

Abstract
Hemoglobin (Hb) E (β-Glu26Lys) remains an enigma in terms of its contributions to red blood cell (RBC) pathophysiological mechanisms; for example, EE individuals exhibit a mild chronic anemia, and HbE/β-thalassemia individuals show a range of clinical manifestations, including high morbidity and death, often resulting from cardiac dysfunction. The purpose of this study was to determine and evaluate structural and functional consequences of the HbE mutation that might account for the pathophysiology. Functional studies indicate minimal allosteric consequence to both oxygen and carbon monoxide binding properties of the ferrous derivatives of HbE. In contrast, redox-sensitive reactions are clearly impacted as seen in the following: 1) the ∼2.5 times decrease in the rate at which HbE catalyzes nitrite reduction to nitric oxide (NO) relative to HbA, and 2) the accelerated rate of reduction of aquometHbE by l-cysteine (l-Cys). Sol-gel encapsulation studies imply a shift toward a higher redox potential for both the T and R HbE structures that can explain the origin of the reduced nitrite reductase activity of deoxyHbE and the accelerated rate of reduction of aquometHbE by cysteine. Deoxy- and CO HbE crystal structures (derived from crystals grown at or near physiological pH) show loss of hydrogen bonds in the microenvironment of βLys-26 and no significant tertiary conformational perturbations at the allosteric transition sites in the R and T states. Together, these data suggest a model in which the HbE mutation, as a consequence of a relative change in redox properties, decreases the overall rate of Hb-mediated production of bioactive NO.
Keywords: Crystal Structure, Hemoglobin, Nitric Oxide, Protein Chemistry, Protein Structure, Spectroscopy, HbE/beta-Thalassemia, Hemoglobin E, Nitrite Reductase, Redox-sensitive Reactions
Introduction
Hemoglobinopathies are inherited disorders of hemoglobin that arise from globin gene mutations, with over 90% due to point mutations. More than 1451 hemoglobinopathies and thalassemias have been identified and are listed in the HbVar database (1). Many of these mutations give rise to diverse molecular diseases, with unique pathogenesis (2). Although many hemoglobinopathies arise from altered oxygen-carrying properties and/or tetramer instability, there are also several hemoglobin variants that do not fall into any of the obvious standard categories. In such cases, the molecular basis for the pathophysiology is at best ambiguous.
The most common worldwide Hb mutation is HbE (β-Glu 26·Lys). This βE-globin allele imparts a selective advantage for resistance to malaria as does HbS and HbC (3, 4). Renewed interest in the mechanisms of HbE diseases and HbE/β-thalassemia originated with the wave of migration from Southeast Asia to Europe and North America (5). HbE hemoglobinopathies, including HbE/β-thalassemia, manifest a heterogeneous clinical spectrum, from asymptomatic to severe. HbEE homozygotes are asymptomatic with a very mild chronic anemia and microcytosis. This clinical presentation has been attributed to the minimally reduced expression of βE messenger RNA (6, 7) that originates from the generation of an alternative splice site created in the reading frame by the mutation of a single nucleotide substitution (GAG→AAG). The consequence of a reduction in βE-globin expression would lead to a small percentage of free α chains, known to be highly unstable.
In contrast to the mild clinical manifestations both of HbEE homozygotes, HbAE traits, and of β-thalassemia trait individuals (e.g. HbA/β-thalassemia), the heterozygous condition of HbE/β-thalassemia is probably the most common severe hemoglobinopathy worldwide, and it presents a diverse clinical spectrum with variations in anemia, growth, development, hepatosplenomegaly, and transfusion requirements (8–11). At birth, infants are asymptomatic, which is attributed to high HbF levels (12). Secondary morbid clinical manifestations arise, in part, as a result of ineffective erythropoiesis and iron overload that resemble those of untreated β-thalassemia major (13). The molecular mechanisms giving rise to the variable pathophysiology of HbE/β0-thalassemia are still largely undetermined (14). Contributions to the phenotype also include the following: (a) the co-existence of α-thalassemia (which diminishes the α/β chain imbalance) (15); (b) the presence of the C→T mutation in position −158 from γG resulting in an increased synthesis of HbF that seems to ameliorate the disease (7, 16); and (c) genetic modifiers that alter the HbE/β0-thalassemia phenotype (17, 18).
A major unanswered question is as follows. How does the HbE mutation contribute to the β0-thalassemia-dependent spectrum of pathophysiologies in HbE disease? Oxygen binding properties of HbE appear to be similar to that of HbA; the HbE tetramer is reported to exhibit normal oxygen binding in the presence of phosphates and at physiological ionic strength (19) but with an increase in oxygen affinity under conditions of low salt concentration (20). Biochemical studies in vitro established a decreased stability of HbE attributed to the positively charged substitution at the site of mutation located near the α1β1 interface (21–24) that has yet to be linked to in vivo RBC alterations and pathophysiology (13). It has been proposed that this instability may only play a role during febrile episodes (25). Regardless, to date, there is no known molecular mechanism or functional consequence of the HbE mutation that accounts for the pathophysiology of HbE diseases.
In this study, structural and functional properties of HbE are compared with those of HbA. High resolution x-ray crystallography and solution phase circular dichroism are used to examine the structural properties of HbE. Oxygen affinity measurements for solution phase samples and CO recombination kinetics for T and R state derivatives encapsulated in sol-gel matrices are used to compare the ligand binding reactivity of the ferrous derivatives of HbE and HbA. The nitrite reductase (NR)3 activity of HbE and HbA is also compared.
The NR reaction is of potential significance in that it yields nitric oxide (NO), a molecule vital for the integrity of the endothelium and cardiac function. Recently, normal hemoglobin function has been broadened beyond that of an oxygen carrier to include a role as a physiologically significant source of bioactive nitric oxide (26–39). One of several proposed NO-generating reactions of hemoglobin is the nitrite reductase reaction in which the five coordinate ferrous heme reacts with the nitrite ion yielding an NO molecule and a met (ferric)-heme. The reaction rate has been shown to be quaternary structure-dependent (36, 40, 41) with evidence supporting the idea that the quaternary state sensitivity arises from the higher redox potential of the T quaternary state versus the R quaternary state (42). The role of redox potential in this reaction has been questioned (43). This study also aims to address this issue with respect to HbA and HbE.
In this investigation, HbA and HbE are compared with respect to nitrite reductase reactivity at pH 7.0 and 7.4. The higher pH is used to enhance the contribution of R state intermediates in the reaction. The pH 7.4 measurements also include the effect of a potent allosteric effector L35 (2-[4-(3,5-dichlorophenylureido)phenoxy]-2-methylpropionic acid) (44), which is used to stabilize T state species and thus provide a means of clearly separating T and R state NR kinetics at pH 7.4.
The reduction of aquomet-heme to ferrous five-coordinate heme l-Cys (45) is protein-dependent (45, 46) and sensitive to the heme redox potential (47). l-Cysteine-mediated reduction of aquometHb to deoxyHb, both in solution and in a sol-gel matrix, is used in this study as a means of evaluating differences in the redox activity of the two proteins for both the T and R state. This comparison is used to help evaluate whether changes in the NR activity of HbE might arise from an alteration in the redox properties of the heme.
Overall, this study reveals, for both the T and R states of HbE and HbA, substantial differences in the rates of the nitrite reductase reaction and the l-Cys-mediated reduction of metHb. These findings are consistent with the HbE mutation causing an increase in the redox potential of both the T and R states for this Hb. This in vitro finding raises the interesting possibility that a decreased overall rate of HbE-mediated production of bioactive NO is a factor in exacerbating the β-thalassemia-induced inflammation. A model is presented regarding the role of HbE as a contributing factor to the HbE/β-thalassemia clinical picture.
EXPERIMENTAL PROCEDURES
Hemoglobin Purification
Human HbE was purified from red blood cells (RBC) obtained from transgenic mice expressing human HbE as described earlier (48). Human HbE purified from transgenic mouse RBC contains solely HbE, lacking contamination from HbA2 as occurs in human red blood cells. Because HbA2 and HbE have a very similar isoelectric point, separation is not feasible. The structural correctness of human HbE expressed by transgenic mice is verified by a variety of techniques, HPLC, mass spectrometry, isoelectric focusing, and absorption spectrophotometry (48).
The hemoglobins were separated and purified from the mouse hemolysates using a DE52 anion exchanger column equilibrated in a 0.2 m glycine buffer adjusted to pH 7.8 with saturated KOH (developer A). A gradient was created with the starting buffer (developer A) and (developer A + 0.005 n NaCl (0.293g/1000 ml)). The eluant was then concentrated and dialyzed for further purification on a CM52 cation exchanger (10 mm potassium phosphate, pH 6.5). After HbE separation and purification, the sample was concentrated and brought to equilibrium in the desired buffer by passage through a Sephadex G-25 column. The sample was then concentrated and further dialyzed against the experimental buffer. Purified human HbE was verified by mass spectrometry, HPLC, and isoelectric focusing as described previously (48). Human HbA was purified as described earlier (41, 49).
Crystallography
Carbonmonoxy (CO) HbE Crystal Growth
Solutions of oxyHbE were converted to COHbE by exposure to CO gas without bubbling. Complete conversion, and the absence of metHbE, was confirmed by the visible absorption spectra at 500–700 nm. The CO liganded form was chosen for study, rather than the oxy form, because of the following: (i) hemoglobin is more stable in the CO form, being less prone to oxidation (50, 51), and (ii) only minor local differences exist between the crystal structures of R state COHbA and oxyHbA (51).
Crystals were grown at ambient temperature by a modification (52) of the batch method (53, 54). The largest and best crystals grew in either 1.65 m potassium phosphate, pH 7.35, 2.5% purified HbE, or 1.60 m potassium phosphate, pH 7.35, 3.0% purified HbE and diffracted to 1.8 Å The Hb stock solution was gently exposed to CO gas without bubbling, added to the CO-precipitating phosphate buffer, and then exposed to CO again. Large crystals (∼1 mm) grew by the next day. The COHbE crystal used for diffraction data collection was formed in 1.6 m potassium phosphate (“high salt”) at pH 7.35. Diffraction from these crystals is consistent with the tetragonal space group P41212 with unit cell dimensions a = b = 53.5 Å, c = 192.0 Å, α = β = γ = 90°, and half of the HbE tetramer in the asymmetric unit.
