

Abstract
Analysis of the changing mRNA expression profile of Mycobacterium tuberculosis though the course of infection promises to advance our understanding of how mycobacteria are able to survive the host immune response. The difficulties of sample extraction from distinct mycobacterial populations, and of measuring mRNA expression profiles of multiple genes has limited the impact of gene expression studies on our interpretation of this dynamic infection process. The development of whole genome microarray technology together with advances in sample collection have allowed the expression pattern of the whole M. tuberculosis genome to be compared across a number of different in vitro conditions, murine and human tissue culture models and in vivo infection samples. This review attempts to produce a summative model of the M. tuberculosis response to infection derived from or reflected in these gene expression datasets. The mycobacterial response to the intracellular environment is characterised by the utilisation of lipids as a carbon source and the switch from aerobic/microaerophilic to anaerobic respiratory pathways. Other genes induced in the macrophage phagosome include those likely to be involved in the maintenance of the cell wall and genes related to DNA damage, heat shock, iron sequestration and nutrient limitation. The comparison of transcriptional data from in vitro models of infection with complex in vivo samples, together with the use of bacterial RNA amplification strategies to sample defined populations of bacilli, should allow us to make conclusions about M. tuberculosis physiology and host microenvironments during natural infection.
Keywords: Intracellular, expression, microarray, transcriptomics, mycobacterium, macrophage, host pathogen interactions
INTRODUCTION
The mycobacterial transcriptome consists of the mRNA content of bacilli in a particular environment at a specific time. This represents, subject to additional levels of mRNA and protein regulation, the products required by mycobacteria for continued survival. Little is known of the events that occur at the gene expression level in host tissues after infection with Mycobacterium tuberculosis. However the development of techniques such as subtractive hybridisation, quantitative RT-PCR and microarray technologies have allowed the transcriptional responses of both host and pathogen to be explored during infection. Thus it is possible to determine the immediate genome-wide response (as measured by mRNA expression) of mycobacteria to a stimulus or environment. The comparison of mycobacterial mRNA expression patterns from intracellular infection models provides a snapshot of the network of interactions between bacilli and the external environment. These transcriptional datasets can be used to (a) help understand the changing metabolism of infecting bacilli, which may in turn provide insights into the host microenvironments encountered by bacilli; (b) identify pathogenic mechanisms used by M. tuberculosis to evade and modulate the immune response; and (c) define the interactions between host and pathogen throughout the multi-factorial and dynamic process of infection. This review outlines the mycobacterial response to infection derived from the mRNA expression patterns of M. tuberculosis extracted from intracellular environments.
M. tuberculosis Pathogenesis
The complex pathogenesis of M. tuberculosis is influenced by a multitude of factors including host and bacterial genetic backgrounds, immunological status and environmental circumstances. The earliest encounter between infecting bacilli and the immune system is likely to be with alveolar macrophages; that some bacilli survive this initial contact provides a relevant starting point to begin investigating host-M. tuberculosis interactions.
Primary infection with M. tuberculosis takes place in the lungs, and may eventually result in pulmonary tuberculosis; however only 5-10% of those individuals infected actually develop clinical disease [1]. On infection a proportion of infecting bacilli are able to survive and successfully multiply within the intracellular environment of the alveolar macrophage. A fundamental step in mycobacterial survival is the halt of phagosome maturation in macrophages after phagocytosis of M. tuberculosis [2]. The bacilli therefore avoid destruction in the fused phagolysosome by acidic hydrolases [3], with the pH of mycobacterial-containing phagosomes remaining at 6.5 [4]. Characterisation of the surface markers of M. tuberculosis-containing phagosomes reveal that many markers of early endosomes and from the plasma membrane are present, for example mycobacterial phagosomes retain the Rab5 molecule but do not acquire the late endosomal marker Rab7, which is believed to be involved in the transition from early to late endosome [5]. Successful control of M. tuberculosis infection is achieved in the lung by the formation of granulomas, where activated macrophages surround the site of infection preventing further dissemination of bacilli and limiting tissue damage [6]. The human granuloma consists of many different cell types including macrophages, giant cells, fibroblasts, T cells and B cells [7], and requires the orchestration of a range of chemokines, cytokines, adhesins and integrins to coordinate the recruitment, migration and retention of cells to the granuloma [8]. A balance is reached where the mycobacterial infection has been controlled, but the bacilli inside the granuloma have not been destroyed [9]. This situation continues until the immunological balance is shifted in such a way that allows the bacilli to begin successfully multiplying again resulting in pulmonary disease [10].
The immuno-modulatory capacity of M. tuberculosis at the interface of bacilli and macrophage has been established for example by the secretion of eukaryotic-like protein kinase G by mycobacteria within the macrophage phagosome [11], and the demonstration that mycobacterial lipids and proteins are trafficked out of infected phagosomes and into uninfected bystander macrophages [12]. The dynamic interaction between mycobacteria and host immune system is also exemplified by the variable contributions of immune cell surface receptors (e.g. TLR receptors) to the successful control of M. tuberculosis at different stages of infection [13]. Further characterisation of the cross-talk between host immune system and M. tuberculosis may reveal novel virulence mechanisms and suggest future innovative treatment strategies.
The metabolic state of bacilli through the course of human infection is largely unknown, especially that of persistent bacilli. However, transcriptional activity has been detected in persistent mycobacteria from in vivo murine models of infection using RT-PCR [14, 15]. The inference that persistent bacilli are metabolically active in some way is especially important in the design of new drugs against M. tuberculosis [16]. The significance of mycobacterial persistence and drug tolerance has been reviewed by Gomez and McKinney [17]. What is clear however is that the metabolic state of infecting bacilli is likely dependent on a number of factors including the oxygen tension and tissue structure of the surrounding environment in the human lung [18, 19].
