

Abstract
We previously reported that several point mutations in the ligand binding domain (LBD) of glucocorticoid receptors (GRs) marginally affect the binding affinity of the synthetic glucocorticoids dexamethasone (Dex) and deacylcortivazol (DAC). However, these mutations dramatically alter the efficacy (Amax) and potency (EC50) of agonists, along with the partial agonist activity (PAA) of the antisteroid Dex-mesylate (DM), for gene induction and repression in a steroid-dependent manner. This was proposed to result, in part, from altered protein-protein interactions in the complex of GR with the coactivator TIF2 despite normal TIF2 binding. To explore the generality of this phenomenon, we now ask whether these mutations also affect the transactivation properties, but not binding, of other GR-bound factors. We find that an elevated concentration of GR, to probe unidentified cofactors, or of the comodulator Ubc9 do not reverse the effects of GR LBD mutations that increase the EC50 and lower the PAA with the GREtkLUC reporter in both CV-1 and U2OS cells. This behavior is more dramatic with Ubc9 and the isolated GR LBD fused to the GAL4 DNA binding domain, despite normal binding of Ubc9 to the mutant GRs. Similar effects, albeit gene, steroid, and transcriptional property specific, are seen with full length GRs and three endogenous genes in U2OS cells. Thus changes in simple steady-state binding capacities of mutant receptors for factors cannot account for the modified transcriptional properties. In all cases, the nuclear translocation of Dex- and DAC-bound wild type and mutant receptors is the same. These results are consistent with the earlier results with TIF2 and support the hypothesis that small changes in the GR LBD can alter the activities of the bound cofactor without modifying cofactor binding. We propose that this separation of binding and the modulation of transactivation parameters occurs for a wide variety of GR-associated cofactors.
The ability of steroid receptors to regulate gene transcription lies at the heart of their roles in differential gene expression during development, differentiation, and homeostasis. Glucocorticoids are particularly effective because they affect virtually every cell in the body and are used to treat a variety of human pathologies including asthma, autoimmune diseases, cancer, and the induction of surfactant in the lungs of premature infants (1, 2). These effects of glucocorticoid steroids are thought to occur via the classical mechanism of passive diffusion into the cell and binding to the intracellular receptor protein. The resulting glucocorticoid receptor (GR1)-steroid complex binds with high affinity to biologically active DNA sequences to recruit other transcriptional cofactors, thereby increasing or decreasing the rates of transcription of gene expression.
In addition to modifying the rates of transcription to increase or decrease the maximal levels of gene product (Amax), many transcriptional cofactors also alter both the concentration of agonist steroid required for half-maximal induction (EC50) and the partial agonist activity of antisteroids (PAA) (reviewed in 3, 4). The EC50 is a measure of steroid potency but it is not the same for all genes regulated by that steroid. Thus, the endogenous subsaturating amount of steroid will cause a different percentage of full induction (or repression) for most responsive genes. The PAA is expressed as percent of maximal activity of an agonist under the same conditions. Therefore, the PAA is a measure of the amount of residual agonist activity displayed by an antisteroid. This parameter is critical when antisteroids are used in endocrine therapies to block the action of endogenous agonist steroids. The PAA also varies with the gene examined. For this reason, it is theoretically possible during endocrine therapy to target the inhibitory action of antisteroids to a subset of the regulated genes, thereby reducing the number of unwanted side effects from that which occurs when all regulated genes are repressed. For all three parameters of GR-regulated gene expression (Amax, EC50, and PAA), changing the level of factor acts as a rheostat to give a continuum of responses for GRs and the other classical steroid receptors (3, 4). These two effects of changing cofactor concentration provide attractive additional mechanisms for achieving differential control of gene expression during development, differentiation, and homeostasis, i.e., by altering the responses in a gene-selective manner to subsaturating, physiological concentrations of agonist steroid and to pharmacologically administered doses of antisteroid.