Low ionic strength crystallization conditions were derived from the Crystal Screen HTP (Hampton Research). In this case, 1 μl of 25 mg/ml HbE (β-Glu26Lys) was mixed in a sitting drop with 1 μl of reservoir solution containing 0.1 m Na-HEPES, pH 7.5, 10% isopropyl alcohol, 20% polyethylene glycol 4000, and samples were equilibrated against reservoir solution at 21 °C for 3 weeks. Before sealing, trays were incubated inside the CO-filled box to convert HbE into the carbonmonoxy form. Diffraction from these crystals is consistent with the space group P212121 with unit cell dimensions a = 60.6 Å, b = 96.0 Å, c = 101.4 Å, α = β = γ = 90°, and one HbE tetramer in the asymmetric unit.
DeoxyHbE Crystal Growth
Crystallization of purified HbE (using a stock solution of HbE in 0.05 m HEPES, pH 7.35) was performed using the Perutz method (52) and as described in detail in Ref. 55 with minor modifications. The buffers and solutions were all deoxygenated in sealed containers with N2. Final mixing (Hb solution with the high salt buffer and iron citrate) was done in an anaerobic chamber. Prior to mixing, deoxygenated sodium dithionite was added to each solution for a final concentration of 3 mm according to Ref. 52. A crystallization well plate was used to obtain diffracting crystals by the vapor diffusion method. 1 μl of 26.5 mg/ml HbE in 50 mm HEPES, pH 7.35, was mixed with 1 μl of reservoir solution containing 1.8 m ammonium phosphate and ammonium sulfate, pH 6.5, 2 mm iron citrate, with 3 mm sodium dithionite and equilibrated against reservoir solution for a week. The crystals belong to a space group P21 with unit cell dimensions a = 53.26 Å, b = 79.63 Å, c = 62.35 Å, and α = γ = 90°, β = 98.66°, and one HbE tetramer in the asymmetric unit.
X-ray Data Collection and Refinement
Before data collection, protein crystals of COHbE were soaked for a few minutes in CO-saturated mother liquor containing 20% glycerol as cryoprotectant. Then crystals were flash-frozen in liquid nitrogen, and diffraction data were collected with CuKα radiation (λ = 1.54 Å) using in-house RaxisIV++ detector coupled with RU300 rotating anode generator (Rigaku, Woodlands, TX). Crystals of deoxyHbE were transferred (inside N2 incubator) to the mother liquor containing 20% glycerol as the cryoprotectant and then flash-cooled in liquid nitrogen. Diffraction data were collected at the X3A beamline (Brookhaven National Laboratory, Upton, NY) using 0.979 Å radiation and a MarCCD 165-mm detector (MarResearch, Hamburg, Germany). Intensities were integrated using the program HKL2000 (56) and reduced to amplitudes using the program TRUNCATE (57). (See Table 1 for statistics.) The structures were determined by molecular replacement with PHASER. Model building and refinement were performed with the programs REFMAC and COOT (58, 59). The quality of the final structure was verified with composite omit maps, and the stereochemistry was checked with the programs WHATCHECK (60) and PROCHEK (61). The LSQKAB and SSM algorithms (56) were used for structural superimpositions (57, 62). The refinement statistics and unit cell parameters are shown in Table 1.
TABLE 1.
Data collection and refinement statistics for the HbE crystal structures
| COHbE low salt | COHbE high salt | Deoxy | |
|---|---|---|---|
| PDB entry | 1YVQ | 1YVT | 3DUT |
| Data collection | |||
| Beamline | Raxis-IV++ | Raxis-IV++ | BNL X3A |
| Wavelength | 1.54 Å | 1.54 Å | 0.979 Å |
| Space group | P212121 | P41212 | P21 |
| Resolution range | 1.8–30.0 | 1.8–30.0 Å | 1.5–30.0 Å |
| Observed reflections | 145,673 | 263,258 | 297,197 |
| Unique reflections | 47,533 | 26,762 | 75,929 |
| Completenessa | 85.2% (84.3%) | 99.7% (100.0%) | 92.2% (68.8%) |
| I/σI | 12.1 (3.5) | 10.9 (3.5) | 12.2 (3.5) |
| R-merge (I)b | 0.044 (0.198) | 0.112 (0.432) | 0.039 (0.348) |
| Structure refinement | |||
| Resolution range | 1.8–20.0 Å | 1.8–20.0 Å | 1.5–20.0 Å |
| Rcrystc | 0.166% | 0.175% | 0.214% |
| Rfreec | 0.218% | 0.214% | 0.245% |
| Protein nonhydrogen atoms | 4575 | 2190 | 4574 |
| Water molecules | 701 | 224 | 488 |
| Average B-factor | 19.4 Å2 | 22.8 Å2 | 15.6 Å2 |
| Root mean square deviations from ideal value | |||
| Bonds | 0.012 Å | 0.011 Å | 0.012 Å |
| Angles | 1.24° | 1.21° | 1.41° |
| Torsion angles | 14.9° | 15.1° | 15.8° |
a Values in parentheses indicate statistics for the high resolution bin.
b Rmerge = ΣΣ j|Ij(hkl) − 〈I(hkl)〉|/ΣΣj|〈I(hkl)〉|, where Ij is the intensity measurement for reflection j, and 〈I〉 is the mean intensity over j reflections.
c Rcryst/(Rfree) = Σ ‖Fo(hkl)| − |Fc(hkl)‖/Σ |Fo(hkl)|, where Fo and Fc are observed and calculated structure factors, respectively. No σ cutoff was applied. 5% of the reflections were excluded from refinement and used to calculate Rfree.
Protein Data Bank Entries
The COHbE structures were deposited with the Protein Data Bank under entry codes 1YVQ (low salt conditions) and 1YVT (high salt conditions), and the deoxyHbE structure was deposited under entry code 3DUT.
Solution Preparation
Samples for solution studies (0.22 mm heme) were prepared as deoxy in an oxygen-free glove box by exhaustive purging with argon, and adding a modest excess dithionite (>1:1 dithionite/heme) as necessary. Samples used to prepare sol-gels were prepared similarly. The thin film nature of the sol-gel preparation requires a higher concentration of Hb than for the solution phase studies resulting in a higher OD for the sol-gel samples compared with the solution phase studies. The visible to near IR optical absorption spectra of samples in BisTris buffer, pH 7.0 and 7.4, were scanned to establish the ligation and redox status of the heme. COHb samples were prepared by exposing the deoxy samples to buffer-saturated CO gas, until the absorption spectrum revealed complete conversion to COHb derivative. Aquomet samples were prepared by oxidizing Hb using K3Fe(CN)6, which was subsequently removed using a spin column prepared from G-10 Sephadex using the same buffer.
Sol-Gel Preparation
Hemoglobin was encapsulated in a thin film of silane-derived sol-gel according to published procedures described previously (41). Basically, tetramethylorthosilicate, hydrolyzed using 2 mm HCl, was mixed in a 1:1 ratio with protein solubilized in 0.05 m BisTris OAc buffer, pH 7.0. The resulting solution was spun in a 1-cm-wide NMR tube until gelation occurred, creating a thin film of an Hb-containing gel on the bottom fourth of the tube. A final concentration of 0.45 mm in heme for the encapsulated Hb is required to produce a high optical quality thin film. After the sol-gel is allowed to age for several days to ensure that polymerization was complete (2–6 days), the bathing buffer of the sol-gel is then changed as needed to suit the specific measurement.
AquometHb T state sol-gel samples were prepared by encapsulating the deoxyHb derivative and, after the aging step, oxidized to the ferric (met) redox state using K3Fe(CN)6. The ferricyanide was flushed out of the sol-gel using several washings once conversion to met was complete. The bathing buffer was then replaced with PBS, pH 7.4. R state aquomet samples were prepared by first encapsulating the CO-ligated Hb derivatives which, after the aging process, were then photolyzed in the presence of oxygen to remove and replace the CO, then purged with argon to remove any residual oxygen, and then oxidized to met using the same technique described for the deoxy samples. Samples encapsulated as the O2-ligated Hb were purged with argon to remove any oxygen before treating with dithionite to form the R state deoxy derivatives. In all cases, optical absorption spectra were used to confirm the ligation and redox status of the encapsulated Hb species. Sol-gel encapsulated R and T state COHb derivatives of HbA and HbE were prepared as described previously in 0.05 m BisTris OAc, pH 7.0, as the bathing buffer (41, 63, 64). The R state COHb samples were prepared by encapsulating the CO derivative directly. The T state samples were prepared by first encapsulating the deoxy derivative. After the sample was allowed to age in 0.05 m BisTris OAc, pH 7.0, it was flushed extensively with CO to form the ligated protein.
Circular Dichroism
Circular dichroism measurements on solution phase samples of deoxy and CO derivatives of HbA and HbE were performed on a Jasco J-815 CD spectrophotometer (Jasco Corp., Tokyo, Japan) in a 1-mm cuvette.
CO Recombination in Sol-gel Encapsulated Hb
CO recombination traces from encapsulated R and T state COHb derivatives were generated as described previously (63, 65, 66). Photodissociation of the CO from the heme is triggered using a 7-ns pulse at 532 nm from a Nd:YAG laser. The recombination trace over several decades in time was generated from a change in absorption using the several milliwatt 442-nm output of a continuous wave HeCd laser as a probe of the evolving populations undergoing CO recombination. The recombination traces are displayed on a log-log plot of normalized absorbance versus time as described previously (67–70).
Nitrite Reductase Activity, pH 7.0
Deoxygenated hemoglobin samples were prepared in solution at pH 7.0 in 0.05 m BisTris, 0.24 mm in heme as described previously (41) with the exception that an excess of dithionite was added to a concentration of 1 mm. Nitrite was added in a slight excess of 1.1:1 to deoxyhemoglobin (Hb) to initiate the reaction. Under these conditions, the nitrite reductase reaction was modified in that the met (Hb+) product generated from Reaction 1
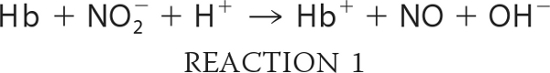 |
which was reduced back to deoxyHb by the excess dithionite. The deoxyHb binds NO yielding the ferrous NO derivative of Hb (NOHb). As a result, the observed reaction consists overwhelmingly of the loss of deoxyHb and the formation of NOHb as described previously (71). Dithionite at this concentration does not reduce nitrite to NO (42, 71).
