

Abstract
Rationale
Cardiomyocytes cultured in a mechanically active three-dimensional configuration can be used for studies that correlate contractile performance to cellular physiology. Current engineered cardiac tissue (ECT) models employ cells derived from either rat or chick hearts. Development of a murine ECT would provide access to many existing models of cardiac disease, and open the possibility of performing targeted genetic manipulation with the ability to directly assess contractile and molecular variables.
Objective
To generate, characterize and validate mouse ECT using a physiologically relevant model of hypertrophic cardiomyopathy.
Methods and Results
We generated mechanically integrated ECT using isolated neonatal mouse cardiac cells derived from both wild type and myosin binding protein C (cMyBP-C) null mouse hearts. The murine ECT's produce consistent contractile forces that follow the Frank-Starling law, accept physiologic pacing. cMyBP-C null ECT show characteristic acceleration of contraction kinetics. Adenoviral-mediated expression of human cMyBP-C in murine cMyBP-C null ECT restores contractile properties to levels indistinguishable from those of wild type ECT. Importantly, the cardiomyocytes used to construct the cMyBP-C−/− ECT have yet to undergo the significant hypertrophic remodeling that occurs in vivo. Thus, this murine ECT model reveals a contractile phenotype that is specific to the genetic mutation rather than to secondary remodeling events.
Conclusions
Data presented here show mouse ECT to be an efficient and cost effective platform to study the primary effects of genetic manipulation on cardiac contractile function. This model provides a previously unavailable tool to study specific sarcomeric protein mutations in an intact mammalian muscle system.
Keywords: Engineered cardiac tissue, Hypertrophic cardiomyopathy, Myosin binding protein-C
INTRODUCTION
Over the last decade, techniques to culture neonatal rat and chick cardiac cells in three-dimensional (3D) environments have been established and refined1–5. 3D engineered cardiac tissue (ECT) has several advantages over traditional monolayer (2D) culture methods in that cardiac cells within the ECT form well aligned, mechanically and electrically integrated strips that more closely resemble functional myocardium. Thus ECT are well suited for a variety of applications including disease modeling, drug discovery, and the study of basic myocardial physiology.
Recent research efforts have focused upon optimizing ECT for applications in tissue replacement therapy or as a platform for pharmacologic screening. Techniques to do this have included the use of neonatal cardiac cells from different species, primarily rat and chick, as well as cardiomyocytes (CM) differentiated from mouse embryonic stem (ES) or induced pluripotent stem (iPS) cells1–3, 6. Additionally, to promote cell survival and maintain differentiation, several different support matrices have been adopted including solid scaffolds7–9, decellularized cardiac tissue scaffolds10 and stackable CM sheets11–13 which use a variety of combinations of different hydrogels such as collagen I, fibrin and fibrinogen1–5, 14–16. These modifications have given rise to ECT in several different configurations, including rings, patches, cylinders, or even intact hearts, each optimized for its envisioned downstream application.
One of the earliest proposed applications of ECT was as a platform to study the physiological effect of gene ablation and genetic mutation on cardiac contractility2. Theoretically, using a knockout background, exogenous genes could be expressed in ECT through adenoviral-mediated gene transfer, and the resulting impact on contractile function readily measured1, 2. However, while cardiac cells from rat myocardium are commonly used, to our knowledge ECT from neonatal mouse cardiac cells has not yet been established and cardiac gene manipulation studies continue to rely on whole animal models and isolated 2D culture approaches. Clearly, establishing murine ECT would provide significant advantages by allowing study of the multiple existing genetic mouse models of cardiac disease. More importantly, murine ECT would permit the physiologic characterization of mutations prior to the development of secondary changes (i.e., hypertrophy, heart failure) that occur in older whole animal and excised tissue models.
One mouse model that has been extensively studied is that of cardiac myosin binding protein-C (cMyBP-C) ablation (cMyBP-C−/−). cMyBP-C plays an important regulatory role in cardiac muscle contraction and mutations in the cMyBP-C encoding gene (MYBPC3) are prevalent causes of familial hypertrophic cardiomyopathy (HCM) in humans17–19. cMyBP-C−/− mice share several of the known phenotypes of the human disease, including pronounced left ventricular and septal hypertrophy, systolic and diastolic dysfunction, and accelerated contractile kinetics20–26.
Here we present data showing successful generation of ECT from cardiac cells derived from wild-type (WT) and cMyBP-C−/− neonatal mouse hearts. We demonstrate that ECT derived from cMyBP-C−/− cardiac cells recapitulate several contractile abnormalities previously observed in both intact hearts and isolated cardiac tissue from cMyBP-C−/− mice20–26. Importantly these abnormalities occur in the absence of detectable hypertrophic remodeling. Additionally, we show that transduction of cMyBP-C−/− mouse ECT with adenovirus encoding WT human cMyBPC completely mitigates the contractile abnormalities in these ECT. Thus we demonstrate a robust and reproducible mouse ECT model which provides a novel tool that bridges the gap between isolated cell culture and the intact heart. We further demonstrate the ability to apply this tool to the study of genetic mouse models of important human cardiac diseases.
METHODS
Animals
Ventricular tissue was harvested from one day old homozygous cMyBP-C−/− mouse pups that were previously generated on the E129X1/SvJ background20, and from one day old WT E129X1/SvJ mice (Taconic). Pups were anesthetized using inhaled isoflourane prior to harvesting of ventricular tissue. This study was approved by the Animal Care and Use Committee of the School of Medicine and Public Health at the University of Wisconsin-Madison in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, Revised 1985).
Isolation of Neonatal Mouse Ventricular Cardiac Cells
Ventricular cardiac cells were isolated using enzymatic dissociation based on methods employed to isolate neonatal rat CMs2, 3, 27, 28. Briefly, excised ventricular tissue was minced, and suspended in 3 mg/ml collagenase type II (Gibco BRL) in KG buffer (pH 7.4) and incubated at 37°C for 20 minutes with gentle agitation. KG buffer consisted of: 127 mmol/L L-glutamic acid potassium salt monohydrate (Sigma); 0.1335% (w/v) NaHCO3 (Gibco); 16.5 mmol/L D-glucose (Sigma); 0.42 mmol/L Na2HPO4 (Sigma); 25 mmol/L HEPES (Sigma). Ventricular tissue was subsequently resuspended and agitated in 0.025% trypsin (Gibco) in KG buffer for 10 minute intervals until dispersed. Cells were pelleted at 200 × g and resuspended in mouse culture media consisting of: 60.3% high glucose DMEM (Gibco); 20% F12 nutrient mix (Gibco) supplemented with 1 mg/mL gentamicin (Sigma), 8.75% fetal bovine serum (HyClone®), 6.25% horse serum (HyClone®), 1% HEPES (Sigma), 1× non-essential amino acid cocktail (Gibco), 3 mmol/L sodium pyruvate (Gibco), 0.00384% (w/v) NaHCO3 (Gibco), 1 μg/mL insulin (Sigma). Cell suspensions were preplated into P100 cell culture dishes and incubated at 37°C for 45 minutes to allow preferential attachment of non-myocyte cell populations and enrichment of CM population. Cardiac cells remaining in suspension were collected, checked for viability by dye-exclusion, counted, and prepared for subsequent ECT construction.