One frequently studied modulatory factor of GR transcriptional properties is TIF2/GRIP1. TIF2 (the name for the human coactivator) is a member of the p160 family of coactivators that includes SRC-1 and AIB1/pCIP/ACTR/TRAP1/RAC3. A central region of TIF2 containing three receptor interacting domains (RIDs) binds to a pocket in the AF2 activating function domain of the GR ligand binding domain (LBD), which is formed by helices 3, 4, 5, and 12 (5). More recently, a second, amino-terminal region of TIF2 has been found to interact with the amino-terminal domain of GR and inhibit corepressor binding to GR (6). Even with two GR-interaction domains for TIF2, there appears to be a limit to how much the Amax, EC50, and PAA of GR-mediated transactivation can be altered with increasing amounts of TIF2 (7). Of further interest is the recent report that several GR LBD mutations in the steroid binding pocket (8) selectively alter both the absolute Amax and EC50 of GR-mediated gene induction in a steroid-specific manner (9). These GR mutations were chosen following a comparison of the x-ray structures of GR LBD bound by two very differently shaped steroids: dexamethasone (Dex) and deacylcortivazol (DAC) (8). It was predicted that these mutations would selectively affect Dex vs. DAC binding. Unexpectedly, the changes in steroid binding of many mutants were minor compared to their effects on Amax, EC50, and PAA (9). This was proposed to result in part from alterations in specific GR surfaces. These perturbed surfaces did not affect TIF2 binding to GR but did modify, via protein-protein induced changes, some TIF2 domains interacting with downstream transcription factors (9). This observation led us to ask if such changes in the efficiency of information transmission through a GR-bound cofactor to the transcriptional machinery was unique to TIF2 or also occurred with other factors known to influence GR transactivation properties. The phenomenon would be of even greater interest if it also occurred for a cofactor that bound to a different region of GR (10). Such a result would suggest that subtle changes in LBD structure can have wider spread topological consequences (11, 12) and are not limited to the GR-TIF2 contact surface(s).
A particularly attractive cofactor with which to address this question is Ubc9, which is the human homolog of a yeast E2-ubiquitin conjugating enzyme. Ubc9 mediates the attachment of a small ubiquitin-like molecule (SUMO) to proteins. However, Ubc9 also alters the Amax, EC50, and PAA of GR-regulated transcription in a manner that is independent of its sumoylation activity (13–15). Like TIF2, Ubc9 binds to the GR LBD (10, 14). Like TIF2, Ubc9 action depends upon domains in both the N-terminal half and LBD of GR, although the LBD interactions are the strongest (14). However, there are two important differences between Ubc9 and TIF2. First, the modulatory effects of TIF2 are most dramatic at low, and disappear at high, GR concentrations (7). In contrast, Ubc9 increases the Amax at low and high concentrations of GR but reduces the EC50, and enhances the PAA, only with high GR concentrations (13–16). Second, the effects of Ubc9 appear to be manifested at a step downstream of TIF2 action (13, 16). This argues that TIF2 and Ubc9 do not interact with the same factors to modulate Amax, EC50, or PAA and could contact different surfaces of the GR LBD.
The purpose of this study was to determine whether the above receptor mutations that alter GR activity with TIF2 also affect the activity of two other cofactors that are known to modulate the transactivation properties of GR but bind to different regions of the GR LBD, and/or interact with other cofactors, than TIF2. The first factor is Ubc9. The second is GR itself. It is well documented that increasing concentrations of GR elevate the PAA and decrease the EC50 of GR gene transactivation (3, 7, 17). GR also forms dimers using surfaces removed from that for binding TIF2 (18). We now find that, when the concentration of GR or Ubc9 is increased, there are minimal consequences of these LBD mutations on Amax but much larger reductions in PAA and EC50. In some circumstances, changes in EC50 depend upon the structure of the bound agonist steroid. These results, in combination with our previous data with TIF2, lead us to conclude that minor changes in GR LBD structure can influence the activity, but not the binding, of a variety of associated transcriptional cofactors in a manner that is sometimes further influenced by the nature of the agonist steroid.
Materials and Methods
Unless otherwise indicated, all cell growth was at 37 °C and all other operations were performed at room temperature or as recommended by the supplier.