The change in absorption was monitored on a Lambda 2 spectrophotometer (PerkinElmer Life Sciences) as a function of time at 430 nm (Sôret band for deoxyHbA). The choice of the 430-nm band to follow the initial rate of decay of the starting deoxy species in solution was based on the relatively large extinction coefficient for deoxyHb at that wavelength, −133,000 m−1 cm−1 as compared with NOHb, ∼8400, and metHb, ∼4000.
The higher optical density of sol-gel samples at 430 nm compared with the solution phase samples precluded the use of the Sôret band to monitor the reaction in the sol-gels. For these samples, the Q-band spectral region (480–650 nm) was scanned at regular 60-s intervals. The data were base-line-corrected at each wavelength, and the change in absorption was plotted as a function of time. The initial linear portion was used to calculate initial rates.
For the sol-gels, the scanned Q-band spectra were also deconvoluted into populations of products using authentic standard basis sets and a program in Mathcad, version 14.0 (Parametric Technology Corp., Needham, MA). The resulting deoxy population was plotted as a function of time to assess the initial reductase rate.
Nitrite Reductase Activity, pH 7.4
The NR reaction was also monitored at pH 7.4 to enhance the R state contribution to the kinetics. Based on the known allosteric properties of metHb and partially liganded Hb, the various intermediates (found in the NR reaction) will switch from T to R more rapidly at the higher pH (72). These R state partially liganded species are purported to be of physiological relevance (73). Deoxygenated hemoglobin samples were prepared in solution at pH 7.4 in 0.05 m BisTris, 0.24 mm in heme with excess dithionite as described for the pH 7.0 measurements. The reaction was conducted in the absence and in the presence of the allosteric effector L35 (2-[4-(3,5-dichlorophenylureido) phenoxy]- 2-methylpropionic acid) (44) at different nitrite/heme ratios. The L35, a gift from Drs. Iraj and Parvis Lalezari, was introduced into the reaction mixture in slight excess of the heme (1:1.1, heme/L35). The L35 was added to enhance the contribution of the T state population and thus allow for a clear distinction between the T and R state NR kinetics at pH 7.4. For this experiment, L35 has the advantage over inositol hexaphosphate and diphosphoglycerate as a T state stabilizer in that it remains a potent effector at the higher pH (74). The NR reaction in solution was followed at 430 nm as described above for the pH 7.0 samples. The sol-gel T and R state samples were prepared in the same manner as at pH 7.0 with the exception that the bathing buffer was changed to pH 7.4.
Reduction of AquometHb by l-Cys
Solution Studies
met-Hb prepared from oxyHbA as described above, was purged with argon as were all buffer solutions. The reactions were performed anaerobically. The thiol stock solution (l-Cys) was prepared in the same buffer (PBS, pH 7.4) prior to addition to the protein. An aliquot of l-Cys (2 mm) was added to the met solution (0.38 mm heme), and the absorption was scanned at regular timed intervals using a 1-mm cuvette. The kinetic data were fit (Origin version 7.5) using the change in the unique 630-nm metHb band as a function of time.
In all instances, the starting aquometHb sample was reduced by the added thiol to form deoxyHb. The conformation-sensitive Q band (∼555 nm for T and ≥558 nm for R) and near IR Band III (∼758 nm for T and ≥ 760 nm for R) were used to confirm both the appearance of the ferrous five-coordinate deoxy derivatives and the quaternary structure status of the resulting deoxy population.
Sol-gel Studies
Reduction by l-Cys of encapsulated samples was performed in PBS, pH 7.4, using 10 mm l-Cys prepared in the same argon-purged buffer. After oxidizing the T state or R state samples of HbE or HbA and flushing out K3Fe(CN)6 as described above, the buffer was changed to PBS, pH 7.4. An aliquot of l-Cys was added to initiate the reaction, and the change in absorbance was monitored as a function of time. As noted above for the solution phase samples, the Q band at 555 nm and near IR band at 758 nm were used to monitor both the formation of the ferrous product and the quaternary status of the products.
RESULTS
Structural Studies
High Resolution Crystal Structures at or Near Physiological pH
Data collection and refinement statistics for the deoxyHbE structure are shown in Table 1. The asymmetric unit of the low salt COHbE crystals contains one full heterotetramer. The overall structure of the tetramer is fairly similar to the previously reported low salt COHbA structure (PDB code 1BBB) and CNHbE structure (PDB 1NQP) with root mean square deviations of 0.36 and 0.21 Å between all tetramer Cα atoms (0.19 and 0.13 Å when β subunits are superimposed separately). Higher overall root mean square deviation between tetramers as opposed to β subunits alone indicate that COHbE and CNHbE are slightly closer in tertiary structure than COHbE and COHbA.
Structural superposition of the COHbE structures (under crystallizing conditions of high and low salt concentrations) and the deoxyHbE (crystallized under high salt concentration) structure with the respective HbA structures (derived from crystals grown under similar conditions) indicates that βLys-26 is not involved in specific interactions with nearby residues, neither from the same subunit nor from the neighboring α subunits (Fig. 1, top panel). Nonetheless, the βLys-26 side chain protrudes into the bulk solvent and interacts with solvent molecules. Because solvent structure varies in the three presented HbE structures and the crystal environment differs for different HbE subunits, the βLys-26 side chain conformation as well as conformations of neighboring βHis-117 and βGlu-22 all show a high degree of variability. The orientation of the β2His-116 (different from that of a cyanmetHbE structure (75)) and the β2His-117 side chains, as well as β2Lys-22, are clearly altered in the COHbE structure (PDB 1YVQ) compared with COHbA (PDB 1BBB) (Fig. 1, bottom panel). These findings help illuminate the reported in vitro HbE instability (see “Discussion”).
FIGURE 1.

Structural superposition of the human (h) HbE structures in the vicinity of βLys-26. Top panel, structural environment of βLys-26 (K26) in the high salt HbE superimposed onto the experimental omit electron density map. α chain is shown in light green, and β chain is shown in gray. Conformations of βLys-26 and nearby residues in high salt (gray), low salt (two subunits, salmon), and deoxy (two subunits, blue) HbE structures are shown. Bottom panel, structural superposition between the COHbA and COHbE structures in the area of β-Glu26Lys mutation. α chain is shown in light green, and β chain is shown in gray. Water molecules in the COHbA structure are shown as red spheres. Side chains corresponding to the COHbA structure are depicted in pink. Note hydrogen bond between βGlu-26 (E26) and βArg-30 (R30).
In contrast, the position of the βGlu-26 side chain in HbA is stabilized through the hydrogen bond with the guanidinium group of βArg-30, which in turn is positioned directly at the αβ chain interface and forms a hydrogen bond with αPhe-117 carbonyl oxygen (Fig. 1, bottom panel). The imidazole ring of βHis-116 forms a hydrogen bond with the αPro-114 carbonyl oxygen.
Evaluation of the extent of local and global structural consequences of β-Glu26Lys mutation was achieved through superpositions between various human HbA and human HbE structures in the R and T states. The β1 subunits of each pair of structures were superimposed, and relative shifts of heme irons in each subunit of the tetramer were measured (Table 2). Apparently, the smallest shifts (0.2–0.5 Å) occurred when the equivalent Hb states crystallized under similar conditions (high salt or low salt) were compared. These numbers suggest background root mean square deviations between the equivalent structures are caused by experimental errors, slight differences in crystallization conditions, and crystal packing. The comparisons point to the known significant influence of salt concentration on Hb tertiary structure, especially in the CO (R state) structures. Hence, structural superpositions between HbE and HbA under equivalent conditions do not reveal statistically significant differences (i.e. no significant conformational changes) at the allosteric transition sites, heme, and central cavity upon comparing these static structures.
TABLE 2.
Relative heme shifts (Å) upon CO-to-deoxy transition in various human hemoglobins
Subunit β1 was used for superposition. Low salt structures are underlined.
| PDB entry |
Subunit |
||||
|---|---|---|---|---|---|
| CO | Deoxy | α1 | β1 | α2 | β2 |
| HbE | |||||
| 1YVT | 3DUT | 1.0 | 1.1 | 3.7 | 6.2 |
| 1YVQ | 3DUT | 1.3 | 1.1 | 6.3 | 9.0 |
| HbA | |||||
| 2DN3 | 2DN2 | 1.6 | 1.0 | 2.8 | 6.6 |
| 1BBB | 2DN2 | 1.7 | 1.0 | 5.5 | 9.2 |
| 1BBB | 1HBB | 2.0 | 0.8 | 5.9 | 10.0 |
| CO | |||||
| 1YVT | 1YVQ | 0.5 | 0.2 | 2.7 | 3.6 |
| 1YVT | 2DN3 | 0.5 | 0.1 | 0.2 | 0.4 |
| 1YVQ | 1BBB | 0.2 | 0.2 | 0.4 | 0.5 |
| 1BBB | 2DN3 | 0.6 | 0.1 | 2.9 | 3.8 |
| 2DN3 | 1LJW | 0.5 | 0.1 | 0.4 | 0.6 |
| Deoxy | |||||
| 3DUT | 2DN2 | 0.4 | 0.1 | 0.8 | 1.0 |
| 3DUT | 1HBB | 0.8 | 0.3 | 0.3 | 1.3 |
| 2DN2 | 1HBB | 0.5 | 0.2 | 0.5 | 0.9 |
Circular Dichroism, a Solution Phase Comparison of HbE and HbA
CD reports on the microenvironment of the heme as well as the secondary structure of the globin. The 285-nm CD region is sensitive to R/T-specific differences at the α1β2 interface, specifically βTrp-37 and αTyr-42, whereas the Sôret bands (410–450) and the 500–600-nm region reports on changes in the heme environment (76). The R/T-sensitive regions of the CD spectra are superimposable such that there was no discernable difference between HbE and HbA for both the deoxy and CO derivatives (data not shown). Very small differences in the heme absorption region (450–700 nm), seen in some comparisons for both derivatives, appear to be insignificant as they are within the range of the very small variation seen from sample to sample for the same protein. The CD results were consistent with there being little if any difference between HbA and HbE with respect to tertiary structure within either the T or R quaternary states.