A greater understanding of how mycobacteria are able to survive the complex environments encountered during infection will aid novel treatment strategies targeted more specifically at intracellular or persistent bacilli. Expression analyses have the power to inform us how the infecting bacilli are responding to the environments in which they reside, telling us a little about the different niches that mycobacteria occupy and how they are able to adapt. The interactions between M. tuberculosis and macrophage have been reviewed recently by Schnappinger at al. [20]; McKinney and Gomez [21]; and the immunology of M. tuberculosis control is described in detail by Flynn and Chan [7]; and Kaufmann [22]. This review focuses on the mycobacterial transcriptional response to the intracellular environment during early infection, and how the expression profile of M. tuberculosis might change with the progression of disease.
The M. tuberculosis Transcriptional Response to the Intracellular Environment
The understanding of mycobacterial interactions with the host has been enhanced by expression studies using (a) in vitro models, such as how bacilli respond to microaerophilic conditions [23, 24], as reviewed by Bacon and Marsh in this issue; (b) tissue culture models of M. tuberculosis infection using murine or human derived macrophages [25, 26]; (c) animal models such as the murine model of M. tuberculosis disease [27]; and (d) from human patient samples [19]. Here we review the limited expression data available from intracellular models of infection, concentrating on the studies using whole genome expression analyses but also incorporating data from quantitative RT-PCR, in situ hybridisation, promoter trap and subtractive/enrichment hybridisation methodologies. Table (1) details the publications to date that have looked at the in vivo expression of multiple M. tuberculosis genes, as opposed to mutant or essentiality studies. Mycobacterial gene expression patterns identified in tissue culture or in vitro models of infection enable us to dissect out the complex interactions between pathogen and host. We review M. tuberculosis expression profiles derived from macrophage infection models in comparison with in vivo transcriptional data. Gene expression data may be classified and interrogated by functional category, chromosome position or overlaid onto metabolic pathways. This third approach is the most informative, helping to define the physiological state of infecting mycobacteria, and has been adopted here wherever possible. The major metabolic themes of intracellular M. tuberculosis gene expression are detailed below and depicted in Fig. (1).
Table 1. A List of Publications Describing the mRNA Expression of Multiple (5 or more) M. tuberculosis Genes from Intracellular Models of Infection, Detailing the Method Used and the Key Pathways Identified or Investigated. (mbRT-PCR – molecular beacon RT-PCR, SCOTS – selective capture of transcribed sequences).
| Reference | Technique | In vivo model | Key pathways |
|---|---|---|---|
| Rachman et al. [19] | Membrane array | Human lung tissue | Cell wall/lipid synthesis Respiratory state |
| Fenhalls et al. [18] | In situ hybridisation | Human lung tissue | Fatty acid metabolism Respiratory state |
| Cappelli et al. [26] | Membrane array | Human peripheral blood derived monocytes |
Transcriptional regulation Cell wall/lipid synthesis |
| Haydel and Clark-Curtiss [62] | SCOTS | Human peripheral blood derived monocytes |
Transcriptional regulation |
| Graham and Clark-Curtiss [32] |
SCOTS | Human peripheral blood derived monocytes |
Fatty acid metabolism Transcriptional regulation |
| Li et al. [33] | Subtractive hybridisation to membrane array |
THP-1 cells | Cell wall/lipid synthesis Transcriptional regulation |
| Dubnau et al. [80] | Promoter trap | THP-1 cells | Fatty acid metabolism Amino acid metabolism |
| Li et al. [41] | Subtractive hybridisation to membrane array |
THP-1 cells | Cell wall/lipid synthesis Iron sequestration |
| Gold et al. [40] | mbRT-PCR | THP-1 cells | Iron sequestration |
| Dubnau et al. [56] | Promoter trap | C57BL/6 murine lung | Antigen expression Transcriptional regulation |
| Shi et al. [34] | mbRT-PCR | C57BL/6 murine lung | Respiratory state |
| Talaat et al. [27] | Microarray | BALB/c, SCID Murine lung | Fatty acid metabolism Iron sequestration |
| Shi et al. [37] | mbRT-PCR | C57BL/6 murine lung | Respiratory state |
| Voskuil et al. [36] | Quantitative RT-PCR | C57BL/6 murine lung | Respiratory state |
| Timm et al. [30] | Quantitative RT-PCR | C57BL/6 Murine lung, human lung tissue |
Iron sequestration Fatty acid metabolism |
| Karakousis et al. [31] | Microarray | Artificial SKH1 murine granuloma | Respiratory state Amino acid metabolism |
| Rachman et al. [28] | Membrane array | Murine bone marrow derived macrophages |
Amino acid metabolism Cell wall/lipid synthesis |
| Schnappinger et al. [25] | Microarray | Murine bone marrow derived macrophages |
Fatty acid metabolism Respiratory state |
Fig. (1).
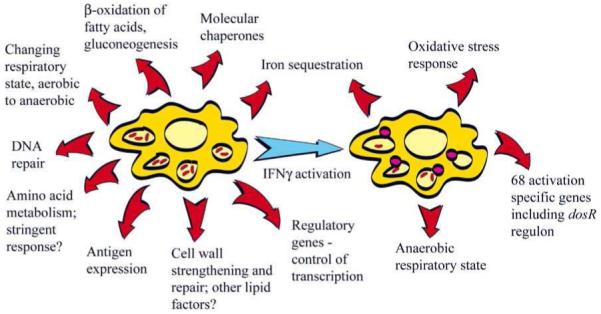
An illustration of the broad transcriptional responses of M. tuberculosis to the intracellular environments of naïve and IFNγ-activated macrophages. Macrophage activation (highlighted by phagolysosome fusion) exposes intracellular bacilli to nitric oxide, and results in decreased mycobacterial survival. Adapted from [21].