Chemicals
Dexamethasone (Dex) was purchased from Sigma (Louis, MO). Dex-21-mesylate (DM) was synthesized as previously described (19). Cortivazol (gift from Roussel UCLAF) was converted to deacylcortivazol (DAC) by C. Thomas (NIDDK, NIH) via hydrolysis of the C21-acetyl group. Renilla TS was a gift from N. M. Ibrahim, O. Fröhlich, and S. R. Price (Emory University School of Medicine, Atlanta, GA).
Transient Transfection and Reporter Analysis
CV-1 or U2OS cells were grown and assayed as previously described (20, 21). Briefly, cells were seeded (20,000 cells per well) one day before transfection in 24-well plates. GRE-tk-luciferase reporter plasmid or pFRLuc (Stratagene-Agilent Technologies, Inc., Santa Clara, CA; for GAL-regulated reporter assay) and other plasmids (total DNA = 300 ng/well) were transiently transfected with FuGENE6 (Roche Molecular Biochemicals, Indianapolis, IN) or Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturers’ protocols. The mutant GRs are described in a previous study (9). The cells were induced by steroids (dissolved in absolute EtOH; final EtOH concentration in assays ± steroid is constant and ≤ 1%) for 16 h and assayed for lucifierase activity using the dual-luciferase reporter assay (Promega, Madison, WI). The luciferase activity was normalized with Renilla TS activity.
Quantitative Real-Time PCR (qRT-PCR)
U2OS cells (200,000 cells per well in six-well plates) were treated with various concentrations of Dex, Dex-mesylate (DM), or DAC for 16 h. Total RNA was extracted using TriZol reagent (Invitrogen) and cDNA was synthesized using SuperScript III, First-Strand Synthesis (Invitrogen). The relative expressional levels of insulin-like growth factor binding protein 1(IGFBP1), inositol hexakisphosphate kinase 3 (I6PK), or ladinin 1 (LAD1) were quantitated by SYBR Green in an ABI 7900HT real-time PCR system (21). The quantitation was normalized with glyceraldehyde-3-phosphate dehydrogenase (#4310884E, Applied biosystems, Carlsbad, CA).
Co-Immunoprecipitation assay and Western Blotting
Cos-7 cells were transiently transfected with plasmids for the wild type GR and its mutants without or with Flag-tagged Ubc9 using FuGENE6 (Roche). After 48 h, cells were induced by Dex for 1 h and then cross-linked at 37 °C with 1 mM dimethyl 3,3′-dithiobispropionimidate (DTBP; Thermo Fisher Scientific Inc., Rockford, IL) as described by Vicent et al. (22). The cell lysates were immunoprecipitated with anti-Flag M2-agarose beads (Sigma) at 0 °C and eluted for immunoblotting by boiling for 10 min in 2X SDS sample buffer (NuPAGE LDS Sample buffer + NuPAGE Sample Reducing Agent). Western blots were prepared, probed at r.t. with mouse anti-Flag monoclonal antibody (Sigma), rabbit anti-β actin polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), rabbit anti-GR antibody (PA1-511A, Affinity Bio-Reagents, Dublin, OH), or mouse anti-GAL4 DBD monoclonal antibody (Santa Cruz), and visualized by ECL detection reagents as described by the manufacturer (GE healthcare, Piscataway, NJ).
Immunocytochemistry
U2OS cells (10,000 cells per well) in 8-well chamber slides (BD Biosciences, San Jose, CA) were transiently transfected GR or its mutants with Lipofectamine 2000 (invitrogen). After 48 h, cells were treated with 1μM Dex, 100nM DAC, or ethanol for 1 h and fixed with 4% paraformaldehyde at r.t. The fixed cells were incubated with rabbit anti-GR antibody and further probed with anti-rabbit IgG-Cy3 (Sigma) at r.t. The slides were mounted using VECTASHIELD with DAPI (Vector Laboratories, Inc., Burlingame, CA) and analyzed by fluorescence microscopy.