Functional Studies
Oxygen Binding Properties
A limited series of oxygen affinity measurements conducted at high and low salt concentrations in phosphate buffer, pH 7.4, revealed the same minor differences in the P50 and Hill coefficient between HbA and HbE as reported previously (19, 20). Overall, the results are consistent with earlier reports that HbE oxygen binding is similar to HbA (19).
CO Recombination in Sol-gels
Fig. 2 shows the evolution of recombination traces for the T and R state forms of sol-gel encapsulated HbE and HbA. There was minimal variation as a function of hemoglobin species between the kinetic traces for CO recombination, following photolysis of CO for the sol-gel-encapsulated COHb derivatives trapped as T state species. Both appear to display functional characteristics previously observed for T state geminate recombination and solvent phase kinetics (63, 64, 77). Once the CO binds to the sol-gel-encapsulated T state deoxyHb derivative, the protein population slowly (days) evolves from the nonequilibrium T state distribution of COHb conformations toward the R state equilibrium distribution of conformations associated with the COHb derivative (64, 69, 70). This CO binding-induced evolution from the T to R state distributions can be followed in the sol-gel where the greatly slowed conformational relaxation allows for facile monitoring of the evolution of the CO recombination trace from the T state pattern to that of the R state. Fig. 2 shows that the rate at which the initially generated nonequilibrium T state population of COHb converts to the equilibrium R state COHb population is very similar for HbE and HbA. This observation indicates similar relative T/R state conformer stability for the two hemoglobins. (Stability, as used here, refers to stabilization of the allosteric T and R structures and not to the intrinsic integrity of the tetramer.) The similar rate of evolution is consistent with the observation that allosteric parameters for HbE and HbA were very similar, consistent with that seen in the comparison of the crystal structures. We have observed that the CO binding-induced T to R transition in the sol-gel under these conditions can be greatly accelerated or slowed depending on the relative stabilities of the T and R structures in a wide range of chemically and mutagenically modified hemoglobins.4 As a consequence, it can be concluded that the similar rates of conformational evolution seen for HbA and HbE are a good indication that the relative stability of the T and R states of these two Hbs is comparable.
FIGURE 2.

Time-dependent change in the CO recombination trace for samples of COHbE and COHbA encapsulated in a thin sol-gel matrix. The samples were initially encapsulated as the deoxy derivatives and subsequently converted to the CO derivative after allowing the sol-gel to age for several days in an oxygen-free environment. The bathing buffer of the sol-gel films is 0.05 m BisTris OAc, pH 7.0. The progression of traces tracks the normalized absorbance (N.A.) changes in the recombination profile, starting from the initial exposure of the deoxyHb samples to CO binding, laser-induced CO photodissociation, and recombination (see “Experimental Procedures”).
Sol-gel samples of HbA and HbE encapsulated after saturation with CO served as functional six-coordinate ferrous R state end points for both proteins (Fig. 2). In this case, there were minor R state differences both in geminate yield and in solvent phase recombination, with HbE manifesting a slightly lower geminate yield and slower solvent phase recombination than HbA. These differences are well within the range of R state variability seen in the geminate and solvent phase recombination (77). Overall, the CO recombination results are consistent with similar reactivity and allosteric stability for the ferrous deoxy derivatives of HbA and HbE with minor differences noted for the R state HbE.
NR Activity in Solution at pH 7.0
Reactions of purified deoxyHbE at neutral pH in the presence of equivalent nitrite (∼1:1 nitrite/heme) and an excess of dithionite were compared with HbA under near-identical conditions. The reaction was monitored using two different approaches. In one case, the initial rates were generated by sitting at the 430 nm absorption maximum for the deoxyHb Sôret band and following the decrease in absorption as a function of time following the addition of nitrite. This method uses the absorption spectrometer in a time drive mode that can record changes in a single wavelength in 2-s intervals. The advantage of this approach is that numerous data points are accumulated over the experimental time course, and the end point is easily determined. Additionally, the reaction is monitored by repetitively scanning of either the Sôret band region or the visible and near IR region of the absorption spectrum subsequent to the addition of the nitrite. The single wavelength monitoring was chosen for generating the actual initial rates because of the higher temporal resolution associated with that method. The successively scanned spectra both allow for verification of Hb species present during the course of the reaction and provide a rough check on the rates obtained by sitting at the Sôret band. Fig. 3 shows decay of the deoxy population using the single wavelength monitoring technique, whereas the right panel in Fig. 3 shows a representative sequence of Sôret band traces showing the decay of the deoxy peak at 430 nm and the build up of the NOHb peak at 418 nm.
FIGURE 3.

Comparison of the initial rate for the nitrite reductase reaction for HbA and HbE in solution at pH 7.0 (0.2 mm heme + ∼1:1 nitrite, 0.05 m BisTris OAc, pH 7.0, with 1 mm dithionite) as reflected in the decrease in the deoxyHb OD at 430 nm. The right panel shows the change in the Sôret absorption band as a function of time for HbA over the same time period. Solution conditions are as described in the text. The down-pointing arrow indicates the time-dependent decrease in absorbance at 430 nm.
It was found (using the absorption change in the Sôret band) that the initial rate of reaction was significantly slower (∼2.5-fold) for deoxyHbE (1.61 × 10−4 ± 0.3 s−1 p = 0.0021) than deoxyHbA (3.92 × 10−4 ± 0.5 s−1, p = 0.0079, n = 4) (Fig. 3 and Table 3). It is important to emphasize that these reactions were conducted in the presence of an excess of sodium dithionite (1 mm). The added dithionite simplifies the reaction and analysis by converting any generated metHb back to the ferrous deoxy derivative. The scanned absorption spectra show that under these conditions with added dithionite, the initial deoxy population is essentially fully converted to ferrous NOHb indicating the following: (a) NO is being generated and (b) the met population is being reduced as it forms as reported previously (71). It has been shown that added dithionite at concentrations below 10 mm (as in this case) does not affect the measured initial rates for the nitrite reductase reaction (42). For the reactions presented here, the dithionite levels are well below this limit.
TABLE 3.
Solution and sol-gel Hb nitrite reductase, initial rates of reaction
BisTris, pH 7.0, + dithionite (>1:1 heme) was used. Rates are means of four samples for the solution phase and two samples for the T state sol-gel (see text for details).
| Sample | Rate | [Nitrite] | λmaxa | Band IIIa |
|---|---|---|---|---|
| s−1 | nm | nm | ||
| Solution deoxyHbE | 1.61 × 10−4 ± 0.3 | 1.1:1 | 555 | 758 |
| Solution deoxyHbA | 3.92 × 10−4 ± 0.5 | 1.1:1 | 555 | 758 |
| [deoxyHbE] T | 7.2 × 10−4 ± 0.7 | 5 mm | 555 | 758 |
| [deoxy HbA] T | 2.1 × 10−3 ± 0.2 | 5 mm | 555 | 758 |
| [deoxyHbE] R | NDb | 5 mm | 560 | 762 |
| [deoxy HbA] R | NDb | 5 mm | 560 | 763 |
a Wavelength maxima of solution and sol-gel-encapsulated Hb samples prior to reaction with nitrite, indicative of the deoxy state. See “Experimental Procedures” for details.
b ND means not determined. Rates for the R-state gels under these conditions and at lower concentrations of nitrite were too fast to accurately determine rates for this reaction.
Nitrite Reductase Activity in Sol-gels at pH 7.0
The sol-gel protocol permits the preparation of samples that remain locked into the T state structure throughout the reaction, whether in the deoxygenated or oxygenated liganded form. Encapsulated T state deoxyHbE samples consistently exhibit a significantly slower initial NR rate than the corresponding encapsulated T state deoxyHbA samples (Fig. 4 and Table 3). The initial rates reported in Table 3 were derived from the initial decay of the deoxy population generated from plots of the deconvoluted time-dependent changes of Hb species contributing to the observed spectra using basis sets as described under “Experimental Procedures.” Similar rates were also obtained using the decay at specific wavelengths. A representative plot of the evolving populations is shown as an inset in Fig. 4.
FIGURE 4.

Time-dependent changes in the visible absorption spectrum reflecting the nitrite reductase reaction for sol-gel-encapsulated Hbs trapped in the T state (0. 4 mm heme + 5 mm nitrite, 0.05 m Bistris OAc, pH 7.4, + 1 mm dithionite). The upward and downward pointing arrows indicate the increasing and decreasing absorbance, respectively. Inset, representative plot of the time-dependent changes in the population of different HbA species resulting from the nitrite reductase reaction of the sol-gel sample. (See “Experimental Procedures” for details). Red circle, NOHb; black square, deoxy; blue triangle, metHb.
The NR reactions at pH 7 for the sol-gel deoxy R state structures for both HbE and HbA were too fast to measure under similar conditions. We estimate that the R state rates are at least a factor of 10 faster. Even at a 10-fold lower concentration of nitrite, the rapidity of the initial decay of the deoxy R state species made it difficult to obtain sufficient data points at early times to produce accurate enough initial R state rates for a reliable HbA/HbE comparison. Nonetheless, the difference in the much slower initial T state rate between HbE and HbA is clearly statistically significant. This T state difference in the initial rate indicates that the HbE/HbA difference in the initial solution phase rate is due, at least in part, to protein-specific differences in the T state reactivity.
Nitrite Reductase Activity in Solution at pH 7.4
Solution phase NR rates at pH 7.4 were measured in an attempt to better expose R state contributions as per the rationale described under “Experimental Procedures.” The same concentration of deoxyHb (0.22 mm in heme) was used as for the pH 7.0 measurements. Three different concentrations of nitrite were used 1:1 nitrite/heme, 2:1 nitrite/heme, and 3:1 nitrite/heme in the absence and in the presence of L35. L35 was used to create kinetic traces reflective of a sustained T state population, thus allowing for a clear T/R differentiation as explained below. Representative time courses for the NR reaction at pH 7.4 are shown in Fig. 5.