β-Oxidation of Fatty Acids
The induction of multiple genes in four of the five reactions required for the β-oxidation of fatty acids (fadD3, 9, 10, 19, fadDE5, 14, 22, 23, 24, 27, 28, 29, 31, echA19, fadB2, 3, fadA5, 6), and the subsequent metabolism of degradation products via the citric and glyoxylate cycles (icl, gltA1, Rv1130) suggests that M. tuberculosis utilises a diverse range of fatty acids as a carbon source during intraphagosomal growth [25, 28]. The induction of these genes involved in fatty acid metabolism on infection of macrophages indicates that the phagosomal compartment that bacilli occupy in at least one stage of infection is low in other sources of carbon such as glucose and glycerol [29]. In addition the up-regulation of pckA, encoding a phosphoenolpyruvate carboxykinase, reveals that fatty acids may be converted through gluconeogenesis into sugars. Genes in these fatty acid metabolism pathways (namely icl and pckA) have also been shown by RT-PCR [30] and microarray analysis [27] to be induced in the mouse lung after infection, and within artificial murine granulomas [31]. The up-regulation of icl has also been described on infection of human-derived macrophage-like cells, using SCOTS [32] and subtractive hybridisation [33], and inside non-necrotic human lung granulomas by in situ hybridisation [18]. The source of these fatty acids is unknown; it may be that the smorgasbord of complex lipids generated by M. tuberculosis is utilised, or host lipids, or a combination of the two depending on the particular microenvironment in which the bacilli find themselves. This pattern of fatty acid utilisation does however appear to be common to all infection models tested. It may therefore represent a fundamental adaptation to intracellular growth and an important target pathway for M. tuberculosis drug development.
Respiratory State
Transcriptional changes have been identified in the aerobic and anaerobic respiratory pathways of M. tuberculosis during infection. Genes related to aerobic respiration such as the type I NADH dehydrogenase (nuoABDFIL), and cytochrome C reductase (qcrC) were identified to be repressed inside the macrophage phagosome; in contrast genes encoding products involved in alternative electron acceptor pathways (frdA a fumarate reductase, narX a nitrate reductase and ndh a type II NADH dehydrogenase) were induced [25]. A similar profile of gene expression has been described in the mouse lung during infection by molecular beacon RT-PCR and has lead Shi et al. [34] to propose that M. tuberculosis may exist in three different respiratory states depending on the microenvironment encountered. During acute infection the energy-efficient type I NADH dehydrogenase (exemplified by the expression of nuoB) together with the aa3-cytochrome C complex (assayed by following the expression of qcrC and ctaD) as a terminal oxidase is utilised. Immune cell activation leading to the generation of nitric oxide blocks this aerobic respiratory pathway and drives M. tuberculosis into a transitional respiratory state characterised by the repression of the aa3 cytochrome C terminal oxidase complex (qcrC, ctaD) and induction of the bd-type terminal oxidase (exemplified by the expression of cydA) and nitrate respiration (assayed by following the expression of narK2). During chronic murine infection the bd-type terminal oxidase is down-regulated and nitrate is used as the final electron acceptor [34]. This pattern is also reflected in the expression of the dosR regulon, a set of around 50 genes induced by hypoxia [23, 35] and nitric oxide treatment [36]. This regulon was induced in M. tuberculosis after stimulation of murine macrophages with IFNγ and was dependent on the presence of a functioning murine NOS2 enzyme [25]. The up-regulation of dosR and around 20 other members of the regulon was also demonstrated within artificial murine granulomas [31]. Furthermore induction of the dosR regulon (assayed using the expression of hspX, Rv2623 and Rv2626c) was described to occur in the mouse lung as bacterial numbers began to fall following the induction of the adaptive immune response [37]; a similar profile was also observed for the expression of Rv1738, fdxA, acg, hspX and Rv2626c in the murine lung [36]. Therefore the dosR regulon, mediated by nitric oxide (and/or hypoxia), is induced in the murine model of infection on activation of the immune response and may be part of a mycobacterial response that leads to persistence in the host. The respiratory and metabolic status of persistent bacilli has recently been comprehensively reviewed by Boshoff and Barry [38].
The respiratory state of bacilli during human disease is less well documented. The expression of narX coding for a probable nitrate reductase was demonstrated to be expressed in M. tuberculosis located in human non-necrotic granulomas [18, 19]; the nitrate reductases narX, narG and the fumarate reductase, frdA were also found to be induced in the human pericavity and distant lung of tuberculosis patients [19]. Additionally the induction of both aerobic and anaerobic genes in human lung tissue [19] suggests that the respiratory status of infecting bacilli is dependent on the microenvironment encountered. The induction of the dosR regulon has not been fully demonstrated in human infection, although hspX has been reported to be up-regulated in the human lung [30]. The role of nitric oxide in the control of M. tuberculosis infection in man is unclear, however the ability of M. tuberculosis to utilise several respiration pathways and the induction of alternative electron acceptors during infection provide possible drug development targets. Transcriptomic analysis of smaller more distinct populations of bacilli during natural infection may help to elucidate the variable respiratory state of M. tuberculosis in the human lung.
Inextricably linked to respiration state are the energy requirements of the infecting bacilli; genes encoding ATP synthase subunits (atpABD-H) were down-regulated in the murine macrophage phagosome and this pattern was repeated in naïve, IFNγ -activated or NOS2-deficient macrophages compared to in vitro growth [25]. Furthermore atpA and atpD were repressed during chronic infection of immuno-competent mice [34]. This repression of ATP-synthases in vivo is likely to simply reflect the different energy demands of bacilli growing logarithmically in vitro or inside the macrophage phagosome. However the differential regulation of genes encoding ATP synthase subunits may be involved in the maintenance of intracellular pH, or may represent the sudden requirement for metabolic activity as the bacilli adapt to a changing environment. This may explain the induction of atpE/F/H at 21 days after infection of immuno-competent compared to severe-immuno deficient (SCID) mice as the bacilli respond to the adaptive immune challenge in the immuno-competent environment [27].