Data analysis
The maximum induced activity (Amax) in cells transiently transfected with GR plasmids was obtained with saturating concentrations of agonist steroid, which was either ≥ 100-fold higher than the EC50 or 10 μM, whichever was lower. For gene induction, the basal activity is that without steroid and maximal activity is that produced by saturating agonist steroid concentrations. The fold induction is (induced value)/(basal activity). The partial agonist activity (PAA) of the antagonist (DM) was calculated by expressing the activity of a saturating concentration of DM (10 μM) as the percent of maximal activity of a saturating concentration of agonist under the same conditions. For dose-response curves, seven concentrations of Dex are used, with each point being the average of triplicate samples ± the standard deviation. One curve of average points yields one value of EC50 (the concentration of agonist required for 50% of the maximal response) via best-fit curve fitting programs with KaleidaGraph (Synergy Software, Reading, PA) following a first-order Hill plot (R2 almost always ≥ 0.95). For bar graphs giving average values of Amax, EC50, and PAA, the average of n replicates (each in triplicate but considered, statistically, as one observation) was plotted ± the standard error of the mean (n observations) unless otherwise noted. Statistical significance was assessed by the two-tailed Student’s t test using InStat 2.03 (GraphPad Software, San Diego, CA). In every case, each average of triplicates was treated as one value of the n experiments. When the difference between the SDs of two populations was significantly different, the Mann-Whitney or Alternate Welch t test was used. A nonparametric test was used if the distribution of values was non-Gaussian.
Results
Activities of mutant GRs with higher GR concentration
One possible reason for the generally lower activities of the previously reported mutant receptors (9) is that the steady-state interactions of GR with unidentified factors is reduced by the LBD mutations. This hypothesis can be tested by increasing the concentration of GR in cells under conditions where GR is a limiting component. It is well known that gene transactivation with elevated levels of wild type GR gives rise to an increased Amax and PAA, along with a lower EC50 (4, 9, 17). This indicates that GR is limiting in these reactions. Mass-action considerations predict that augmenting the levels of GR will force the steady-state reaction towards GR complex formation with normally binding cofactors. Thus, raising the concentration of subsaturating amounts of presumed low avidity binders (e.g., mutant GRs) will increase the complex formation with a cofactor much more than when elevating the amount of strong binders (e.g., wild type GRs). In the present case, elevated levels of the weakly binding mutant GRs would drive the Amax, PAA, and EC50 back to wild type levels if our hypothesis is correct.
Four rat GR LBD mutations (Q588K, A625I, L626I, and R629Y) were selected for the current study. The R629Y mutant, though, has negligible transactivation activity under many conditions (9) and thus was not included in the experiments in the first phase of the current study. The three other mutants, plus the wild type receptor, were transiently transfected into CV-1 cells at low and high concentrations of receptor plasmid. The effect of varying receptor concentration on the induction properties of a co-transfected, synthetic reporter gene (GREtkLUC) was then determined. The properties examined were the Amax and EC50 for the agonist dexamethasone (Dex) and the PAA for the antiglucocorticoid Dex-21-mesylate (DM) (Fig. 1). Most values of Amax and PAA with low levels of mutant receptors (0.1 ng of receptor plasmid) in CV-1 cells are significantly less than those for wild type GR while the EC50s are higher. Transfections with 10–20 times higher concentrations of each receptor plasmid cause the expected (9, 17) increases in Amax and PAA, and decreases in EC50. It should be noted that the 4-fold increase in Amax is less than the 10- to 20-fold increase in plasmid for the wild type GR. This suggests that the GR levels are approaching saturation under the high GR conditions (7). The Amax and PAA of the mutants increase more than for the wild type GR. Thus, the Amax of each mutant GRs is now closer to wild type levels. This is what might be expected if the mutant GRs have reduced binding of a factor(s) required for Amax. However, the PAA with the antiglucocorticoid DM is still significantly lower than that of the wild type GR. More receptor is actually less effective in reducing the EC50 of the mutant GRs compared to the wild type receptor (note log scale). Thus, the ratio of EC50s for mutant vs. wild type receptors increases from 12 to 29 with low amounts of receptor to 33 to 81 with high receptor concentration. Importantly, Western blots indicate that all mutants are expressed in Cos-7 cells at about the same level as wild type GR (see Fig. 6 below). Therefore, raising the level of mutant GRs cannot overcome the effects of the LBD mutations to decrease the PAA and increase the EC50 for GR-mediated gene induction. We conclude that these reduced transcriptional activities of the mutant receptors are not attributable to lower binding capacities of important endogenous cofactors.