Generation and Characterization of Human MYBPC3 Expressing Adenovirus (adWT)
An adenovirus encoding N-terminally Myc-tagged full-length human cMyBP-C (adWT) was generated using the ViraPower™ Adenoviral Expression System (Invitrogen), according to the manufacturer's protocol. The transduction efficiency of adWT was assessed in neonatal murine cardiac cells in 2D culture. Control viruses expressing the LacZ reporter-gene (adLacZ) and lacking any significant reading frame (adC−) were similarly prepared (see data supplement for detailed methodology).
ECT construction
WT and cMyBP-C−/− neonatal mouse ventricular cells suspended in mouse media were rotated at 50 rpm on a gyratory shaker (195 mm diameter) for 6–8 hours at 37°C and 5% CO2 to allow aggregation of small, uniform clusters of viable cells. For production of cMyBP-C−/− adWT ECT, cMyBP-C−/− cardiac cells were transduced with adWT at a multiplicity of infection (MOI) of 20 during the rotational culture period. Following rotational culture, approximately 8.0 × 105 CMs were suspended in 83.3 μL mouse media and added to 116.7 μL of an ECT matrix mixture consisting of 66.7 μL 2 mg/mL acid soluble rat-tail collagen type-I (pH 3.0) (Sigma), 8.3 μL 10× MEM (Gibco), 8.3 μL reconstitution buffer (200 mmol/L NaHCO3 (Gibco); 200 mmol/L HEPES (Sigma); 100 mmol/L NaOH (Sigma)) and 33.3 μL Matrigel (BD Biosciences). 200 μL of the cell/matrix suspension were cast into 20 mm × 3 mm cylinder constructs using a Flexcell Tissue Train silicone membrane culture plate (Flexcell International) and incubated under preprogrammed vacuum conditions for 120 minutes (37°C, 5% CO2) to form cylindrical-shaped ECT constructs that were attached at each end to fibrinous tabs. Upon matrix polymerization, mouse media was added to the ECT within the 6-well culture dish. ECT constructs were maintained in culture for 7 days with media changes every other day.
RESULTS
Structure of Murine ECT
Spontaneous contraction began in an uncoordinated manner on the second day after ECT construction with gradual transition to coordinated rhythmic contraction by day 5. During seven days in culture, ECT diameter decreased from the mold width of 3 mm to approximately 1.1 mm as the construct formed and compacted (figure 1A and B). Cells were distributed unevenly through the ECT with elongated CMs predominantly located near the surface, while non-CM cell populations were distributed evenly throughout the ECT (figure 1C–F). CM-dense areas were often noted at the bottom of the ECT, presumably due to gravitational settling of the CMs during matrix polymerization2, 29, 30. CMs in this region were particularly well defined, elongated in shape, and rich in mitochondria, with the sarcomeres aligned to the long axis of the ECT (figure 1 C–J). Gap junctions were frequently noted between adjacent CMs in these areas (figure 1J). Additionally, isolated round-shaped CMs with disorganized sarcomeric orientation were often observed closer to the center of the structure. There were no genotype-dependent differences in the final diameter (WT 1.13 ± 0.04 mm; cMyBP-C−/− 1.11 ± 0.07 mm; cMyBP-C−/− adWT 1.05 ± 0.03 mm) (figure 1A and B), gross structure or cellular distribution of ECT (Online Figure V).
Figure 1. Structure of Mouse ECT.

Ultrastructure of a mouse ECT in a 6-well dish (A) and mounted to a force transducer in a perfusion chamber (B). H&E-stained longitudinal-(C and E) and cross-sections (D and F) through a mouse ECT. Immunofluoresce in a thin section through a mouse ECT detected by rabbit anti-cMyBP-C primary antibody and AlexaFluor 647 goat anti-rabbit secondary antibody (red fluorescence) in addition to DAPI nuclear counterstaining (blue fluorescence) (G and H). Transmission electron micrographs of a longitudinal- (I) and a cross-section (J) through a mouse ECT showing 4 cells with aligned sarcomeres and numerous tight junctions. Scale bars or objectives used for visualization indicated in the bottom right corner of each panel.
Physiological Response of WT Murine ECT
ECT generated from rat and chick hearts recapitulate several aspects of the contractile phenotype of intact cardiac tissue1–3, 29, 31, 32. We therefore examined the contractile responses of murine ECT to standard physiologic challenges. Maximal twitch force (FMax) of mouse ECT increased in response to increasing stretch applied to ECT in accordance with the Frank-Starling law (figure 2A and B). Furthermore, ECT responded appropriately to an increase in perfusion temperature from 24°C to 37°C with a significant increase in FMax and the rate of contraction, assessed as the elapsed time between the electrical pulse and peak developed twitch force (Ct100). Increasing temperature similarly increased the rate of relaxation, assessed from elapsed time between peak developed twitch force and 50% twitch force decay (Rt50) (figure 2C and D). FMax decreased as stimulation frequency was increased from 4 Hz to 8 Hz in mouse ECT incubated at 37°C. This negative force-frequency relationship was less pronounced at slower pacing frequencies (4–6 Hz) (figure 2E and F). The magnitude of twitch force generated by, and physiological responsiveness of, murine ECT was similar to that of similar ECT produced from rat CMs33.
Figure 2. Responsiveness of Mouse ECT to Physiological Stimuli.

Representative force trace from a mouse ECT electrically paced at 2 Hz, showing increased twitch force as the length is increased corresponding to the Frank-Starling relationship (A and B); representative trace showing the effect of temperature on twitch force in a mouse ECT electrically paced at 4 Hz showing an increased twitch force, as well as accelerated kinetics of contraction and relaxation when perfusion temperature is increased from 24°C to 37°C (C and D); representative trace showing the effect on force due to increasing pacing frequency from 4Hz to 8Hz in a mouse ECT at 37°C (E and F).
Effect of cMyBP-C Ablation on Contractile Function
cMyBP-C ablation increases the rates of contraction and relaxation in both intact hearts and isolated papillary muscle20–26, with an increase in power output noted in skinned cardiac fibers21. We therefore investigated the contractile properties of cMyBP-C−/− ECT. Contractile function was assessed in WT and cMyBP-C−/− ECT that were electrically stimulated at murine physiological frequencies of 6 Hz, 7 Hz and 8 Hz (figure 3). FMax was significantly greater in cMyBP-C−/− ECT than in WT ECT at all frequencies (figure 3B and Online Table 1). Expression levels of several sarcomeric components were similar between WT and cMyBP-C−/− ECT at both the transcript and protein levels (figures 4, 5, S6, S7 and S8), indicating that increased force production by cMyBP-C−/− ECT are unlikely to be due to differences in CM number. Additionally, Ct100 and Rt50 were significantly shorter in cMyBP-C−/− than in WT ECT at all stimulation frequencies (figure 3C and D; Online Table 1), indicating accelerated kinetics of contraction and relaxation in cMyBP-C−/− ECT.
Figure 3. Effect of cMyBP-C Ablation on ECT Function.