Iron Sequestration
The lack of available iron during infection is a problem encountered by many pathogens, which is further exploited by the induction of iron-sequestering complexes as part of the host immune response [39]. M. tuberculosis genes involved in the generation of mycobactin, a siderophore-like iron-binding molecule (mbtA-J) were identified to be induced in IFNγ-activated murine macrophages [25]; a subset of genes up-regulated in low iron conditions were also induced in naïve murine macrophages. The induction of genes involved in the control of mycobacterial iron sequestration and storage has also been demonstrated to occur during murine infection; Timm et al. [30] reported the induction of mbtB in murine lungs, which coincided with the control of bacterial growth by the immune response. In addition the expression of bfrA (encoding an iron-storing bacterioferritin) was down-regulated [30]. This gene expression profile correlates well with the up-regulation of mbtD (involved in mycobactin synthesis), hupB (coding for an iron-regulated protein), and fdxA (encoding ferredoxin) at 21 days post infection in immuno-competent compared to severely immuno-deficient mice [27]. The M. tuberculosis genes mbtB, mbtI and Rv3402c regulated by IdeR (an iron-dependent regulator) were also induced on infection of human macrophage-like THP-1 cells [40]. Furthermore furB, encoding a probable ferric/zinc uptake regulator was also up-regulated on THP-1 infection [41]. The relative increase in the expression of genes involved in iron sequestration in activated macrophages (or immuno-competent mice) suggests that the bacilli find themselves in an iron-limited environment after immune activation, or that the mycobacterial requirement for iron increases in the microenvironment in which the bacilli reside.
In addition to iron, other moieties required for successful mycobacterial growth are likely to be limited at various stages of infection. Rachman et al. [19] observed an induction of amino acid transport-related genes in M. tuberculosis infected human lung sections. Indeed a number of genes identified to be induced by nutrient starvation were also demonstrated to be up-regulated after infection of naïve or activated murine macrophages [42, 25]. The requirement of relA (encoding (p)ppGpp synthase and hydrolase activities) as part of the stringent response has been demonstrated in long-term anaerobic models [43] and during murine infection [31]. A comparison of the expression profiles of a relA knockout mutant with WT M. tuberculosis under starvation conditions has identified around 160 genes that may be regulated by (p)ppGpp, including genes involved in anaerobic respiration (narH, narI) and fatty acid metabolism (aceAb, acpM, fas, kasA, kasB) [44]. Further exploration of the mycobacterial stringent response may reveal insights into how M. tuberculosis bacilli are able to adapt to nutrient-limited surroundings during infection.
Mycobacterial Lipid Moieties
The mycobacterial cell wall acts as the interface between the infecting bacilli and the host environment, therefore it is unsurprising to find genes involved in the synthesis and modification of cell wall associated fatty acids up-regulated in vivo. Genes involved in mycolic acid modification (desA1/2 and umaA) were found to be induced inside the murine phagosome [25]. Additionally the desaturases encoded by desA1 and desA3 were up-regulated in infected human lung together with mmaA3/A4 and umaA [19]. The induction of these genes is likely connected with the maintenance of the hydrophobic nature of the cell wall barrier from damage during macrophage infection. A second set of genes coding for polyketide synthases involved in the biosynthesis of mycobacterial complex lipids have also been demonstrated to be induced during macrophage infection. Mycocerosic acid synthase (mas) and the related genes fadD28 [41] and fadD26 [25] were up-regulated in the macrophage phagosome; these gene products are part of the biosynthetic pathway of phthiocerol dimycocerosate which is required for successful mycobacterial infection [45]. In addition the induction of pks2 [32], necessary for sulpholipid generation [46], and pks6 [33], involved in the expression of an unknown polar lipid [47], in human macrophages further highlights the potential importance of mycobacterial complex lipids during infection. Whether these lipid moieties are part of a response to strengthen the lipid-rich mycobacterial cell wall, or play a more active role in host-pathogen interactions is currently unknown [12].
DNA Repair
Mycobacterial genes involved in DNA recombination and repair were up-regulated in the intraphagosomal environment of both naïve and activated murine macrophages (alkA, recX, recC, dinF and radA). Interestingly this pattern of gene expression was also observed on infection of NOS2-deficient macrophages, indicating that the probable M. tuberculosis response to DNA damage occurs in the absence of nitric oxide [25]. A similar subset of genes mfd, dinF/G and polA were also induced on murine macrophage infection [28]. Furthermore genes involved in mycobacterial DNA repair have also been demonstrated to be up-regulated on infection of human macrophages (uvrA) [32], and the human lung (dinX, dinF, gyrA/B) [19].
Molecular Chaperones
The induction of molecular chaperones involved in the folding and translocation of polypeptides has been observed in M. tuberculosis under a number of stress conditions, exemplified by the heat shock response [48]. Rachman et al. [19] identified a number of genes related to chaperone biosynthetic pathways (groEL/ES, suhB, dnaJ1/2, dnaK, hspR and hspX) to be up-regulated in the human lung during M. tuberculosis infection. Similarly the induction of groEL2 was reported after infection of human macrophage cell lines [41]. The activation of genes coding for molecular chaperones and DNA repair proteins may be part of a common stress response induced by multiple stimuli, for example different drug treatments [49, 50], the details of which are yet to be elucidated.
Antigen Expression
The induction of genes encoding members of the highly immunogenic PE/PE-PGRS and PPE families in vivo was demonstrated in human pulmonary tissue [19] and murine macrophages [25]. Interestingly the induction of some PE/PPE genes occurs alongside the repression of others in both macrophage and murine models [51], and this pattern of PE/PPE differential expression is repeated across many different growth/stress conditions [52]. The functional significance of these gene families, which command approximately 10% of the coding capacity of M. tuberculosis H37Rv is uncertain [53], however there is evidence that some of these proteins are found at the cell surface and may mediate interactions with the host [54]. Elucidation of the expression pattern of these genes, which may act as variable surface antigens during infection [55], may influence future vaccine design strategies. Other potential M. tuberculosis antigens such as members of the ESAT-6 family (esxH, esxO, esxV) were also identified to be expressed in the murine lung [56]. Shi et al. [57] identified differences in the transcription of genes encoding M. tuberculosis secreted antigens throughout murine lung infection, and Dahl et al. [44] have reported that the expression of many of these antigenic proteins may be under the control of relA as part of the mycobacterial stringent response. These observations highlight how gene expression approaches may be used to supplement antigenic studies and influence vaccine design strategy.