Fig. 1.

Variation in induction parameters of mutant GRs in CV-1 cells with more GR. Triplicate data points ± transiently transfected GR plasmid (Low GR = 0.05 ng/well, High GR = 0.5–1 ng/well) were analyzed to determine the Amax, PAA, and EC50 as described in Materials and Methods (error bars = S.E.M. from 4 independent experiments). P-values for mutant vs. wild type GR under the same conditions are * < 0.05 and ** < 0.005. Under conditions such as the present where very few differences exist between the conditions with different receptors, the prevailing model of steroid hormone action is that the EC50 of gene induction is directly proportional to the affinity of steroid binding to its cognate receptor. Therefore, the predicted EC50 of Dex with the mutant GRs, indicated by the thick horizontal lines, can be calculated by multiplying the EC50 of Dex with wild type GR by the relative increase in the Kd for cell-free Dex binding to the mutant receptors. This relative increase in Kd was previously determined (9) by Scatchard analysis to be 1 for wt GR, 2.8 for Q588K, 5.9 for A625I, and 2.4 for L626I.
Fig. 6.

Binding of Ubc9 to wild type and mutant GRs. Co-immunoprecipitation of full length (A) and truncated (B) wild type and mutant GRs with Ubc9. Extracts were prepared of Cos-7 cells, which were transiently transfected with GR and Flag/Ubc9 (or empty Flag vector, indicated by *) and subsequently incubated with the indicated steroids, were treated with anti-Flag and subjected to Western blotting as described in Materials and Methods.
The generality of the above behavior was addressed by using a different cell line, i.e., U2OS cells. As seen in Fig. 1 for CV-1 cells, the PAA was again lower, and the EC50 higher, for GREtkLUC reporter induction by the mutant GRs in U2OS cells (Fig. 2A). Increased GR concentration affected the PAA of wild type and mutant GRs about equally, so that the mutant GRs still display substantially less activity than wild type GR in U2OS cells. The EC50s (note log scale) of mutant receptors generally went from 5 to 10-fold greater than the EC50 for wild type GR at low receptor concentrations to 20 to 120-fold greater at high receptor levels.
Fig. 2.

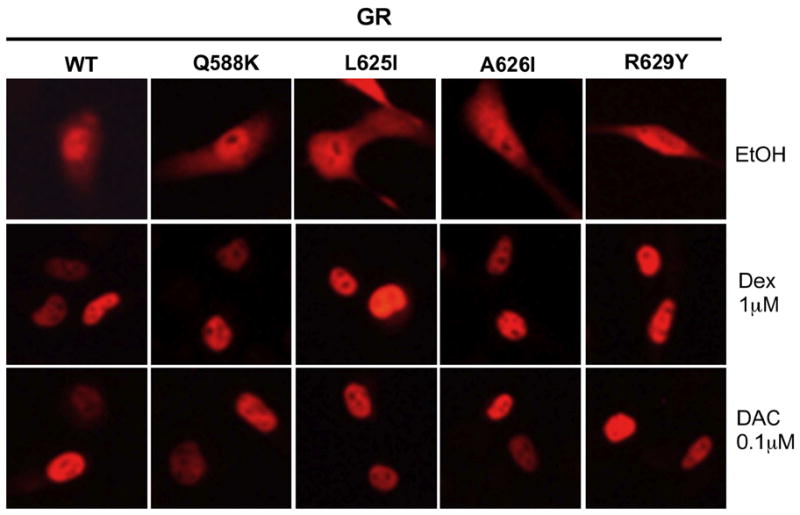
Properties of mutant GRs in U2OS cells. (A) Variation in induction parameters of mutant GRs in U2OS cells with elevated GR. Data from triplicate transiently transfected cells were generated, analyzed, and plotted along with predicted EC50 values as in Fig. 1 (error bars = S.E.M. from 5 independent experiments). P-values for mutant vs. wild type GR under the same conditions are * < 0.05 and ** < 0.005. (B) Whole cell localization of wild type and mutant GRs following treatment with Dex and DAC. Cells were treated as described in Materials and Methods with the indicated steroids. Cells were visualized with DAPI stain while GR was identified by immunofluorescence.
