scholar]
- 2. Matson S. W., Kaiser-Rogers K. A. (1990) Annu. Rev. Biochem. 59, 289–329 [DOI] [PubMed] [Google Scholar]
- 3. Patel S. S., Picha K. M. (2000) Annu. Rev. Biochem. 69, 651–697 [DOI] [PubMed] [Google Scholar]
- 4. Egelman E. H., Yu X., Wild R., Hingorani M. M., Patel S. S. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 3869–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim D. E., Narayan M., Patel S. S. (2002) J. Mol. Biol. 321, 807–819 [DOI] [PubMed] [Google Scholar]
- 6. Tabor S., Richardson C. C. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matson S. W., Tabor S., Richardson C. C. (1983) J. Biol. Chem. 258, 14017–14024 [PubMed] [Google Scholar]
- 8. Singleton M. R., Sawaya M. R., Ellenberger T., Wigley D. B. (2000) Cell 101, 589–600 [DOI] [PubMed] [Google Scholar]
- 9. Singleton M. R., Dillingham M. S., Wigley D. B. (2007) Annu. Rev. Biochem. 76, 23–50 [DOI] [PubMed] [Google Scholar]
- 10. Crampton D. J., Mukherjee S., Richardson C. C. (2006) Mol. Cell 21, 165–174 [DOI] [PubMed] [Google Scholar]
- 11. Satapathy A. K., Kochaniak A. B., Mukherjee S., Crampton D. J., van Oijen A., Richardson C. C. (2010) Proc. Natl. Acad. Sci. U. S. A. 107, 6782–6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith D. M., Benaroudj N., Goldberg A. (2006) J. Struct. Biol. 156, 72–83 [DOI] [PubMed] [Google Scholar]
- 13. Zhang X., Wigley D. B. (2008) Nat. Struct. Mol. Biol. 15, 1223–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mogni M. E., Costa A., Ioannou C., Bell S. D. (2009) Biochemistry 48, 8774–8775 [DOI] [PubMed] [Google Scholar]
- 15. Washington M. T., Rosenberg A. H., Griffin K., Studier F. W., Patel S. S. (1996) J. Biol. Chem. 271, 26825–26834 [DOI] [PubMed] [Google Scholar]
- 16. Lee S. J., Qimron U., Richardson C. C. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8908–8913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mendelman L. V., Notarnicola S. M., Richardson C. C. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 10638–10642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Satapathy A. K., Crampton D. J., Beauchamp B. B., Richardson C. C. (2009) J. Biol. Chem. 284, 14286–14295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Notarnicola S. M., Park K., Griffith J. D., Richardson C. C. (1995) J. Biol. Chem. 270, 20215–20224 [DOI] [PubMed] [Google Scholar]
- 20. Crampton D. J., Ohi M., Qimron U., Walz T., Richardson C. C. (2006) J. Mol. Biol. 360, 667–677 [DOI] [PubMed] [Google Scholar]
- 21. Picha K. M., Patel S. S. (1998) J. Biol. Chem. 273, 27315–27319 [DOI] [PubMed] [Google Scholar]
- 22. Ahnert P., Patel S. S. (1997) J. Biol. Chem. 272, 32267–32273 [DOI] [PubMed] [Google Scholar]
- 23. Sawaya M. R., Guo S., Tabor S., Richardson C. C., Ellenberger T. (1999) Cell 99, 167–177 [DOI] [PubMed] [Google Scholar]
- 24. Crampton D. J., Guo S., Johnson D. E., Richardson C. C. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 4373–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.