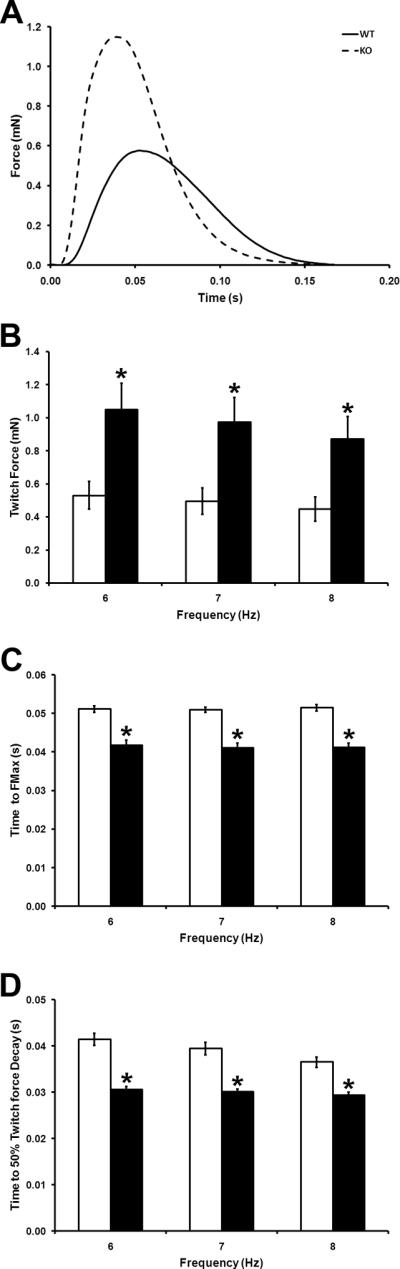
(A) Representative twitches of WT and cMyBP-C−/− ECT paced at 6Hz. (B) Maximal twitch force (FMax) in WT and cMyBP-C−/− ECT paced at 6Hz, 7Hz and 8Hz. (C) Time to peak developed twitch force in WT and cMyBP-C−/− ECT paced at 6Hz, 7Hz and 8Hz. (D) Time from peak developed twitch force to 50% force decay in WT and cMyBP-C−/− ECT paced at 6Hz, 7Hz and 8Hz. WT ECT- open bars; cMyBP-C−/− ECT- filled bars. * P < 0.05 vs. WT (Student's t-test; WT n = 7, cMyBP-C−/− n = 6).
Figure 4. Expression of Hypertrophic Response Genes in WT and cMyBP-C−/− ECT.

mRNA expression levels of MYBPC3 (A) Nppa (B), Nppb (C), Myh7 (D) and Myh6 in WT (open bars) and cMyBP-C−/− (filled bars) ECT. The Myh7:MyH6 ratio is shown in F. * P < 0.05 vs. WT (Student's t-test; WT n = 5; cMyBP-C−/− n = 5).
Figure 5. Expression of Human cMyBP-C in KO CMs and ECT.
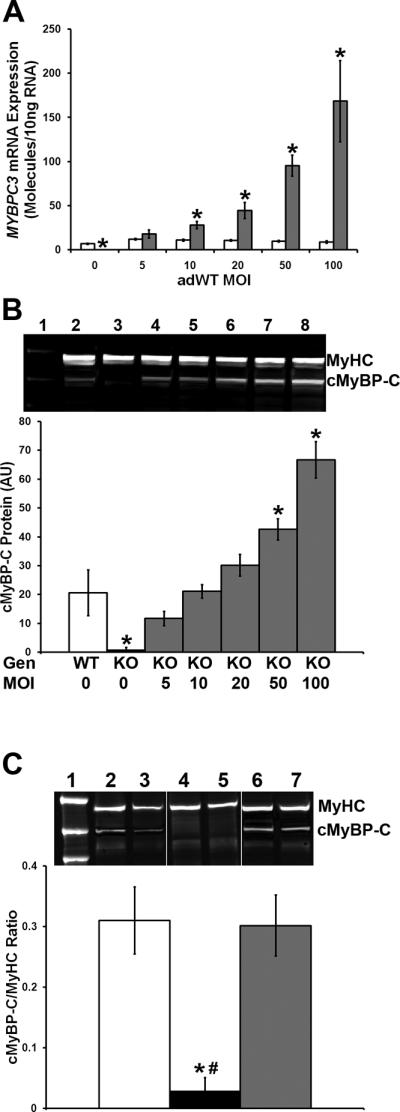
(A) Mouse and human MYBPC3 mRNA levels as assessed by qRT-PCR in WT CMs transduced with adWT virus at 0, 5, 10, 20, 50 and 100 MOI. Open bars = mouse Mybpc3 transcript level; grey bars = human MYBPC3 transcript level. * P < 0.05 vs. mouse Mybpc3 (Student's t-test; n ≥ 6 per group). (B) Total cMyBP-C and total MyHC protein levels in cMyBP-C−/− CMs transduced with adWT virus. Lane 1 - molecular weight marker; lane 2 - untransduced WT CMs; lanes 3–8 – cMyBP-C−/− CMs transduced with adWT virus at 0, 5, 10, 20, 50 and 100 MOI. The graph shows protein levels as of total cMyBP-C (mouse and human) in WT (open bar) and cMyBP-C−/− (grey bars) CMs. * P < 0.05 vs. cMyBP-C in WT (one way ANOVA, Bonferroni's post-hoc analysis; n = 4 per group). (C) Total cMyBP-C and MyHC protein levels in WT, cMyBP-C−/− and cMyBP-C−/− adWT ECT. Lane 1 – molecular weight marker; lanes 2 and 3 – WT ECT; lanes 4 and 5 - cMyBP-C−/− ECT; lanes 6 and 7 - cMyBP-C−/− adWT ECT. The graph shows protein levels of total cMyBP-C (mouse and human) in WT (open bar), cMyBP-C−/− (filled bar) and cMyBP-C−/− adWT ECT (grey bar). * P < 0.05 vs. cMyBP-C in WT; # P < 0.05 vs. cMyBP-C in cMyBP-C−/− adWT (one way ANOVA, Bonferroni's post-hoc analysis; WT N = 6; cMyBP-C−/− N = 4; cMyBP-C−/− adWT N = 7).
cMyBP-C−/− ECT Represent `Pre-Hypertrophic' HCM
Much of the contractile data characterizing the effects of genetic manipulation on heart function are necessarily derived from adult mice, which typically have already undergone notable hypertrophic remodeling. A potential advantage of ECT in studies of genetically manipulated CMs is the relative immaturity of the CMs at the time of isoloation for ECT construction. To evaluate the status of our source CMs, we first measured the heart-to-body weight ratio in newborn (P1) and juvenile pups from WT and cMyBP-C−/− litters. Heart-to-body weight ratios were similar in neonatal WT and cMyBP-C−/− mouse pups (Online Figure IA). By postnatal day 10 (P10), heart to body weight ratios were significantly higher in the cMyBP-C−/− hearts and remained elevated at postnatal day 35 (P35).