FIGURE 5.

Comparison of the initial rate for the nitrite reductase reaction for HbA in solution in the presence and absence of L35. DeoxyHbA + 1:1 nitrite ± L35. Red trace, 0.22 mm heme + L35 (1:1 heme); blue trace, 0.22 mm heme without L35. Solution conditions are as follows: 0.05 m BisTris, pH 7.4; arrows represent approximate time points when Band III peaks were generated (see Fig. 6).
In the presence of L35, the NR trace remains almost flat and linear with essentially no change or break in the trace from time 0 to 2000 s. For samples having the highest concentration of nitrite (data not shown), a break was observed toward the very end of the reaction. The trace for samples, without the addition of L35, started out looking very similar to the L35-containing samples. However, after a relatively short (nitrite concentration-dependent) initial slow phase, a clear break occurred that was followed by a much more rapid NR process.
The deoxy-heme-derived low optical density Band III (∼760 nm) in the near IR region of the absorption spectrum is conformation-sensitive (78–80). The Band III peak position is ∼758 nm for the deoxy T state, ∼762 nm for the quasi-stable/relaxed deoxy R state, and ∼765 nm for the deoxy R state that retains the tertiary structure of the fully liganded R structure. All of the solution phase deoxy samples started out at ∼758 nm, consistent with a T state designation. As the deoxy population decays during the NR reaction, the intensity of Band III decreases. For the L35-containing samples, the peak position of Band III does not noticeably shift as the intensity decreases during the time course of the NR reaction (data not shown). In contrast, the Band III spectra generated from the L35-free samples at time points just before, during, and just after the break in the trace (denoted by arrows in Fig. 5) show both a progressive decrease in Band III due to the loss of deoxy-heme and a progressive broadening of the band with a clear red shift from the initial T state peak position at ∼758 nm toward longer (redder) wavelengths (Fig. 6). This red shift is consistent with the appearance of a population of R state deoxy-hemes (78–80) at the time point where the kinetics begin to accelerate.
FIGURE 6.

Changes in the normalized absorption spectrum of Band III in the reaction of deoxyHbA with nitrite and at the appearance of the change in slope for this reaction. Black line, deoxy T state at time 0; red line, Band III at time point at the change in slope. Inset shows the time progression of Band III.
The kinetic traces for the samples with and without the L35 and the time-dependent red shifting of Band III are consistent with assigning the fast NR phase to the emerging R state population that starts to appear at the point of the break. The rates for the fast NR phase obtained using the three different nitrite/heme ratios are presented in Table 4. These rates are typically on the order of 10-fold faster than for the slower T state NR process obtained using the same nitrite ratios in the presence of L35. It is important to note that at each concentration of nitrite, both the fast and the slow rates for HbE are lower by a factor of 2 or more relative to HbA. This same pattern was observed for the slow phase at pH 7.0.
TABLE 4.
R state nitrite reduction rates
BisTris, pH 7.4, + dithionite (>1 dithionite, 1 heme) was used. Solution data for deoxy 0.22 mm (heme) + nitrite in the ratio is as noted. R state values are averages of 2–4 runs. The sol-gel data for [deoxy Hb] was 0.45 mm (heme). 1:1 nitrite in 3 ml of bathing buffer, 0.05 m Bis-Tris, pH 7.4, + dithionite was used for solution.
| HbE | R state average |
|---|---|
| s−1 | |
| Nitrite ratio | |
| 1:1 | 3.4 × 10−4 ± 2.0 |
| 2:1 | 8.2 × 10−4 ± 2.0 |
| 3:1 | 1.46 × 10−3 ± 0.5 |
| HbA | |
| Nitrite ratio | |
| 1:1 | 5.93 × 10−4 ± 0.4 |
| 2:1 | 1.57 × 10−3 ± 0.2 |
| 3:1 | 2.79 × 10−3 ± 0.04 |
| [deoxyHbE] R | 1.7 × 10−3 ± 0.3 |
| [deoxyHbA] R | 4.8 × 10−3 ± 0.6 |
Sol-gel measurements of the R state deoxy gel samples at pH 7.4 are also consistent with slower rates for HbE (Table 4 and representative spectra are shown in Fig. 7). The peak positions for the Q band and Band III for the initial deoxy samples were ∼559 and 766 nm, respectively, for both R state deoxy samples. In contrast, the corresponding peak positions for samples encapsulated as the T state deoxy derivative are consistently 555 and ∼756–758 nm. The 559- and 766-nm values were consistent with samples that were fully R state, with the tertiary structure of the initial species in the fully liganded R state. The sol-gel results support the claim, based on the solution phase study, that the R state NR rate was slower for HbE. Thus, it appears that, for both the T and R state forms of deoxy hemoglobin, the NR rates are slower for HbE compared with HbA.
FIGURE 7.

Absorption changes of the R state deoxyHbA sol-gel + 0.5 mm nitrite as a function of time; 0.05 m BisTris, pH 7.4, 0.45 mm (heme). Inset A is the distribution of deoxy as a function of time; inset B is the initial position of Band III.
Redox Properties
Reduction Rates of MetHb by l-Cys in Solution Phase
The redox potential has been invoked as a major determinant of Hb nitrite reductase activity (42, 81). Relative to the R state, T state-stabilized hemoglobins are characterized by a higher redox potential (E½) (40), which allegedly contributes to the lower R state rate for the NR reaction (40). In lieu of traditional redox potential measurements that require large amounts of material and are conducted under conditions of questionable physiological relevance, we chose to use the redox-sensitive rate of l-Cys-induced reduction of aquometHb (45, 46) as a relative indicator of reduction potential (i.e. propensity to reduce). This approach also allows for redox comparisons of sol-gel-encapsulated samples, which for obvious reasons cannot be performed in standard redox potential measurements requiring a solution phase.
In solution, the rate of l-Cys-induced formation of deoxyHb from metHb is faster for HbE compared with HbA (2.3 × 10−3 ± 0.4 s vis à vis 2.1 × 10−4 ± 0.01 s) (Fig. 8 and Table 5). A possible origin for the difference in the rates of the thiol-induced reduction is a difference in the R/T equilibrium between the two Hbs. If there is a substantial protein-specific difference in the amount of R and T metHb species in solution, the difference in redox property of the R and T state could account for the observed protein-specific difference in reduction rates. Alternatively, there could be protein-specific differences in the redox properties of the T and R states that could account for the observed solution phase results as well. Sol-gel encapsulation was used to trap the T and R state populations of metHb under the same pH conditions thereby allowing for a direct relative comparison of the rates of reduction for the T and R state derivatives of both proteins.
FIGURE 8.

Kinetic trace of the reduction of metHb by l-cysteine in solution: (metHb (0. 4 mm heme) + 2 mml-Cys, PBS, pH 7.4). l-Cys is added to samples of metHb anaerobically to initiate reaction. The samples are then scanned at 2-min intervals, and base-line corrected. The change in 630 nm (unique metHb band) is monitored as a function of time. Inset, a representative plot showing the increase and decrease in the spectral bands of HbA as a function of time are indicated by up- and down-pointing arrows, respectively.
TABLE 5.
Rates of reduction (s−1) by l-Cys of metHb in solution and sol-gel (s−1)
| Sample | Solutiona | T stateb | R stateb | Metb |
|---|---|---|---|---|
| HbE | 2.30 × 10−3 ± 0.4 | 6.3 × 10−5 ± 0.4 | 1.9 × 10−4 ± 0.1 | 6.2 × 10−5 ± 0.4 |
| 3.9 × 10−3 ± 0.2 | ||||
| HbA | 2.10 × 10−4 ± 0.01 | 4.2 × 10−5 ± 0.6 | 1.3 × 10−4 ± 0.1 | 3.6 × 10−5 ± 0.6 |
| 3.0 × 10−3 ± 0.4 | ||||
| Mb (horse) | 1.51 × 10−5 ± 0.01 |
a Solution phase is 0.38 mm (heme) + 2 mm l-Cys, PBS, pH 7.4. Data were analyzed by monitoring the change in the met absorbance band at 630 nm over time. n = 3 for HbE and n = 2 for HbA.
b Sol-gel is 0.45 mm (heme) + 10 mm l-Cys, PBS, pH 7.4. Sol-gel data were analyzed as described for solution.
Reduction Rates of MetHb by l-Cys in Sol-gel
Both T and R state sol-gel samples were oxidized to metHb in PBS, pH 7.4 buffer, just prior to reaction with l-Cys as described under “Experimental Procedures.” Reduction of the T state met samples with l-Cys followed biphasic kinetics for both HbE and HbA, with HbE being slightly faster in both rates. Reduction of the R state met samples also manifested differences again with HbE faster than HbA. Sol-gel samples of HbE encapsulated initially as aquomet were also reduced more rapidly than the correspondingly encapsulated aquometHbA samples (Fig. 9 and Table 5).
FIGURE 9.

Kinetic trace of the reduction of metHb by l-Cys in sol-gels. 0.4 mm (heme) + 10 mm l-Cys, PBS, pH 7.4. Samples of Hb encapsulated as T (deoxy) or R (ligated to CO) state are converted to met as described under “Experimental Procedures.” l-Cys is added to initiate reaction and monitored as in solution (Fig. 5). The shorter x axis scale (seconds) for the T state samples was compared with the x axis used on the R state samples serves to highlight the difference in the rate between the T and R states and the biphasic nature of the data. a, HbA; b, HbE.
It is important to note that under the conditions employed and during the time frame of these experimental measurements, relative structural instability is not indicated for aquometHbE compared with aquometHbA. In addition, a comparison of the time course for autooxidation of oxygenated forms of HbE and HbA revealed comparable autoxidation rates based on metHb formation occurring over time frames that exceed those of the reduction and reductase measurements (data not shown).