In both CV-1 and U2OS cells, the EC50 for gene induction at elevated levels of receptor is 20 to 120-fold higher for the mutant GRs than for wild type GR. These differences are much greater than can be explained by the 2 to 6-fold increase in Kd for steroid binding to the mutant GRs (9). The expected value of the EC50, if it reflected only the reduced affinity of Dex for each mutant receptor, is indicated by the thick horizontal line through each bar of the EC50 graphs. Furthermore, these differences are exacerbated by high receptor concentrations. These differences should be diminished if they were due simply to a decreased ability of mutant GRs to form dimers or interact with other required factors. Thus, some other explanation is required.
This inability of increased GR concentration to counter the effects of the LBD mutations on PAA and EC50 could be due to unequal nuclear binding of GR complexes. However, immunocytochemistry shows equal nuclear localization by 1 μM Dex and by 100 nM DAC (discussed below) in U2OS cells under the bioassay conditions of Fig. 2 (Fig. 2B). Thus the altered transactivation properties of the mutant GRs, which are disproportionate to changes in steroid binding affinity, are not due to altered nuclear binding of GR complexes.
Activities of mutant GRs with added Ubc9
Ubc9 binds to GR and has been identified as acting downstream of GR (13–16). For studies with Ubc9, we have included the fourth mutant, R629Y. Ubc9 is known to increase the Amax of high concentrations of GR (13–16). Thus, the marginal activity at high levels of the R629Y mutant might also be augmented by added Ubc9. As shown in Fig. 3 for CV-1 cells, the ability of added Ubc9 to increase the Amax of mutant and wild type GRs is about the same. This is true for both those mutants that have similar levels of activity and, as for R629Y, dramatically reduced activity. The PAA of the antiglucocorticoid DM with the four mutant GRs is well below that for wild type GRs. Elevated Ubc9 does produce a larger increase with mutant GRs than wild type GR. However, this comparison is theoretically flawed. The scale for PAA is non-linear and cannot exceed 100% of a full agonist. Clearly, factors causing a 10-fold increase in the PAA of one antagonist (e.g., from 5 to 50%) can not produce the same 10-fold increase of another antagonist the displays 50% PAA before the addition of factors. Thus, the increase in PAA of DM with wild type GR to about 100% in Fig. 3 is as large as possible even if the fold-increase is less than that seen with most of the mutant receptors. In contrast, the EC50 for Dex induction of GREtkLUC by the wild type and mutant GRs are all decreased by about 2-fold by added Ubc9. It should be noted that no thick horizontal bar is present for the R629Y mutant because its affinity for Dex was below detection (9), thus making the calculation of a theoretical EC50 impossible.
Fig. 3.

Variation in induction parameters of mutant GRs in CV-1 cells with increased Ubc9. Data from triplicate transiently transfected cells (1 ng GR ± 150 ng Ubc9 plasmid) were generated, analyzed, and plotted along with predicted EC50 values as in Fig. 1 (error bars = S.E.M. from 6–7 independent experiments). The absence of any measurable affinity of Dex for R629Y (9) precluded the calculation of a predicted EC50, so no thick horizontal bar is included. P-values for mutant vs. wild type GR under the same conditions are * < 0.05, ** < 0.005, and *** < 0.0005.