We used qRT-PCR to examine expression of early (atrial natriuretic peptide (Nppa) and brain natriuretic peptide (Nppb)) and long term (beta-myosin heavy chain (Myh7) and alpha-myosin heavy chain (Myh6)) hypertrophic marker genes. In P1 cMyBP-C−/− hearts expression levels of Myh6 and Myh7 were not differentially regulated (Online Figure IB and C), while expression of Nppa and Nppb were already significantly upregulated (Online Figure ID–F). This suggests activation of early hypertrophic pathways in the “pre-hypertrophic” myocardium. The expression profiles of hypertrophic marker genes confirmed the presence of overt cardiac hypertrophy in P10 and P35 cMyBP-C−/− mice (Online Figure I).
Because ECT are isolated from P1 pups, the initial expression levels of early and late-hypertrophic genes would presumably be the same between the newly formed ECT and the P1 hearts of the respective genotype. However, over the 10 days in vitro ECT might progress along the same pathway as the intact hearts, which in the cMyBP-C−/− pups showed significant activation of hypertrophic signaling. We therefore examined WT and cMyBP-C−/− ECT for hypertrophic signals by qRT-PCR. All marker genes (early and late) were expressed at similar levels in WT and cMyBP-C−/− ECT (figure 4), indicating relative quiescence of hypertrophic signaling in cMyBP-C−/− ECT.
Adenoviral-Mediated Expression of Human cMyBP-C Restores Contractile Function in cMyBPC−/− ECT
2D cultures of WT ventricular myocytes were transduced with adenovirus expressing Myc-tagged WT human cMyBP-C (adWT). qRT-PCR was used to determine the expression of endogenous (murine) and the exogenous (human) cMyBP-C over a range of MOIs. Expression of human and murine cMyBP-C were similar at an MOI of 5, whether using a standard curve method (figure 5A) or expressed as a fraction of beta-actin expression levels using the delta-delta Ct method (data not shown). Furthermore, expression of the human MYBPC3 transgene did not have a significant effect on endogenous mouse Mybpc3 mRNA levels, irrespective of MOI (figure 5A). Expression of human MYBPC3 was, however, significantly higher than that of endogenous mouse Mybpc3 at greater than 5 MOI (figure 5A).
We then examined protein expression by Western blot analysis using an anti-cMyBP-C antibody. Total cMyBP-C and myosin heavy chain (MyHC) protein levels were assessed in untransduced WT cardiac cells and cMyBP-C−/− cardiac cells transduced at MOI of 0, 5, 10, 20, 50 and 100 in 2D culture. Human cMyBP-C protein levels were expressed as a fraction of MyHC level and compared to endogenous mouse cMyBP-C levels in untransduced WT cardiac cells. cMyBP-C levels in transduced cMyBP-C−/− cells at MOI of 5, 10 or 20 did not significantly differ from cMyBP-C levels in untransduced WT cells. Transduction at higher MOI resulted in significantly higher cMyBP-C levels (figure 5B). Transduction of cardiac cells with adWT adenovirus did not significantly affect cell survival, as assessed by reduction of 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT assay), at any MOI used (Online Figure IIIB). Additionally, LacZ reporter-gene expression in mouse neonatal cardiac cells transduced with the adLacZ control virus was assessed by beta-galactosidase assay 48 hours post-transduction. Transduction efficiency approached 100% at an MOI of 5 while transduction at higher MOI resulted in more intense staining, likely due to beta-galactosidase buildup in the in the cytoplasm (Online Figure II). Transduction with adLacZ had a modestly adverse effect on cell survival, as assessed by an MTT assay, which was statistically different from nontransduced cells at MOI of 10 and 100 (Online Figure IIIA).
We chose to use an MOI of 20 for subsequent generation of ECT, as even marginally lower cMyBP-C protein levels have been shown to cause HCM in humans17. Transduction was performed during the 8-hour rotational culture. Quantitative Western blots confirmed the presence of equivalent cMyBP-C levels in untransduced WT ECT and cMyBP-C−/− adWT ECT, while signal was absent from untransduced cMyBP-C−/− ECT (figure 5C).
To confirm that exogenous human cMyBP-C normally inserted into the sarcomere, we used immunofluorecence microscopy to detect endogenous and human cMyBP-C. WT ECT showed a double cross-striated pattern, characteristic of C-zone specific incorporation of cMyBP-C into the sarcomere (figure 6 B and C). The double cross-striated cMyBP-C staining pattern was restored after adWT transduction in cMyBP-C−/− ECT (figure 6H and I). This labeling pattern was absent in untransduced cMyBP-C−/− ECT constructs incubated with the same anti-cMyBP-C antibody (figure 6E and F). These data indicate that exogenous human cMyBP-C is appropriately incorporated into the mouse sarcomere in ECT constructs.
Figure 6. Incorporation of Human cMyBP-C into the Mouse Sarcomere.

Immunofluorescence in thin sections through a WT (A – C), cMyBP-C−/− (D – F) and cMyBP-C−/− adWT ECT (G – I). Desmin was detected by a mouse anti-desmin primary antibody and AlexaFluor 568 goat anti-mouse secondary antibody (red fluorescence) (A, D, G). cMyBP-C was detected by rabbit anti-cMyBP-C primary antibody and AlexaFluor 488 goat anti-rabbit secondary antibody (green fluorescence) (B, E, H). Panels C, F and I – desmin and cMyBP-C immunofluorescence overlay. DAPI was used as a nuclear counterstain in all panels (blue fluorescence). Scale bars indicate 10 μm.
One of the major difficulties with genetically engineered mice is determining whether the phenotype is due entirely to deficiency of the specific gene or secondary remodeling events. We therefore sought to determine whether adenoviral-mediated transduction of human cMyBP-C could rescue the physiologic deficits identified in the cMyBP-C−/− ECT. FMAX in cMyBP-C−/− adWT ECT was restored to levels similar to that of WT ECT at all stimulation frequencies and was significantly lower than that of cMyBP-C−/− ECT (figure 7B and Online Table 1). Furthermore Ct100 and Rt50 in cMyBP-C−/− adWT ECT were similar to those of WT ECT and significantly longer than those of cMyBP-C−/− ECT at all three pacing frequencies (figure 7C, D and Online Table 1), while transduction of cMyBP-C ECT with the adC− control virus failed to alter these parameters (Online Figure IV). We found no genotype-dependent differences in total protein or RNA yields (Online Figure VIA and B) while, expression levels of total myosin heavy chain protein and Myh6 transcript levels were not influenced by ECT genotype (Online Figure VIC and D), indicating that functional differences are unlikely to be due to total cell or CM number. Additionally, expression levels of several genes encoding sarcomeric proteins and/or proteins involved in excitation-contraction coupling, were similar in WT, cMyBP-C−/− and cMyBP-C−/− adWT ECT (Online Figure VIII). These findings confirm that exogenous human cMyBP-C can incorporate into the mouse sarcomere and restore normal physiological function. Furthermore, these data reinforce that the physiologic abnormalities identified in the cMyBP-C−/− ECT are specifically due to the deficiency of cMyBP-C, and not to compensatory mechanisms
Figure 7. Effect of Expression of Human cMyBP-C on ECT Contractile Function.