DISCUSSION
The findings presented demonstrate the first reported in vitro functional differences for HbE relative to HbA. The data imply that the functional difference arises from a difference in redox properties between the two hemoglobins. An explanation for the observed changes in nitrite reductase activity and l-Cys-mediated reduction based on perturbed steric effects within the R and T state is unlikely, given the similarity between HbA and HbE with respect to geminate and solvent phase recombination profiles and the similarity between HbE and HbA high resolution crystal structures. In brief, the presented results appear most consistent with an explanation of the decreased nitrite reductase reactivity of HbE arising from an increase in the HbE redox potential relative to HbA.
Our crystallographic studies show that the hydrogen bonding changes at the site of mutation do not have a significant impact on either the allosterically important α1β2 interface or the local tertiary structure at the site of the heme. These structural findings are further supported by the lack of significant differences in the CD spectra between HbA and HbE. Ligand binding properties and allosteric parameters all indicate very minor differences in CO or O2 reactivity between HbE and HbA. Based on the structural comparisons and the CO recombination measurements, it is not surprising that the oxygen affinity is not significantly affected by the mutation according to earlier reports (19, 20) and confirmed by our results.
Structural Impact of the β26 Mutation
The structural findings do reflect upon earlier reported in vitro instability of this mutant (21–24). As shown in Fig. 1, the βLys-26 substitution that results in a loss of H-bonds with a concomitant re-orientation of the nearest neighbor βHis-116 and the βHis-117 side chains and that of βLys-22 may also account for the established in vitro integral instability of the HbE tetramer. Specifically, the in vitro instability of HbE may be attributed to the loss of the H-bond between the βGlu-26 side chain with the guanidinium group of βArg-30, and the subsequent H-bond loss of the latter with the αPhe-117 carbonyl oxygen (positioned directly at the αβ chain interface); and next to the electrostatic changes that originate from βArg-30, βHis-116, βHis-117, and βHis-122. Moreover, the charge repulsion in the microenvironment of β26 and the changes in the orientation of βHis-116 and βHis-117 are indicated as a major key to HbE instability and propensity for oxidation.
Evidence that βHis-116 plays a critical role in stabilizing α1β1 interactions and inhibiting Hb oxidation was presented previously (82), using recombinant engineered Hb with β116 amino acid substitutions. This resulted in increased autooxidation rates and decreased subunit association, all of which are characteristic of HbE. βHis-116 nearest neighbor mutations result in clinical consequences; Hb Roma (βA115V) was identified in a heterozygous patient with mild erythrocytic hypochromia and microcytosis (83). Compared with HbA, the hemolysate (75% HbA (with minor hemoglobins)) and 25% Hb Roma, in the presence and absence of 2,3-bisphosphoglycerate, exhibited increased oxygen affinity and mild destabilization.
Overall, the conformational changes observed at βHis-116 and βHis-117, communicated by the βLys-26 substitution with the ensuing electrostatic change, may be the major attribute or at least a significant contributor that confers heightened in vitro instability of HbE. This is in contrast to a cyanmetHbE structure derived at high pH (75) wherein the authors attributed HbE thermal instability to the specific water structure observed in their cyanmetHbE structure derived at high pH. However, because water structure changes dramatically with crystallization conditions (as shown herein by our structures and in comparison with published hemoglobin structures), their conclusion becomes unconvincing as the source of HbE instability.
Functional Impact of the β26 Mutation
In the last decade, new hemoglobin functionalities have been proposed. For example, Hb has been implicated in the mechanism for hypoxic vasodilation (31, 35, 84). Although there is still considerable controversy as to the nature of the Hb-based mechanism, there is ample evidence that Hb can act as an allosterically controlled source of deliverable NO (84–87). One such proposed mechanism is based on deoxyhemoglobin functioning as a nitrite reductase converting nitrite to NO (37, 38, 88). It is clear that the generation of bioactive NO under physiological conditions is not likely to be accounted for by just the nitrite reductase reaction. Other reactions and mechanisms are still under investigation (30, 36, 89–91). Nonetheless, there are clear indications of the role of this reaction as seen in studies that show that the efficacy of potential Hb-based oxygen carriers are correlated with nitrite reductase activity (92–94).
The nitrite reductase rate is clearly slower for deoxyHbE than for deoxyHbA. An obvious question is as follows. Through what mechanism does the HbE mutation create this change in reactivity? The presented results implicate a change in the intrinsic nitrite reductase activity of both T and R state forms of HbE. The decreased NR rates for deoxy-hemes in both the T and R states of HbE relative to HbA, without evidence of a measurable change in the allosteric properties of HbE, point to a mechanism that directly affects the hemes in both quaternary states. This observation, along with the finding that there is an absence of significant differences in the x-ray structure, CO recombination kinetics, and oxygen binding properties, indicates that it is neither the R/T equilibrium nor the ligand binding properties that are the basis for the decrease in NR activity for HbE T and R states. Instead, the HbE/HbA differences in the redox properties suggest that the most likely mechanism for this alteration in the NR activity is a general increase in the redox potential for HbE that impacts the redox activity of both quaternary states.
In brief, the key impact of the HbE mutation upon function is manifested in the altered nitrite reductase activity observed in vitro. This finding has implications for pathophysiology and provides the basis for the proposal of a mechanism that may explain the contribution of HbE to pathophysiological consequences observed in HbE/β-thalassemia.
Implications for Pathophysiology
HbE/β-thalassemia patients manifest endothelial dysfunction (95). Endothelial dysfunction is associated with reduced production of NO through the endothelial NOS pathway and increased production of reactive oxygen species. Although still a subject of controversy, NO at the right concentration is likely to play a role in not only maintaining vascular tone but also scavenging reactive oxygen species. It has been proposed that lowered levels of NO in the RBC can affect the stability of the RBC and thus increase the rate of hemolysis, which can be a major source of inflammation (96, 97).
Impaired NO bioavailability is reported to be linked to endothelial dysfunction and vascular pathology in hemoglobinopathies such as sickle cell disease and thalassemia (98–101). Kato and Taylor (98) summarize NO-regulated pathways in vascular homeostasis that include vasoconstriction, platelet-platelet interaction and attachment, and release of procoagulant factors and growth factors. Animal models of sickle cell disease lend support to the NO status as a contributing factor in these pathologies (102, 103). Although the exact role of NO in sickle cell disease and thalassemia remains controversial (104), the in vitro findings of a reduced nitrite reductase activity for HbE merits further consideration as a contributing factor to the pathophysiology, especially that of HbE/β-thalassemia.
Two HbE properties are plausible contributors to the clinical setting as follow: (i) the observed nitrite reductase properties of HbE are consistent with a reduced capacity to generate bioactive NO in an RBC, and (ii) HbE has an enhanced affinity for RBC membrane ((HbE > HbC > HbS > HbA)) (105–107). Based on these two properties, we propose the following mechanism for how HbE exacerbates βE-thalassemia.
The RBCs in HbE/β-thalassemia patients are subject to stresses that contribute to RBC instability. Excess α chains, arising from heterozygous thalassemia, produce heme, hemichromes, labile iron that yields reactive oxygen species that bind to the lipid bilayer and band 3. This situation would further compromise the RBC membrane that is already corrupted with the high binding affinity of HbE. The increased presence of reactive species may heighten HbE instability (in the cytosol and bound to the membrane) that in turn would further elevate free globin chains and levels of heme, hemichromes, labile iron to over-ride the intrinsic RBC protective redox enzymes, and give rise to a stressed RBC leading to further membrane abnormalities and hemolysis. Membrane dysfunction, including band 3 (the anion transporter) would further interfere with the normal NO RBC physiology and “RBC NO metabolin” as proposed in Ref. (35).
This backdrop of a stressed RBC coupled with the absence or reduction of the alleged protective attributes of NO should lead to a chronic enhanced RBC hemolysis and negative vascular consequences. An implication of this proposed NO-associated mechanism is that therapeutic designs to elevate levels of bioactive NO, for example, by the use of S-nitrosoglutathione, might offset some of the clinical consequences of HbE/β-thalassemia.
The previously established enhanced binding of HbE to the RBC membrane may have additional consequences to further explain the HbE/β-thalassemia clinical picture. It has been proposed that Hb-containing RBC membrane fragments (i.e. microparticles) are taken up by macrophages, which result in increased levels of inflammatory factors, such as cytokines (96, 97). Enhanced Hb binding to RBC membrane fragments, yielded upon hemolysis, could result in a more potent trigger of inflammation and increased levels of potentially toxic non-transferrin-carried iron. Inflammation and non-heme iron overload are clinical features of HbE/β-thalassemia (108, 109). Thus, the novel finding of HbE as an altered nitrite reductase, coupled with the known enhanced binding of HbE to the RBC membrane in consideration of the aforementioned caveats, provides a new model to explain the severe pathophysiological consequences of HbE/β-thalassemia. RBC and transgenic animal experiments are being designed to test the validity of this model.
In summary, the in vitro findings are most consistent with a decreased ability of HbE to generate NO from nitrite as a result of an increased HbE redox potential arising from small local tertiary structure changes in the heme environment engendered by the βLys-26 substitution. Overall, this in vitro study reveals structural and functional properties of HbE with implications that provide a framework for a new paradigm of molecular and cellular mechanisms that may contribute to HbE-associated pathophysiology.
This work was supported, in whole or in part, by National Institutes of Health Grants R21 DK064123 (to R. E. H.) and R21 HL106421 (to R. E. H. and J. M. F.). This work was also supported by American Heart Association Heritage Affiliate Grant-in-aid 0755906T (to R. E. H.) and FJC, a foundation of philanthropic funds (to J. M. F.).
J. M. Friedman, C. J. Roche, D. Dansker, and U. Samuni, unpublished results.
- NR
- nitrite reductase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PDB
- Protein Data Bank.