The Regulation of Mycobacterial In Vivo Gene Expression
A greater understanding of the regulation of M. tuberculosis gene expression may be gained by looking at the differential expression of genes known to control mycobacterial transcription. The induction of low iron responsive (ideR) and heat shock responsive (hspR) transcriptional regulators are likely at least in part to coordinate the expression of iron sequestration factors and molecular chaperones as discussed above. The transcription factors sigB and sigE were found to be induced in the murine phagosome [25]. One possible stimulus for the increased expression of these regulatory proteins has been demonstrated to be cell wall stress (treatment with detergent [58]), something that is likely to be relevant throughout infection. Furthermore sigB, sigC and sigH were observed to be up-regulated within artificial murine granulomas [31] and sigE, sigG and sigH have been demonstrated to be induced inside human macrophages [26, 32]. In addition, the regulatory proteins sigF and sigL may also be necessary for successful in vivo survival [33, 59, 60]. The up-regulation of a number of two component systems in vivo has also been reported, including dosR which mediates the M. tuberculosis response to hypoxia [23], and which is induced together with much of its regulon inside artificial murine granulomas [31] as previously described. Moreover the expression of mtrA, regX3, phoP, prrA, mprA, kdpE, trcR and tcrX have all been detected after infection of human-derived macrophages with M. tuberculosis [61, 62]. Further characterisation of the gene subsets controlled by these global regulators and of the stimuli that influence their expression will help reveal the networks of gene regulation required for M. tuberculosis pathogenicity.
What the M. tuberculosis Intracellular Transcriptome Can Tell us About the Phagosomal Environment
The comparison of expression data from in vitro and in vivo models is a valuable tool in understanding both the nature of the intracellular environment that M. tuberculosis encounters during infection and how bacilli respond to these stresses. By comparing overlapping gene expression patterns from in vitro and in vivo models it is possible to speculate on the conditions that infecting bacilli face. The phagosomal environment of naïve macrophages is likely to be low in glucose or glycerol based carbon sources, low in amino acids, and low in iron as compared to nutrient starvation and low iron in vitro conditions [42, 63, 64]. On activation (of murine macrophages with IFNγ) the bacilli are likely to be exposed to nitric oxide, oxidative stress and possibly a hypoxic environment [25], although a shift to anaerobic respiration in bacilli may be due to the presence of NO, rather than the absence of oxygen [36]. Schnappinger et al. [25] defined 68 genes that were preferentially induced after infection of IFNγ-activated compared to naïve murine macrophages. Nearly half of these genes were identified as part of the dosR regulon, a set of around 50 genes induced by hypoxia [23, 35] and nitric oxide treatment [36]. Additionally many of the activation-specific genes were also up-regulated by nitric oxide or hydrogen peroxide treatment in vitro, and the induction of these genes was not observed in NOS2-deficient macrophages [25]. A subset of iron-responsive genes was also induced to a greater degree in activated murine macrophages. The intraphagosomal M. tuberculosis expression profile also encompasses subsets of genes that have been characterised as part of the mycobacterial responses to various stresses, such as DNA damage, detergent treatment, low pH and heat shock [48, 58, 65, 66]. A summary of the possible microenvironments that M. tuberculosis encounters during infection is depicted in Fig. (2).
Fig. (2).
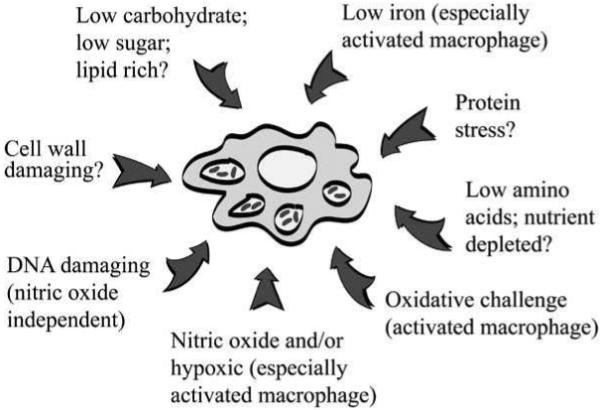
An illustration of the possible microenvironments that M. tuberculosis may encounter on infection as inferred from the transcriptional data discussed in this review.
Mycobacterial gene expression is regulated by many factors and controlled by a complex network of sensory systems and feedback loops. It is clear that some pathways are induced by multiple stimuli; for example genes associated with the mycobacterial response to low iron are also induced by nitric oxide, H2O2 or hypoxia [25, 35]. Furthermore the expression of dosR (integeral to the M. tuberculosis response to macrophage activation and persistence as modelled in vitro) is induced by hypoxia, nitric oxide (s-nitrosoglutathoine), ethanol and H2O2 treatments but not heat or cold-shock [67]. Further investigation is required to identify which environmental conditions are influencing M. tuberculosis gene expression in an effort to better understand the multi-factorial infection process.
INFECTION MODELS AND MAN
The comparison of M. tuberculosis gene expression profiles from tissue culture models derived from murine or human sources is challenging. This is partly due to differences in the survival of bacilli after macrophage infection between murine and human models. Schnappinger et al. [25] described little or no growth of M. tuberculosis in naïve murine bone marrow macrophages and a decrease in mycobacterial survival after 72 h in IFNγ-activated macrophages, compared to successful M. tuberculosis growth in human-derived monocytes and THP-1 cells [68, 69]. This may be due to variation in immunologically relevant elements, such as TLR expression [70], or the production of antimicrobial moeities such as nitric oxide. Indeed the role of nitric oxide in the control of human mycobacterial infection has been controversial; although it is likely that nitric oxide does play some part in the human immune response to M. tuberculosis [71]. The induction of nitric oxide from cells in vitro may however depend on where and how the immune cells were extracted, matured and infected [72].
The extrapolation of expression data from tissue culture models to animal models and natural M. tuberculosis infection is obviously complicated by many factors. Amongst the most apparent are the sampling of broad populations of bacilli from different microenvironments from in vivo infection models and the differences in pathology between murine and human infection [73]. There are however many similarities to be drawn between the mycobacterial responses to macrophage activation (derived from tissue culture models) and responses to the onset of the adaptive immune system in murine lungs; as expression profiling methods progress it should be possible to further explore human M. tuberculosis infection.