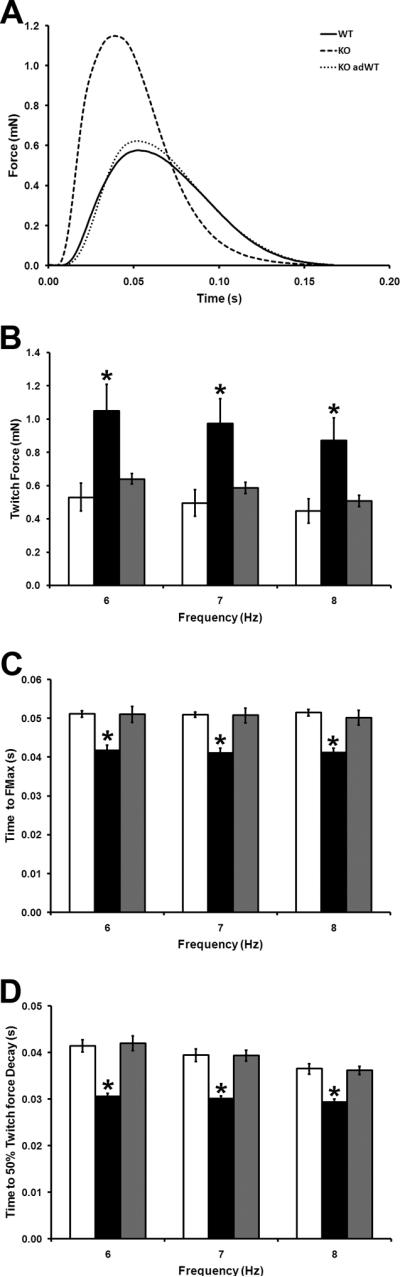
(A) Representative twitches WT, cMyBP-C−/− and cMyBP-C−/− adWT ECT paced at 6Hz. (B) Maximal twitch force (FMax) in WT, cMyBP-C−/− and cMyBP-C−/− adWT ECT paced at 6Hz, 7Hz and 8Hz. (C) Time to peak developed twitch force in WT, cMyBP-C−/− and cMyBP-C−/− adWT ECT paced at 6Hz, 7Hz and 8Hz. (D) Time from peak developed twitch force to 50% force decay in WT, cMyBP-C−/− and cMyBP-C−/− adWT ECT paced at 6Hz, 7Hz and 8Hz. WT ECT- open bars; cMyBP-C−/− ECT- filled bars; cMyBP-C−/− adWT ECT- grey bars. * P < 0.05 vs. WT and cMyBP-C−/− adWT (one way ANOVA, Bonferroni's post-hoc analysis ; WT n = 7, cMyBP-C−/− n = 6; cMyBPC−/− adWT n = 6).
DISCUSSION
Our data presented here demonstrates the feasibility of applying 3D culture methods in the study of both murine and human cardiac physiology, as well as validating a novel method to obtain fundamental contractile data from `unremodeled' cardiomyocytes carrying mutations that lead to HCM. Furthermore, we demonstrate the feasibility of introducing sarcomeric proteins into a multicellular cardiac muscle preparation with the potential to develop a robust screening system for genetic cardiac disease.
Generation of Murine ECT
To our knowledge this is the first report describing ECT generated from neonatal mouse cardiac cells. As described here, the murine ECT provides a powerful, high-throughput tool to test how specific alterations in cardiac genes contribute to abnormal cardiac physiology and produce disorders such as hypertrophic and dilated cardiomyopathies. While our approach is similar to those used previously for the generation of rat ECT2, 4, we found several adaptations that were essential for the generation of well organized, functioning mouse ECT. First, viability and quality of resulting ECT were improved when cells were harvested from mouse pups less than 24 hours old. In agreement with previous studies3, 4, we also observed that incubation of mouse cardiac cells with gentle gyration (rotational culture) allowing aggregation of the isolated cells is beneficial to establishing mechanically active ECT. However, while previous studies maintained cells in rotational culture for more than 12 hours 3, 4, we found that a culture period of 6–8 hours yeilded more uniform small clusters of cells and reduced excessive cell death. Third, seeding neonatal mouse CMs at a density of 8 × 105 per ECT with a concentration of 4 × 106 cells/ml is necessary to establish robust muscle strips that extend the length of the construct. This optimal seeding density is similar to that used to produce fibrin-based rat ECT (4.1 × 106 cells/mL)5, but higher than previously used for the generation of chick (1.3 × 106 cells/mL)1 and rat (3.2 × 106 cells/mL)2 ECT in collagen matrices. Generally, cardiac cells from 4 to 5 neonatal hearts were required for each ECT generated. Finally, we found that addition of F12 nutrient supplement mixture to the culture media during the extended 7–10 day culture period was essential for optimal cell viability.
Mouse ECT produced measurable force, accepted electrical pacing, and responded appropriately to physiological stimuli including increases in passive tension and temperature. This is not surprising given that CMs in mouse ECT align with the long axis of the construct and form myofibrils spanning the length of the ECT with preserved sarcomeres, abundant mitochondria, and numerous gap- and tight-junctions. This level of organization observed in mouse ECT is distinctly absent from the stellate-shaped CMs commonly observed in monolayer cultures. As in other species1–3, 29, 31, 32, mouse CMs in the ECT responded appropriately to increasing stretch in accordance with the Frank-Starling law. Similar to observations in intact myocardial tissue34–36, increasing the perfusion temperature to 37°C significantly accelerated the rates of contraction and relaxation in mouse ECT. The ability to acquire measurements under physiologic conditions is crucial to drawing physiologically relevant conclusions about contractile kinetics.
Unlike intact neonatal and adult mouse cardiac tissue, mouse ECT did not display a positive force-frequency response6, 37–40. Indeed, the force frequency tended to be negative in mouse ECT, a phenomenon that was more striking at higher pacing frequencies. Similar negative force-frequency relationships were previously noted in rat ECT2, 6, 32. This negative forcefrequency relationship may be a result of the dimensions of ECT constructs. Previously, Janssen et al.36 showed a positive force-frequency response in ultra-thin rat trabeculae, but a flat or negative relationship in trabeculae with a diameter of more than 100 μm, specifically at higher pacing frequencies. They speculated that diffusion of oxygen and nutrients, as well as removal of toxic metabolites may become limiting at higher pacing frequencies when diffusion distances exceeds ~50–75 μm, resulting in a negative force frequency relationship36. Since mouse ECT are generally about 1 mm in diameter and well organized CM-rich cell layers often exceed 100 μm, diffusion distance may account for the negative force-frequency response in ECT derived from mice as well as from other species.
Further refinement of the mouse ECT model is desirable to achieve a more defined growth environment. Current methods of culturing rat ECT have successfully reduced or eliminated serum from the growth media by substituting a complex cocktail of supplements14. Attempts to replace serum in the mouse cultures have thus far not been successful. Additionally, alternatives to the collagen matrix and Matrigel have been explored in rat ECT5, 14–16. Most promising is the use of fibrin which supports better cell survival and more uniform final tissue configuration. Fibrin also appears to undergo more extensive remodeling by the CMs and nonmyocytes within the ECT, likely resulting in a more physiologic extracellular matrix than achieved using collagen5, 15. The distribution of CMs around the outer margin of the ECT is commonly observed in collagen-based ECT2, 29. This likely reflects both the gradient of nutrients and oxygen, which diminish 50–100 μm from the outer surface of the construct36, 41, as well as a settling of cells as the collagen matrix slowly polymerizes29. Development of a fibrin-based matrix might reduce this phenomenon as the fibrin polymerizes more quickly maintaining a better distribution of CMs throughout the full cross-section of the ECT5. Finally, the murine ECTs are typically avascular, though co-cultured endothelial cells have been observed to selfassemble into rudimentary capillary structures14. Recent advances in promoting vascularzation of 3D tissue constructs42–44 may facilitate greater CM densities in collagen based murine ECT.