REFERENCES
- 1. Giardine B., van Baal S., Kaimakis P., Riemer C., Miller W., Samara M., Kollia P., Anagnou N. P., Chui D. H., Wajcman H., Hardison R. C., Patrinos G. P. (2007) Hum. Mutat. 28, 206 [DOI] [PubMed] [Google Scholar]
- 2. Forget B. H., Higgs D. R. (2009) in Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (Steinberg M. H., Forget B. G., Higgs D. R., Weatherall D. J. eds) 2nd Ed., pp. 25–26, Cambridge University Press, New York [Google Scholar]
- 3. Chotivanich K., Udomsangpetch R., Pattanapanyasat K., Chierakul W., Simpson J., Looareesuwan S., White N. (2002) Blood 100, 1172–1176 [PubMed] [Google Scholar]
- 4. Williams T. N. (2006) Curr. Opin. Microbiol. 9, 388–394 [DOI] [PubMed] [Google Scholar]
- 5. Vichinsky E. (2007) American Society Hematology Education Program, pp. 79–83 [DOI] [PubMed] [Google Scholar]
- 6. Traeger J., Wood W. G., Clegg J. B., Weatherall D. J. (1980) Nature 288, 497–499 [DOI] [PubMed] [Google Scholar]
- 7. Winichagoon P., Fucharoen S., Wilairat P., Chihara K., Fukumaki Y. (1995) Southeast Asian J. Trop. Med. Public Health 26, Suppl. 1, 241–245 [PubMed] [Google Scholar]
- 8. Weatherall D. J. (2000) J. Pediatr. Hematol. Oncol. 22, 551 [DOI] [PubMed] [Google Scholar]
- 9. Fucharoen S., Ketvichit P., Pootrakul P., Siritanaratkul N., Piankijagum A., Wasi P. (2000) J. Pediatr. Hematol. Oncol. 22, 552–557 [DOI] [PubMed] [Google Scholar]
- 10. Singer S. T., Kuypers F. A., Olivieri N. F., Weatherall D. J., Mignacca R., Coates T. D., Davies S., Sweeters N., Vichinsky E. P. (2005) Br. J. Haematol. 131, 378–388 [DOI] [PubMed] [Google Scholar]
- 11. Premawardhena A., De Silver S., Arambepola M., Olivieri N. F., Vichinsky E. P., Merson L., Muraco G., Allen A., Fisher C., Peto T., Weatherall D. J. (2005) Ann. N.Y. Acad. Sci. 1054, 33–39 [DOI] [PubMed] [Google Scholar]
- 12. Rees D. C. (2000) J. Pediatr. Hematol. Oncol. 22, 567–572 [DOI] [PubMed] [Google Scholar]
- 13. Fucharoeon S. (2001) in Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (Steinberg M. H., Forget B. G., Higgs D. R., Nagel R. L., Bunn H. F. eds) Cambridge University Press, New York [Google Scholar]
- 14. Olivieri N. F., De Silva S., Premawardena A., Sharma S., Viens A. M., Taylor C. M., Brittenham G. M., Weatherall D. J. (2000) J. Pediatr. Hematol. Oncol. 22, 593–597 [DOI] [PubMed] [Google Scholar]
- 15. Winichagoon P., Fucharoen S., Weatherall D., Wasi P. (1985) Am. J. Hematol. 20, 217–222 [DOI] [PubMed] [Google Scholar]
- 16. Watanapokasin Y., Winichagoon P., Fuchareon S., Wilairat P. (2000) Hemoglobin 24, 105–116 [DOI] [PubMed] [Google Scholar]
- 17. Nuntakarn L., Fucharoen S., Fucharoen G., Sanchaisuriya K., Jetsrisuparb A., Wiangnon S. (2009) Blood Cells Mol. Dis. 42, 32–35 [DOI] [PubMed] [Google Scholar]
- 18. Nuinoon M., Makarasara W., Mushiroda T., Setianingsih I., Wahidiyat P. A., Sripichai O., Kumasaka N., Takahashi A., Svasti S., Munkongdee T., Mahasirimongkol S., Peerapittayamongkol C., Viprakasit V., Kamatani N., Winichagoon P., Kubo M., Nakamura Y., Fucharoen S. (2010) Hum. Genet. 127, 303–314 [DOI] [PubMed] [Google Scholar]
- 19. Bunn H. F., Meriwether W. D., Balcerzak S. P., Rucknagel D. L. (1972) J. Clin. Invest. 51, 2984–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gacon G., Wajcman H., Labie D., Najman A. (1974) FEBS Lett. 41, 147–150 [DOI] [PubMed] [Google Scholar]
- 21. Macdonald V. W., Charache S. (1983) J. Lab. Clin. Med. 102, 762–772 [PubMed] [Google Scholar]
- 22. Shinar E., Rachmilewitz E. A. (1990) Semin. Hematol. 27, 70–82 [PubMed] [Google Scholar]
- 23. Shinar E., Rachmilewitz E. A. (1990) Ann. N.Y. Acad. Sci. 612, 118–126 [DOI] [PubMed] [Google Scholar]
- 24. Chiu D. T., van den Berg J., Kuypers F. A., Hung I. J., Wei J. S., Liu T. Z. (1996) Free Radic. Biol. Med. 21, 89–95 [DOI] [PubMed] [Google Scholar]
- 25. Rees D. C., Clegg J. B., Weatherall D. J. (1998) Blood 92, 2141–2146 [PubMed] [Google Scholar]
- 26. Luchsinger B. P., Rich E. N., Gow A. J., Williams E. M., Stamler J. S., Singel D. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McMahon T. J., Stamler J. S. (1999) Methods Enzymol. 301, 99–114 [DOI] [PubMed] [Google Scholar]
- 28. Gow A. J., Stamler J. S. (1998) Nature 391, 169–173 [DOI] [PubMed] [Google Scholar]
- 29. Jia Y., Xu L., Turner D. J., Martin J. G. (1996) J. Appl. Physiol. 80, 404–410 [DOI] [PubMed] [Google Scholar]
- 30. Angelo M., Singel D. J., Stamler J. S. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 8366–8371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luchsinger B. P., Rich E. N., Yan Y., Williams E. M., Stamler J. S., Singel D. J. (2005) J. Inorg. Biochem. 99, 912–921 [DOI] [PubMed] [Google Scholar]
- 32. Nagababu E., Ramasamy S., Abernethy D. R., Rifkind J. M. (2003) J. Biol. Chem. 278, 46349–46356 [DOI] [PubMed] [Google Scholar]
- 33. Nagababu E., Ramasamy S., Rifkind J. M. (2006) Nitric Oxide 15, 20–29 [DOI] [PubMed] [Google Scholar]
- 34. Nagababu E., Ramasamy S., Rifkind J. M. (2007) Biochemistry 46, 11650–11659 [DOI] [PubMed] [Google Scholar]
- 35. Kim-Shapiro D. B., Schechter A. N., Gladwin M. T. (2006) Arterioscler. Thromb. Vasc. Biol. 26, 697–705 [DOI] [PubMed] [Google Scholar]
- 36. Gladwin M. T., Grubina R., Doyle M. P. (2009) Acc. Chem. Res. 42, 157–167 [DOI] [PubMed] [Google Scholar]
- 37. Gladwin M. T., Kim-Shapiro D. B. (2008) Blood 112, 2636–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grubina R., Huang Z., Shiva S., Joshi M. S., Azarov I., Basu S., Ringwood L. A., Jiang A., Hogg N., Kim-Shapiro D. B., Gladwin M. T. (2007) J. Biol. Chem. 282, 12916–12927 [DOI] [PubMed] [Google Scholar]
- 39. Rifkind J. M., Nagababu E., Cao Z., Barbiro-Michaely E., Mayevsky A. (2009) Adv. Exp. Med. Biol. 645, 27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang Z., Shiva S., Kim-Shapiro D. B., Patel R. P., Ringwood L. A., Irby C. E., Huang K. T., Ho C., Hogg N., Schechter A. N., Gladwin M. T. (2005) J. Clin. Invest. 115, 2099–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roche C. J., Dantsker D., Samuni U., Friedman J. M. (2006) J. Biol. Chem. 281, 36874–36882 [DOI] [PubMed] [Google Scholar]
- 42. Grubina R., Basu S., Tiso M., Kim-Shapiro D. B., Gladwin M. T. (2008) J. Biol. Chem. 283, 3628–3638 [DOI] [PubMed] [Google Scholar]
- 43. Bonaventura C., Henkens R., Alayash A. I., Crumbliss A. L. (2007) IUBMB Life 59, 498–505 [DOI] [PubMed] [Google Scholar]
- 44. Lalezari I., Lalezari P., Poyart C., Marden M., Kister J., Bohn B., Fermi G., Perutz M. F. (1990) Biochemistry 29, 1515–1523 [DOI] [PubMed] [Google Scholar]
- 45. Takeoka S., Sakai H., Kose T., Mano Y., Seino Y., Nishide H., Tsuchida E. (1997) Bioconjug. Chem. 8, 539–544 [DOI] [PubMed] [Google Scholar]
- 46. Romero F. J., Ordoñez I., Arduini A., Cadenas E. (1992) J. Biol. Chem. 267, 1680–1688 [PubMed] [Google Scholar]
- 47. Lohr N. L., Keszler A., Pratt P., Bienengraber M., Warltier D. C., Hogg N. (2009) J. Mol. Cell. Cardiol. 47, 256–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Q., Bouhassira E. E., Besse A., Suzuka S. M., Fabry M. E., Nagel R. L., Hirsch R. E. (2004) Blood Cells Mol. Dis. 33, 303–307 [DOI] [PubMed] [Google Scholar]
- 49. Hirsch R. E., Lin M. J., Vidugiris G. J., Huang S., Friedman J. M., Nagel R. L., Vidugirus G. V. (1996) J. Biol. Chem. 271, 372–375 [DOI] [PubMed] [Google Scholar]
- 50. Baldwin J. M. (1980) J. Mol. Biol. 136, 103–128 [DOI] [PubMed] [Google Scholar]
- 51. Shaanan B. (1983) J. Mol. Biol. 171, 31–59 [DOI] [PubMed] [Google Scholar]
- 52. Perutz M. F., Greer J. (1970) Biochem. J. 119, 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hirsch R. E., Lin M. J., Nagel R. L. (1988) J. Biol. Chem. 263, 5936–5939 [PubMed] [Google Scholar]
- 54. Vekilov P. G., Feeling-Taylor A., Hirsch R. E. (2003) Methods Mol. Med. 82, 155–176 [DOI] [PubMed] [Google Scholar]
- 55. Safo M. K., Abraham D. J. (2003) Methods Mol. Med. 82, 1–19 [DOI] [PubMed] [Google Scholar]