CONCLUSIONS
The transcriptional response of M. tuberculosis to the intracellular environment has highlighted a number of mechanisms that mycobacteria may utilise to successfully survive in vivo. This transcriptional data has also revealed a little about the microenvironments that bacilli may encounter. The future comparison of expression patterns from distinct mycobacterial populations from in vivo, tissue culture and in vitro models of infection promises to uncover a great deal more about M. tuberculosis pathogenesis, which will hopefully lead to novel treatment strategies.
One of the most exciting features of transcriptional profiling using DNA microarrays is the expression data obtained for genes of unknown function that would be overlooked using a bottom-up experimental strategy. As more microarray datasets become available the co-expression of genes encoding hypothetical proteins with known transcriptional pathways may allow novel gene functions to be described and explored. The transcriptional data discussed herein mostly concerns the induction of mycobacterial transcription. Nevertheless the repression of genes may be just as important for the outcome of host pathogen interactions during infection, as described recently for regulation of the mce1 operon in murine macrophages [74]. The expression of mycobacterial genes discussed in this review is (in most cases) relative to in vitro logarithmically growing bacilli; this comparison highlights the differences between logarithmic growth in complete media and in vivo growth but does not necessarily reveal which mechanisms are key to mycobacterial survival (as discussed [75]), or indeed how essential particular genes are for successful infection. This review has focused purely on the mRNA expression data available for the intracellular survival of M. tuberculosis, ignoring the huge arsenal of data on the functional significance of particular genes using techniques such as signature tag mutagenesis [76, 77], and knockout/down studies [78, 79]; and of course areas including mRNA degradation, protein expression, processing and regulation.
We are currently able to study the transcriptome of a limited number of host-mycobacterial environments; the challenge for the next few years will be to integrate these datasets with expression data from additional infection models to elucidate host and tissue specific responses. The investigation of (relatively) simple in vitro or tissue culture models should help us to understand the transcriptional events from complex environments such as the murine or human lung. In addition the introduction of bacterial RNA amplification methods will allow the investigation of smaller, highly defined populations of bacilli from in vivo. Whole genome technologies offer exciting prospects for the future, such as the integration of expression data with transcriptional regulation data from emerging technologies such as ChIP/chip to identify global networks of expression that are important during infection; or in combination with gene knockout/knockdown technologies to investigate the effects of individual gene inactivation on the M. tuberculosis transcriptome during infection. All of this should aid in the understanding of M. tuberculosis natural infection and contribute to drug development programmes.
ACKNOWLEDGEMENTS
SJW was funded by an EU STREP Sixth Framework Programme Priority (LHP-CT-2004-012187). SJW and PDB would like to acknowledge the Wellcome Trust and its Functional Genomics Resources Initiative for funding the multi-collaborative microbial pathogen microarray facility at St. George’s (BμG@S).
ABBREVIATIONS
- RT-PCR
Reverse transcription-polymerase chain reaction
- TLR
Toll-like receptor
- SCOTS
Selective capture of transcribed sequences
- NOS2
Nitric oxide synthase 2
REFERENCES
- [1].Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Infect. Immun. 2002;70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Clemens DL, Horwitz MA. J. Exp. Med. 1995;181:257–270. doi: 10.1084/jem.181.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pieters J, Gatfield J. Trends Microbiol. 2002;10:142–146. doi: 10.1016/s0966-842x(02)02305-3. [DOI] [PubMed] [Google Scholar]
- [4].Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, Allen RD, Gluck SL, Heuser J, Russell DG. Science. 1994;263:678–681. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- [5].Deretic V, Fratti RA. Mol. Microbiol. 1999;31:1603–1609. doi: 10.1046/j.1365-2958.1999.01279.x. [DOI] [PubMed] [Google Scholar]
- [6].Saunders BM, Frank AA, Orme IM. Immunology. 1999;98:324–328. doi: 10.1046/j.1365-2567.1999.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Flynn JL, Chan J. Annu. Rev. Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- [8].Saunders BM, Cooper AM. Immunol. Cell Biol. 2000;78:334–341. doi: 10.1046/j.1440-1711.2000.00933.x. [DOI] [PubMed] [Google Scholar]
- [9].Russell DG. Nat. Rev. Mol. Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- [10].Chan J, Flynn J. Clin. Immunol. 2004;110:2–12. doi: 10.1016/s1521-6616(03)00210-9. [DOI] [PubMed] [Google Scholar]
- [11].Walburger A, Koul A, Ferrari G, Nguyen L, Prescianotto-Baschong C, Huygen K, Klebl B, Thompson C, Bacher G, Pieters J. Science. 2004;304:1800–1804. doi: 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- [12].Beatty WL, Ullrich HJ, Russell DG. Eur. J. Cell Biol. 2001;80:31–40. doi: 10.1078/0171-9335-00131. [DOI] [PubMed] [Google Scholar]