Specific physiologic functions of cMyBP-C
Mutations in MYBPC3 are a common cause of human familial HCM with over 150 distinct mutations implicated thus far17–19. The impact of most of these mutations on cardiac function remains unknown, mainly due to the prohibitive cost and time required to generate mouse models expressing specific gene mutations. The murine ECT cMyBP-C−/− model in which human mutations can be introduced and characterized therefore provides a readily accessible tool to screen disease causing mutations. The cost is comparatively low, and the time from design to physiologic testing is approximately 3 months.
Our initial application of the murine ECT to study human disease has focused on the cMyBP-C−/− mouse precisely because it is a well-characterized mouse model of cardiomyopathy. Overall cMyBP-C ablation does not affect birth frequency or long-term survival of cMyBP-C−/−mice20. However, at the whole organ level cMyBP-C ablation causes significant cardiac hypertrophy, myocytic disarray, fibrosis and impaired left ventricular fractional shortening and ejection fractions20, 26, 45. The accelerated rates of contraction and relaxation we have identified in cMyBP-C−/− ECT are similar to contraction kinetics observed in isolated skinned myocardium from cMyBP-C−/− hearts20–26.
The increased FMAX in cMyBP-C−/− ECT contrasts with the impaired left ventricular fractional shortening and ejection fractions in the intact cMyBP-C−/− heart, and FMax observed in isolated skinned fibers that is similar between the WT and MyBP-C−/− preparations 20, 22, 26, 45. It is important to note however, that in these studies functional measurements were generally performed in tissue derived from older hearts which had already undergone hypertrophic remodeling, fibrosis and changes in extracellular matrix elasticity. Our data are, however, consistent with findings by Korte et al. (2003)21, who showed an increase in power output in skinned cMyBP-C−/− fibers. This is compatible with a model in which cMyBP-C ablation increases the probability of crossbridge formation as was previously proposed24–26 and supports the assertion that ECT may accurately reflect the inherent contractile properties of the cardiomyocytes prior to remodeling and not the consequences of remodeling and other secondary events. These observations suggest that force measurements made in ECT faithfully reflect the contractile parameters of CMs from which they were derived, in the absence of adaptive/maladaptive remodeling and other confounding factors. Our findings also suggest that the ECT model may be well suited to examining the chain of events that leads from the initial genetic mutation and contractile phenotype to the manifestation of hypertrophy and heart failure. Future experiments will focus on further evaluation of this model of early MYBP-C deficiency with careful assessment of other factors that may contribute to contractile performance such as the phosphorylation status of sarcomeric proteins. While we would not expect there to be differences between overall cMyBP-C−/− and WT ECT protein phosphorylation, this remains to be fully explored since potential differences could have important effects upon both FMax and contractile kinetics.
Rescue of normal physiology by adenovirus-mediated expression of wild-type human protein confirms that our findings are due specifically to absence of cMyBP-C. Importantly, virally expressed protein incorporated normally into the sarcomere. By transducing cells during the rotational cultures, we ensured that exogenous human cMyBP-C in cMyBP-C−/− adWT ECT was expressed at levels that were nearly identical to those of the endogenous protein in WT ECT without causing a significant reduction is cell survival. Immunohistochemical analysis of cMyBPC−/− adWT ECT showed a uniform transduction efficiency of most of the cells throughout the construct with exogenous human cMyBP-C appropriately incorporated into the sarcomeric C-zones. A similar approach will be employed in future studies to assess the contractile effects of expressing human HCM mutant cMyBP-C on the cMyBP-C−/− background. Based on these data we suggest that other sarcomeric proteins causative of human HCM can be similarly expressed and appropriately incorporated into murine ECT models derived from corresponding knock-out animals.
In conclusion, we have developed a mouse ECT model for investigations of cardiomyocyte physiology and pathophysiology. We have shown that cMyBPC−/− ECT recapitulate the accelerated contractile kinetics observed in isolated muscle from cMyBPC−/− mice, in the absence of hypertrophic remodeling, thus confirming the fundamental contractile phenotype of cMyBP-C ablation. Furthermore we demonstrated the ability to introduce human sarcomeric proteins onto a knockout murine background. Murine ECT represents a new tool that bridges the gap between traditional isolated 2D CM cell culture and intact animal models. This technique will allow generation and characterization of novel genetic models more efficiently and at significantly less cost compared to traditional animal models. This model could also provide an opportunity to study mutations that result in embryonic lethality in traditional animal models. More importantly, this model permits determination of the basic contractile phenotype in the absence of significant remodeling. Collectively, the strengths of this technique have potential to increase our understanding of integrated cardiac physiology in health and disease.
Supplementary Material
Novelty and Significance.
What is known:
3D engineered cardiac tissue constructs (ECT) replicate important myocardial physiology in vitro.
ECT using murine cardiomyocytes has not been previously available.
Development of multicellular murine cardiac preparations would extend the utility of existing mouse models of genetic cardiac disease and facilitate the study of rare mutations.
What new information does this article contribute?
Demonstration of the feasibility of creating functionally intact murine ECT in a robust, reproducible and cost effective manner.
Ability of the ECT to recapitulate the contractile phenotype of the MyBP-C−/− mouse model.
Demonstrates the ability to appropriately express a human sarcomeric protein in a mouse cardiomyocyte and derive contractile function data.
Hypertrophic cardiomyopathy, which affects nearly 1:500 humans, is tremendously heterogeneous, with mutations in several different proteins linked to the eventual development of hypertrophy. Currently many of the identified mutations that are linked to human hypertrophic cardiomyopathy have not been studied in any detail. Most of our understanding of the functional impact of specific mutations comes from mouse models in which the gene of interest has been knocked out. These knock-out models typically take years and thousands of dollars to develop. They are an impractical tool to screen multiple mutations within a given gene. We have developed a method to culture mouse cardiomyocytes in a 3-dimensional tissue construct forming a functional strip of cardiac muscle tissue. This model allows us to study contractile function and molecular events in existing mouse models of cardiac disease prior to the development of significant remodeling events. Furthermore we can rapidly and efficiently express specific mutant proteins in an intact cardiac tissue system and identify the impact of the mutation on contractile function and the development of the hypertrophic phenotype. This technology will identify important genotype-phenotype relationships and identify potential pathways to target for therapeutic intervention.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Dr. Ben August of The Electron Microscope Facility and Ms Beth Gray, Research Animal Resources Center, University of Wisconsin-Madison, for their assistance. The authors are grateful to Dr. Michael Wilhelm for critical review of the manuscript. pET-Mybpc3WT was a kind gift from Dr Ina Rybakova.