- 56. Otwinowski Z., Minor W. (1997) Macromolecular Crystallography, Part a, Vol. 276, pp. 307–326, Academic Press, New York [Google Scholar]
- 57. Collaborative Computational Project, Number 4 (1994) Acta Crystallogr. D Biol. Crystallogr. D50, 760–763 [Google Scholar]
- 58. Storoni L. C., McCoy A. J., Read R. J. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 [DOI] [PubMed] [Google Scholar]
- 59. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 60. Hooft R. W., Vriend G., Sander C., Abola E. E. (1996) Nature 381, 272 [DOI] [PubMed] [Google Scholar]
- 61. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 62. Krissinel E., Henrick K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 63. Khan I., Shannon C. F., Dantsker D., Friedman A. J., Perez-Gonzalez-de-Apodaca J., Friedman J. M. (2000) Biochemistry 39, 16099–16109 [DOI] [PubMed] [Google Scholar]
- 64. Samuni U., Roche C., Dantsker D., Friedman J. (2008) in Dioxygen Binding and Sensing Proteins. A Tribute to Beatrice and Jonathan Wittenberg (Bolognesi M., di Prisco G., Verde C. eds) pp. 133–159, Springer, New York [Google Scholar]
- 65. Dantsker D., Samuni U., Friedman A. J., Yang M., Ray A., Friedman J. M. (2002) J. Mol. Biol. 315, 239–251 [DOI] [PubMed] [Google Scholar]
- 66. Samuni U., Roche C. J., Dantsker D., Friedman J. M. (2007) J. Am. Chem. Soc. 129, 12756–12764 [DOI] [PubMed] [Google Scholar]
- 67. Samuni U., Dantsker D., Khan I., Friedman A. J., Peterson E., Friedman J. M. (2002) J. Biol. Chem. 277, 25783–25790 [DOI] [PubMed] [Google Scholar]
- 68. Samuni U., Juszczak L., Dantsker D., Khan I., Friedman A. J., Pérez-González-de-Apodaca J., Bruno S., Hui H. L., Colby J. E., Karasik E., Kwiatkowski L. D., Mozzarelli A., Noble R., Friedman J. M. (2003) Biochemistry 42, 8272–8288 [DOI] [PubMed] [Google Scholar]
- 69. Samuni U., Dantsker D., Juszczak L. J., Bettati S., Ronda L., Mozzarelli A., Friedman J. M. (2004) Biochemistry 43, 13674–13682 [DOI] [PubMed] [Google Scholar]
- 70. Samuni U., Roche C. J., Dantsker D., Juszczak L. J., Friedman J. M. (2006) Biochemistry 45, 2820–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Salhany J. M. (2008) Biochemistry 47, 6059–6072 [DOI] [PubMed] [Google Scholar]
- 72. Antonini E., Brunori M. (1971) Hemoglobin and Myoglobin in Their Reactions with Ligands, 1st Ed., North-Holland Publishing Co., Amsterdam [Google Scholar]
- 73. Crawford J. H., Isbell T. S., Huang Z., Shiva S., Chacko B. K., Schechter A. N., Darley-Usmar V. M., Kerby J. D., Lang J. D., Jr., Kraus D., Ho C., Gladwin M. T., Patel R. P. (2006) Blood 107, 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Peterson E. S., Shinder R., Khan I., Juczszak L., Wang J., Manjula B., Acharya S. A., Bonaventura C., Friedman J. M. (2004) Biochemistry 43, 4832–4843 [DOI] [PubMed] [Google Scholar]
- 75. Sen U., Dasgupta J., Choudhury D., Datta P., Chakrabarti A., Chakrabarty S. B., Chakrabarty A., Dattagupta J. K. (2004) Biochemistry 43, 12477–12488 [DOI] [PubMed] [Google Scholar]
- 76. Geraci G., Parkhurst L. J. (1981) Methods Enzymol. 76, 262–275 [DOI] [PubMed] [Google Scholar]
- 77. Khan I., Dantsker D., Samuni U., Friedman A. J., Bonaventura C., Manjula B., Acharya S. A., Friedman J. M. (2001) Biochemistry 40, 7581–7592 [DOI] [PubMed] [Google Scholar]
- 78. Chavez M. D., Courtney S. H., Chance M. R., Kiula D., Nocek J., Hoffman B. M., Friedman J. M., Ondrias M. R. (1990) Biochemistry 29, 4844–4852 [DOI] [PubMed] [Google Scholar]
- 79. Sassaroli M., Rousseau D. L. (1987) Biochemistry 26, 3092–3098 [DOI] [PubMed] [Google Scholar]
- 80. Huang J., Juszczak L. J., Peterson E. S., Shannon C. F., Yang M., Huang S., Vidugiris G. V., Friedman J. M. (1999) Biochemistry 38, 4514–4525 [DOI] [PubMed] [Google Scholar]
- 81. Bonaventura C., Taboy C. H., Low P. S., Stevens R. D., Lafon C., Crumbliss A. L. (2002) J. Biol. Chem. 277, 14557–14563 [DOI] [PubMed] [Google Scholar]
- 82. Adachi K., Yang Y., Lakka V., Wehrli S., Reddy K. S., Surrey S. (2003) Biochemistry 42, 10252–10259 [DOI] [PubMed] [Google Scholar]
- 83. Manconi B., De Rosa M. C., Cappabianca M. P., Olianas A., Carelli Alinovi C., Mastropietro F., Ponzini D., Amato A., Pellegrini M. (2010) Biochim. Biophys. Acta 1800, 327–335 [DOI] [PubMed] [Google Scholar]
- 84. Allen B. W., Stamler J. S., Piantadosi C. A. (2009) Trends Mol. Med. 15, 452–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Isbell T. S., Gladwin M. T., Patel R. P. (2007) Am. J. Physiol. Heart Circ. Physiol. 293, H2565–H2572 [DOI] [PubMed] [Google Scholar]
- 86. Gladwin M. T. (2005) Am. J. Respir. Cell Mol. Biol. 32, 363–366 [DOI] [PubMed] [Google Scholar]
- 87. Gow A. J. (2005) Am. J. Respir. Cell Mol. Biol. 32, 479–482 [DOI] [PubMed] [Google Scholar]
- 88. Basu S., Grubina R., Huang J., Conradie J., Huang Z., Jeffers A., Jiang A., He X., Azarov I., Seibert R., Mehta A., Patel R., King S. B., Hogg N., Ghosh A., Gladwin M. T., Kim-Shapiro D. B. (2007) Nat. Chem. Biol. 3, 785–794 [DOI] [PubMed] [Google Scholar]
- 89. Cao Z., Bell J. B., Mohanty J. G., Nagababu E., Rifkind J. M. (2009) Am. J. Physiol. Heart Circ. Physiol. 297, H1494–H1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Roche C. J., Friedman J. M. (2010) Nitric Oxide 22, 180–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Salgado M. T., Nagababu E., Rifkind J. M. (2009) J. Biol. Chem. 284, 12710–12718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kluger R. (2010) Curr. Opin. Chem. Biol. 14, 538–543 [DOI] [PubMed] [Google Scholar]
- 93. Lui F. E., Dong P., Kluger R. (2008) Biochemistry 47, 10773–10780 [DOI] [PubMed] [Google Scholar]
- 94. Cabrales P., Friedman J. (2007) Transfusion Alternatives in Transfusion Medicine 9, 281–293 [Google Scholar]
- 95. Kukongviriyapan V., Somparn N., Senggunprai L., Prawan A., Kukongviriyapan U., Jetsrisuparb A. (2008) Pediatr. Cardiol. 29, 130–135 [DOI] [PubMed] [Google Scholar]
- 96. Hod E. A., Zhang N., Sokol S. A., Wojczyk B. S., Francis R. O., Ansaldi D., Francis K. P., Della-Latta P., Whittier S., Sheth S., Hendrickson J. E., Zimring J. C., Brittenham G. M., Spitalnik S. L. (2010) Blood 115, 4284–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Puddu P., Puddu G. M., Cravero E., Muscari S., Muscari A. (2010) Can. J. Cardiol. 26, 140–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kato G. J., Taylor J. G., 6th (2010) Br. J. Haematol. 148, 690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kato G. J., Wang Z., Machado R. F., Blackwelder W. C., Taylor J. G., 6th, Hazen S. L. (2009) Br. J. Haematol. 145, 506–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Morris C. R. (2008) Hematology American Society Hematology Education Program, 177–185, National Institutes of Health, Bethesda [Google Scholar]
- 101. Morris C. R., Gladwin M. T., Kato G. J. (2008) Curr. Mol. Med. 8, 620–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hsu L. L., Champion H. C., Campbell-Lee S. A., Bivalacqua T. J., Manci E. A., Diwan B. A., Schimel D. M., Cochard A. E., Wang X., Schechter A. N., Noguchi C. T., Gladwin M. T. (2007) Blood 109, 3088–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kaul D. K., Zhang X., Dasgupta T., Fabry M. E. (2008) Am. J. Physiol. Heart Circ. Physiol. 295, H39–H47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bunn H. F., Nathan D. G., Dover G. J., Hebbel R. P., Platt O. S., Rosse W. F., Ware R. E. (2010) Blood 116, 687–692 [DOI] [PubMed] [Google Scholar]
- 105. Shaklai N., Yguerabide J., Ranney H. M. (1977) Biochemistry 16, 5585–5592 [DOI] [PubMed] [Google Scholar]
- 106. Shaklai N., Sharma V. S., Ranney H. M. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 65–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chen Q., Balazs T. C., Nagel R. L., Hirsch R. E. (2006) FEBS Lett. 580, 4485–4490 [DOI] [PubMed] [Google Scholar]
- 108. Conran N., Costa F. F. (2009) Clin. Biochem. 42, 1824–1838 [DOI] [PubMed] [Google Scholar]
- 109. Chakraborty I., Mitra S., Gachhui R., Kar M. (2010) Ann. Acad. Med. Singapore 39, 13–16 [PubMed] [Google Scholar]