- [13].Quesniaux V, Fremond C, Jacobs M, Parida S, Nicolle D, Yeremeev V, Bihl F, Erard F, Botha T, Drennan M, Soler MN, Le Bert M, Schnyder B, Ryffel B. Microbes Infect. 2004;6:946–959. doi: 10.1016/j.micinf.2004.04.016. [DOI] [PubMed] [Google Scholar]
- [14].Hu Y, Mangan JA, Dhillon J, Sole KM, Mitchison DA, Butcher PD, Coates AR. J. Bacteriol. 2000;182:6358–6365. doi: 10.1128/jb.182.22.6358-6365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pai SR, Actor JK, Sepulveda E, Hunter RL, Jr., Jagannath C. Microb. Pathog. 2000;28:335–342. doi: 10.1006/mpat.2000.0353. [DOI] [PubMed] [Google Scholar]
- [16].McKinney JD. Nat. Med. 2000;6:1330–1333. doi: 10.1038/82142. [DOI] [PubMed] [Google Scholar]
- [17].Gomez JE, McKinney JD. Tuberculosis (Edinb) 2004;84:29–44. doi: 10.1016/j.tube.2003.08.003. [DOI] [PubMed] [Google Scholar]
- [18].Fenhalls G, Stevens L, Moses L, Bezuidenhout J, Betts JC, Helden P.v., Lukey PT, Duncan K. Infect. Immun. 2002;70:6330–6338. doi: 10.1128/IAI.70.11.6330-6338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rachman H, Strong M, Ulrichs T, Grode L, Schuchhardt J, Mollenkopf H, Kosmiadi GA, Eisenberg D, Kaufmann SH. Infect. Immun. 2006;74:1233–1242. doi: 10.1128/IAI.74.2.1233-1242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schnappinger D, Schoolnik GK, Ehrt S. Microbes Infect. 2006;8:1132–1140. doi: 10.1016/j.micinf.2005.10.027. [DOI] [PubMed] [Google Scholar]
- [21].McKinney JD, Gomez JE. Nat. Med. 2003;9:1356–1357. doi: 10.1038/nm1103-1356. [DOI] [PubMed] [Google Scholar]
- [22].Kaufmann SH. Nat. Rev. Immunol. 2001;1:20–30. doi: 10.1038/35095558. [DOI] [PubMed] [Google Scholar]
- [23].Park HD, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, Schoolnik GK, Sherman DR. Mol. Microbiol. 2003;48:833–843. doi: 10.1046/j.1365-2958.2003.03474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Muttucumaru DG, Roberts G, Hinds J, Stabler RA, Parish T. Tuberculosis (Edinb) 2004;84:239–246. doi: 10.1016/j.tube.2003.12.006. [DOI] [PubMed] [Google Scholar]
- [25].Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. J. Exp. Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cappelli G, Volpe E, Grassi M, Liseo B, Colizzi V, Mariani F. Res. Microbiol. 2006;157:445–455. doi: 10.1016/j.resmic.2005.10.007. [DOI] [PubMed] [Google Scholar]
- [27].Talaat AM, Lyons R, Howard ST, Johnston SA. Proc. Natl. Acad. Sci. USA. 2004;101:4602–4607. doi: 10.1073/pnas.0306023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rachman H, Strong M, Schaible U, Schuchhardt J, Hagens K, Mollenkopf H, Eisenberg D, Kaufmann SH. Microbes Infect. 2006;8:747–757. doi: 10.1016/j.micinf.2005.09.011. [DOI] [PubMed] [Google Scholar]
- [29].McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr., Russell DG. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- [30].Timm J, Post FA, Bekker LG, Walther GB, Wainwright HC, Manganelli R, Chan WT, Tsenova L, Gold B, Smith I, Kaplan G, McKinney JD. Proc. Natl. Acad. Sci. USA. 2003;100:14321–14326. doi: 10.1073/pnas.2436197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Karakousis PC, Yoshimatsu T, Lamichhane G, Woolwine SC, Nuermberger EL, Grosset J, Bishai WR. J. Exp. Med. 2004;200:647–657. doi: 10.1084/jem.20040646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Graham JE, Clark-Curtiss JE. Proc. Natl. Acad. Sci. USA. 1999;96:11554–11559. doi: 10.1073/pnas.96.20.11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li MS, Waddell SJ, Monahan IM, Mangan JA, Martin SL, Everett MJ, Butcher PD. FEMS Microbiol. Lett. 2004;233:333–339. doi: 10.1016/j.femsle.2004.02.028. [DOI] [PubMed] [Google Scholar]
- [34].Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. Proc. Natl. Acad. Sci. USA. 2005;102:15629–15634. doi: 10.1073/pnas.0507850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bacon J, James BW, Wernisch L, Williams A, Morley KA, Hatch GJ, Mangan JA, Hinds J, Stoker NG, Butcher PD, Marsh PD. Tuberculosis (Edinb) 2004;84:205–217. doi: 10.1016/j.tube.2003.12.011. [DOI] [PubMed] [Google Scholar]
- [36].Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK. J. Exp. Med. 2003;198:705–713. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shi L, Jung YJ, Tyagi S, Gennaro ML, North RJ. Proc. Natl. Acad. Sci. USA. 2003;100:241–246. doi: 10.1073/pnas.0136863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Boshoff HI, Barry CE., 3rd Nat. Rev. Microbiol. 2005;3:70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- [39].Weinberg ED. Life Sci. 1992;50:1289–1297. doi: 10.1016/0024-3205(92)90279-x. [DOI] [PubMed] [Google Scholar]
- [40].Gold B, Rodriguez GM, Marras SA, Pentecost M, Smith I. Mol. Microbiol. 2001;42:851–865. doi: 10.1046/j.1365-2958.2001.02684.x. [DOI] [PubMed] [Google Scholar]
- [41].Li MS, Monahan IM, Waddell SJ, Mangan JA, Martin SL, Everett MJ, Butcher PD. Microbiology. 2001;147:2293–2305. doi: 10.1099/00221287-147-8-2293. [DOI] [PubMed] [Google Scholar]
- [42].Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Mol. Microbiol. 2002;43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- [43].Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, Barry CE., 3rd J. Bacteriol. 2000;182:4889–4898. doi: 10.1128/jb.182.17.4889-4898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dahl JL, Kraus CN, Boshoff HI, Doan B, Foley K, Avarbock D, Kaplan G, Mizrahi V, Rubin H, Barry CE., 3rd Proc. Natl. Acad. Sci. USA. 2003;100:10026–10031. doi: 10.1073/pnas.1631248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cox JS, Chen B, McNeil M, Jacobs WR., Jr. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- [46].Sirakova TD, Thirumala AK, Dubey VS, Sprecher H, Kolattukudy PE. J. Biol. Chem. 2001;276:16833–16839. doi: 10.1074/jbc.M011468200. [DOI] [PubMed] [Google Scholar]