SOURCES OF FUNDING This work was supported by NIH K08 HL074224-01 (JCR); American Heart Association BGIA 0765303U (JCR), NIH R37 HL089200 (RLM), and funding from the Children's Cardiomyopathy Foundation (JCR).
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- 3D
Three dimensional
- 2D
Two dimensional
- ECT
Engineered cardiac tissue
- HCM
Hypertrophic cardiomyopathy
- ES
Embryonic stem (cells)
- iPS
Induced pluripotent stem (cells)
- CM
Cardiomyocyte
- WT
Wild type
- cMyBP-C
Cardiac myosin binding protein-C
- Mybpc3
Mouse cardiac myosin binding protein-C gene
- MYBPC3
Human cardiac myosin binding protein-C gene
- cMyBP-C−/−
Homozygous cMyBP-C knockout mice/cells/ECT
- adWT
Adenovirus encoding human cMyBP-C
- MOI
Multiplicity of infection
- Myh6
Mouse alpha myosin heavy chain gene
- Myh7
Mouse beta myosin heavy chain gene
- Actb
Mouse beta actin gene
- Nppa
Mouse atrial natriuretic peptide gene
- Nppb
Mouse brain natriuretic peptide gene
- qRT-PCR
Quantitative real-time polymerase chain reaction
- FMax
Maximal twitch force
- Ct100
Time to maximal twitch force
- Rt50
Time to 50% twitch force decay
- SEM
Standard error of the mean
- MTT
3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- TEM
Transmission electron microscope
- P1
Postnatal day 1
- P10
Postnatal day 10
- P35
Postnatal day 35
Footnotes
DISCLOSURES The authors have no disclosures.
Please refer to Supplemental Data for additional methods.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE LIST
- 1.Eschenhagen T, Fink C, Remmers U, Scholz H, Wattchow J, Weil J, Zimmermann W, Dohmen HH, Schafer H, Bishopric N, Wakatsuki T, Elson EL. Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: A new heart muscle model system. FASEB J. 1997;11:683–694. doi: 10.1096/fasebj.11.8.9240969. [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann WH, Fink C, Kralisch D, Remmers U, Weil J, Eschenhagen T. Three-dimensional engineered heart tissue from neonatal rat cardiac myocytes. Biotechnol Bioeng. 2000;68:106–114. [PubMed] [Google Scholar]
- 3.Tobita K, Liu LJ, Janczewski AM, Tinney JP, Nonemaker JM, Augustine S, Stolz DB, Shroff SG, Keller BB. Engineered early embryonic cardiac tissue retains proliferative and contractile properties of developing embryonic myocardium. Am J Physiol Heart Circ Physiol. 2006;291:H1829–1837. doi: 10.1152/ajpheart.00205.2006. [DOI] [PubMed] [Google Scholar]
- 4.Clause KC, Tinney JP, Liu LJ, Gharaibeh B, Huard J, Kirk JA, Shroff SG, Fujimoto KL, Wagner WR, Ralphe JC, Keller BB, Tobita K. A three-dimensional gel bioreactor for assessment of cardiomyocyte induction in skeletal muscle-derived stem cells. Tissue Eng Part C Methods. 2010;16:375–385. doi: 10.1089/ten.tec.2009.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen A, Eder A, Bonstrup M, Flato M, Mewe M, Schaaf S, Aksehirlioglu B, Schworer A, Uebeler J, Eschenhagen T. Development of a drug screening platform based on engineered heart tissue. Circ Res. 2010;107:35–44. doi: 10.1161/CIRCRESAHA.109.211458. [DOI] [PubMed] [Google Scholar]
- 6.Xi J, Khalil M, Shishechian N, Hannes T, Pfannkuche K, Liang H, Fatima A, Haustein M, Suhr F, Bloch W, Reppel M, Saric T, Wernig M, Janisch R, Brockmeier K, Hescheler J, Pillekamp F. Comparison of contractile behavior of native murine ventricular tissue and cardiomyocytes derived from embryonic or induced pluripotent stem cells. FASEB J. 2010;24:2739–2751. doi: 10.1096/fj.09-145177. [DOI] [PubMed] [Google Scholar]
- 7.Carrier RL, Papadaki M, Rupnick M, Schoen FJ, Bursac N, Langer R, Freed LE, Vunjak-Novakovic G. Cardiac tissue engineering: Cell seeding, cultivation parameters, and tissue construct characterization. Biotechnol Bioeng. 1999;64:580–589. doi: 10.1002/(sici)1097-0290(19990905)64:5<580::aid-bit8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 8.Engelmayr GC, Jr., Cheng M, Bettinger CJ, Borenstein JT, Langer R, Freed LE. Accordion-like honeycombs for tissue engineering of cardiac anisotropy. Nat Mater. 2008;7:1003–1010. doi: 10.1038/nmat2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radisic M, Park H, Shing H, Consi T, Schoen FJ, Langer R, Freed LE, Vunjak-Novakovic G. Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proc Natl Acad Sci U S A. 2004;101:18129–18134. doi: 10.1073/pnas.0407817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, Taylor DA. Perfusion-decellularized matrix: Using nature's platform to engineer a bioartificial heart. Nat Med. 2008;14:213–221. doi: 10.1038/nm1684. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu T, Yamato M, Isoi Y, Akutsu T, Setomaru T, Abe K, Kikuchi A, Umezu M, Okano T. Fabrication of pulsatile cardiac tissue grafts using a novel 3-dimensional cell sheet manipulation technique and temperature-responsive cell culture surfaces. Circ Res. 2002;90:e40. doi: 10.1161/hh0302.105722. [DOI] [PubMed] [Google Scholar]
- 12.Sekine H, Shimizu T, Yang J, Kobayashi E, Okano T. Pulsatile myocardial tubes fabricated with cell sheet engineering. Circulation. 2006;114:I87–93. doi: 10.1161/CIRCULATIONAHA.105.000273. [DOI] [PubMed] [Google Scholar]
- 13.Bel A, Planat-Bernard V, Saito A, Bonnevie L, Bellamy V, Sabbah L, Bellabas L, Brinon B, Vanneaux V, Pradeau P, Peyrard S, Larghero J, Pouly J, Binder P, Garcia S, Shimizu T, Sawa Y, Okano T, Bruneval P, Desnos M, Hagege AA, Casteilla L, Puceat M, Menasche P. Composite cell sheets: A further step toward safe and effective myocardial regeneration by cardiac progenitors derived from embryonic stem cells. Circulation. 2010;122:S118–123. doi: 10.1161/CIRCULATIONAHA.109.927293. [DOI] [PubMed] [Google Scholar]
- 14.Naito H, Melnychenko I, Didie M, Schneiderbanger K, Schubert P, Rosenkranz S, Eschenhagen T, Zimmermann WH. Optimizing engineered heart tissue for therapeutic applications as surrogate heart muscle. Circulation. 2006;114:I72–78. doi: 10.1161/CIRCULATIONAHA.105.001560. [DOI] [PubMed] [Google Scholar]
- 15.Huang YC, Khait L, Birla RK. Contractile three-dimensional bioengineered heart muscle for myocardial regeneration. J Biomed Mater Res A. 2007;80:719–731. doi: 10.1002/jbm.a.31090. [DOI] [PubMed] [Google Scholar]
- 16.Bian W, Liau B, Badie N, Bursac N. Mesoscopic hydrogel molding to control the 3d geometry of bioartificial muscle tissues. Nat Protoc. 2009;4:1522–1534. doi: 10.1038/nprot.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that mybpc3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Seidman JG, Seidman CE. Narrative review: Harnessing molecular genetics for the diagnosis and management of hypertrophic cardiomyopathy. Ann Intern Med. 2010;152:513–520. W181. doi: 10.1059/0003-4819-152-8-201004200-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ, Elliott PM. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441. doi: 10.1161/CIRCGENETICS.108.821314. [DOI] [PubMed] [Google Scholar]
- 20.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-c knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 21.Korte FS, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-c. Circ Res. 2003;93:752–758. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 22.Stelzer JE, Dunning SB, Moss RL. Ablation of cardiac myosin-binding protein-c accelerates stretch activation in murine skinned myocardium. Circ Res. 2006;98:1212–1218. doi: 10.1161/01.RES.0000219863.94390.ce. [DOI] [PubMed] [Google Scholar]
- 23.Stelzer JE, Fitzsimons DP, Moss RL. Ablation of myosin-binding protein-c accelerates force development in mouse myocardium. Biophys J. 2006;90:4119–4127. doi: 10.1529/biophysj.105.078147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stelzer JE, Patel JR, Moss RL. Protein kinase a-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cmybp-c. Circ Res. 2006;99:884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 25.Stelzer JE, Patel JR, Walker JW, Moss RL. Differential roles of cardiac myosin-binding protein c and cardiac troponin i in the myofibrillar force responses to protein kinase a phosphorylation. Circ Res. 2007;101:503–511. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- 26.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase a phosphorylation of cardiac myosin binding protein c modulates cardiac function. Circ Res. 2008;103:974–982. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atkins DL, Marvin WJ., Jr. Chronotropic responsiveness of developing sinoatrial and ventricular rat myocytes to autonomic agonists following adrenergic and cholinergic innervation in vitro. Circ Res. 1989;64:1051–1062. doi: 10.1161/01.res.64.6.1051. [DOI] [PubMed] [Google Scholar]
- 28.Lloyd TR, Marvin WJ., Jr. Sympathetic innervation improves the contractile performance of neonatal cardiac ventricular myocytes in culture. J Mol Cell Cardiol. 1990;22:333–342. doi: 10.1016/0022-2828(90)91466-k. [DOI] [PubMed] [Google Scholar]
- 29.Asnes CF, Marquez JP, Elson EL, Wakatsuki T. Reconstitution of the frank-starling mechanism in engineered heart tissues. Biophys J. 2006;91:1800–1810. doi: 10.1529/biophysj.105.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chlopcikova S, Psotova J, Miketova P. Neonatal rat cardiomyocytes--a model for the study of morphological, biochemical and electrophysiological characteristics of the heart. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2001;145:49–55. [PubMed] [Google Scholar]
- 31.Eschenhagen T, Didie M, Heubach J, Ravens U, Zimmermann WH. Cardiac tissue engineering. Transpl Immunol. 2002;9:315–321. doi: 10.1016/s0966-3274(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 32.Kim do E, Lee EJ, Martens TP, Kara R, Chaudhry HW, Itescu S, Costa KD. Engineered cardiac tissues for in vitro assessment of contractile function and repair mechanisms. Conf Proc IEEE Eng Med Biol Soc. 2006;1:849–852. doi: 10.1109/IEMBS.2006.259753. [DOI] [PubMed] [Google Scholar]
- 33.De Lange WJ, Tong CW, Ralphe JC. Electrical pacing of rat engineered cardiac tissue (ect) improves cardiac function by increasing cardiomyocyte differentiation and survival. Pediatric Research. 2009;66:470. [Google Scholar]
- 34.de Tombe PP, ter Keurs HE. Force and velocity of sarcomere shortening in trabeculae from rat heart. Effects of temperature. Circ Res. 1990;66:1239–1254. doi: 10.1161/01.res.66.5.1239. [DOI] [PubMed] [Google Scholar]
- 35.Janssen PM. Kinetics of cardiac muscle contraction and relaxation are linked and determined by properties of the cardiac sarcomere. Am J Physiol Heart Circ Physiol. 2010;299:H1092–1099. doi: 10.1152/ajpheart.00417.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janssen PM, Stull LB, Marban E. Myofilament properties comprise the rate-limiting step for cardiac relaxation at body temperature in the rat. Am J Physiol Heart Circ Physiol. 2002;282:H499–507. doi: 10.1152/ajpheart.00595.2001. [DOI] [PubMed] [Google Scholar]
- 37.Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role of camkiidelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci U S A. 2010;107:10274–10279. doi: 10.1073/pnas.1005843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pillekamp F, Halbach M, Reppel M, Rubenchyk O, Pfannkuche K, Xi JY, Bloch W, Sreeram N, Brockmeier K, Hescheler J. Neonatal murine heart slices. A robust model to study ventricular isometric contractions. Cell Physiol Biochem. 2007;20:837–846. doi: 10.1159/000110443. [DOI] [PubMed] [Google Scholar]
- 39.Tong CW, Gaffin RD, Zawieja DC, Muthuchamy M. Roles of phosphorylation of myosin binding protein-c and troponin i in mouse cardiac muscle twitch dynamics. J Physiol. 2004;558:927–941. doi: 10.1113/jphysiol.2004.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka H, Shigenobu K. Effect of ryanodine on neonatal and adult rat heart: Developmental increase in sarcoplasmic reticulum function. J Mol Cell Cardiol. 1989;21:1305–1313. doi: 10.1016/0022-2828(89)90676-7. [DOI] [PubMed] [Google Scholar]
- 41.Radisic M, Malda J, Epping E, Geng W, Langer R, Vunjak-Novakovic G. Oxygen gradients correlate with cell density and cell viability in engineered cardiac tissue. Biotechnol Bioeng. 2006;93:332–343. doi: 10.1002/bit.20722. [DOI] [PubMed] [Google Scholar]
- 42.Chiu LL, Radisic M, Vunjak-Novakovic G. Bioactive scaffolds for engineering vascularized cardiac tissues. Macromol Biosci. 2010;10:1286–1301. doi: 10.1002/mabi.201000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiu LL, Radisic M. Scaffolds with covalently immobilized vegf and angiopoietin-1 for vascularization of engineered tissues. Biomaterials. 2010;31:226–241. doi: 10.1016/j.biomaterials.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 44.Radisic M, Marsano A, Maidhof R, Wang Y, Vunjak-Novakovic G. Cardiac tissue engineering using perfusion bioreactor systems. Nat Protoc. 2008;3:719–738. doi: 10.1038/nprot.2008.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brickson S, Fitzsimons DP, Pereira L, Hacker T, Valdivia H, Moss RL. In vivo left ventricular functional capacity is compromised in cmybp-c null mice. Am J Physiol Heart Circ Physiol. 2007;292:H1747–1754. doi: 10.1152/ajpheart.01037.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.