- [47].Waddell SJ, Chung GA, Gibson KJ, Everett MJ, Minnikin DE, Besra GS, Butcher PD. Lett. Appl. Microbiol. 2005;40:201–206. doi: 10.1111/j.1472-765X.2005.01659.x. [DOI] [PubMed] [Google Scholar]
- [48].Stewart GR, Wernisch L, Stabler R, Mangan JA, Hinds J, Laing KG, Young DB, Butcher PD. Microbiology. 2002;148:3129–3138. doi: 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- [49].Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE., 3rd J. Biol. Chem. 2004;279:40174–40184. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- [50].Waddell SJ, Stabler RA, Laing K, Kremer L, Reynolds RC, Besra GS. Tuberculosis (Edinb) 2004;84:263–274. doi: 10.1016/j.tube.2003.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dheenadhayalan V, Delogu G, Sanguinetti M, Fadda G, Brennan MJ. J. Bacteriol. 2006;188:3721–3725. doi: 10.1128/JB.188.10.3721-3725.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Voskuil MI, Schnappinger D, Rutherford R, Liu Y, Schoolnik GK. Tuberculosis (Edinb) 2004;84:256–262. doi: 10.1016/j.tube.2003.12.014. [DOI] [PubMed] [Google Scholar]
- [53].Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- [54].Brennan MJ, Delogu G, Chen Y, Bardarov S, Kriakov J, Alavi M, Jacobs WR., Jr. Infect. Immun. 2001;69:7326–7333. doi: 10.1128/IAI.69.12.7326-7333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Banu S, Honore N, Saint-Joanis B, Philpott D, Prevost MC, Cole ST. Mol. Microbiol. 2002;44:9–19. doi: 10.1046/j.1365-2958.2002.02813.x. [DOI] [PubMed] [Google Scholar]
- [56].Dubnau E, Chan J, Mohan VP, Smith I. Infect. Immun. 2005;73:3754–3757. doi: 10.1128/IAI.73.6.3754-3757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shi L, North R, Gennaro ML. Infect. Immun. 2004;72:2420–2424. doi: 10.1128/IAI.72.4.2420-2424.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Manganelli R, Voskuil MI, Schoolnik GK, Smith I. Mol. Microbiol. 2001;41:423–437. doi: 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
- [59].Geiman DE, Kaushal D, Ko C, Tyagi S, Manabe YC, Schroeder BG, Fleischmann RD, Morrison NE, Converse PJ, Chen P, Bishai WR. Infect. Immun. 2004;72:1733–1745. doi: 10.1128/IAI.72.3.1733-1745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dainese E, Rodrigue S, Delogu G, Provvedi R, Laflamme L, Brzezinski R, Fadda G, Smith I, Gaudreau L, Palu G, Manganelli R. Infect. Immun. 2006;74:2457–2461. doi: 10.1128/IAI.74.4.2457-2461.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zahrt TC, Deretic V. Proc. Natl. Acad. Sci. USA. 2001;98:12706–12711. doi: 10.1073/pnas.221272198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Haydel SE, Clark-Curtiss JE. FEMS Microbiol. Lett. 2004;236:341–347. doi: 10.1016/j.femsle.2004.06.010. [DOI] [PubMed] [Google Scholar]
- [63].Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. Infect. Immun. 2002;70:3371–3381. doi: 10.1128/IAI.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hampshire T, Soneji S, Bacon J, James BW, Hinds J, Laing K, Stabler RA, Marsh PD, Butcher PD. Tuberculosis (Edinb) 2004;84:228–238. doi: 10.1016/j.tube.2003.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rand L, Hinds J, Springer B, Sander P, Buxton RS, Davis EO. Mol. Microbiol. 2003;50:1031–1042. doi: 10.1046/j.1365-2958.2003.03765.x. [DOI] [PubMed] [Google Scholar]
- [66].Fisher MA, Plikaytis BB, Shinnick TM. J. Bacteriol. 2002;184:4025–4032. doi: 10.1128/JB.184.14.4025-4032.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kendall SL, Movahedzadeh F, Rison SC, Wernisch L, Parish T, Duncan K, Betts JC, Stoker NG. Tuberculosis (Edinb) 2004;84:247–255. doi: 10.1016/j.tube.2003.12.007. [DOI] [PubMed] [Google Scholar]
- [68].Stokes RW, Doxsee D. Cell Immunol. 1999;197:1–9. doi: 10.1006/cimm.1999.1554. [DOI] [PubMed] [Google Scholar]
- [69].Munoz-Elias EJ, McKinney JD. Nat. Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lauw FN, Caffrey DR, Golenbock DT. Trends Immunol. 2005;26:509–511. doi: 10.1016/j.it.2005.08.006. [DOI] [PubMed] [Google Scholar]
- [71].Nathan C, Shiloh MU. Proc. Natl. Acad. Sci. USA. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chan ED, Chan J, Schluger NW. Am. J. Respir. Cell Mol. Biol. 2001;25:606–612. doi: 10.1165/ajrcmb.25.5.4487. [DOI] [PubMed] [Google Scholar]
- [73].Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, Tufariello J, Flynn J, Chan J. Cell Microbiol. 2006;8:218–232. doi: 10.1111/j.1462-5822.2005.00612.x. [DOI] [PubMed] [Google Scholar]
- [74].Casali N, White AM, Riley LW. J. Bacteriol. 2006;188:441–449. doi: 10.1128/JB.188.2.441-449.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kendall SL, Rison SC, Movahedzadeh F, Frita R, Stoker NG. Trends Microbiol. 2004;12:537–544. doi: 10.1016/j.tim.2004.10.005. [DOI] [PubMed] [Google Scholar]
- [76].Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. Mol. Microbiol. 1999;34:257–267. doi: 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- [77].Sassetti CM, Boyd DH, Rubin EJ. Proc. Natl. Acad. Sci. USA. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Parish T, Gordhan BG, McAdam RA, Duncan K, Mizrahi V, Stoker NG. Microbiology. 1999;145:3497–3503. doi: 10.1099/00221287-145-12-3497. [DOI] [PubMed] [Google Scholar]
- [79].Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, Schnappinger D. Nucleic Acids Res. 2005;33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Dubnau E, Fontan P, Manganelli R, Soares-Appel S, Smith I. Infect. Immun. 2002;70:2787–2795. doi: 10.1128/IAI.70.6.2787-2795.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]