The modulatory activity of additional Ubc9 in CV-1 cells (Fig. 3) is recapitulated in U2OS cells (Fig. 4). The changes with exogenous Ubc9 in Amax, PAA (given the above mentioned constraints), and EC50 are about the same for wild type and mutant GRs. It should be noted that the EC50 for gene induction in both cell lines by each of the mutant receptors except R629Y, which does not have a measurable affinity for Dex (9), is still 9 to 16-fold higher than predicted from the Dex binding affinity of each receptor (indicated by thick horizontal line for each bar graph). This difference persists in the presence of exogenous Ubc9. Thus, the increased EC50, decreased PAA, and often decreased Amax of the mutant receptors cannot be rectified simply by a higher concentration of Ubc9; and, these behaviors are not unique to a single cell line.
Fig. 4.

Variation in induction parameters of mutant GRs in U2OS cells with a higher amount of Ubc9. Data from triplicate transiently transfected cells were generated, analyzed, and plotted along with predicted EC50 values as in Fig. 3 (error bars = S.E.M. from 5 independent experiments). P-values for mutant vs. wild type GR under the same conditions are * < 0.05 and ** < 0.005.
Role of AF domains and steroid structure in determining mutant GR properties
The above data establish that more of either GR or Ubc9 are active in altering several induction parameters (Amax, PAA, and EC50) of both mutant vs. wild type GRs. However, we remain confronted with the conundrum of why the LBD mutations have a much larger effect on PAA and EC50 than Amax. One possible explanation derives from the facts that the major determinant of Amax is the activation function domain in the amino terminal half of GR (i.e., AF1) while the C-terminal AF2 domain is sufficient to regulate the PAA and EC50 with added GR, Ubc9, or TIF2 (14). Furthermore, as seen with TIF2, the effects of these mutations are greatly reduced when GR is bound by the bulky glucocorticoid, deacylcortivazol (DAC) (9). DAC looks (Fig 5A) and binds to GR very differently from Dex (8, 23). Therefore, we predicted both that the ability of the mutant receptors to respond to added Ubc9 would be decreased upon removal of the AF1 domain and that the Amax and EC50 of such truncated GRs would be less affected by the mutations when bound by DAC than by Dex. To address this question, we used the chimeric receptor GAL/GR525C, which involves the GAL4 DNA binding domain fused to the GR LBD (amino acids 525 to the C-terminus; Fig. 5B). We then determined the transactivation properties of GAL/GR525C with a GAL regulated reporter, FRLuc. We limited our comparisons to a single, representative mutant (A625I). While the other mutants may display different properties, this has generally not been the case so far except for R629Y (see Figs. 1–4 and ref. 9).
Fig. 5.
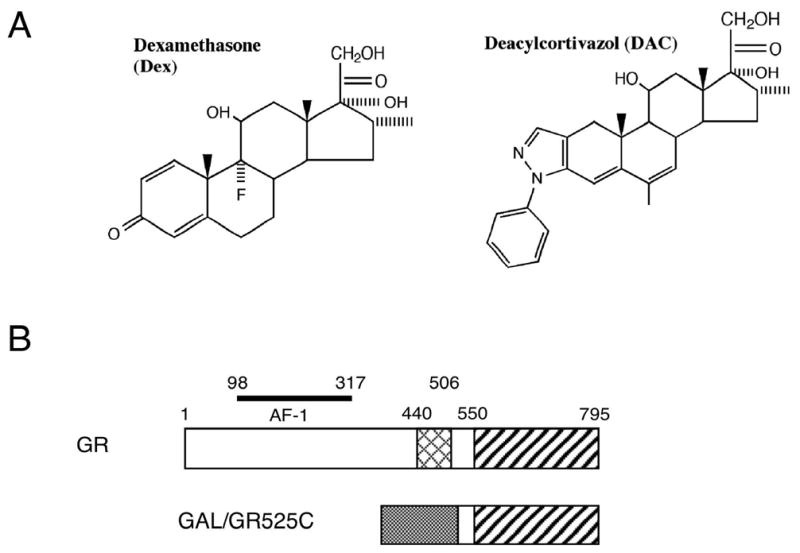

Role of GR LBD in determining induction properties of Dex- vs. DAC-bound mutant vs. wild type GRs with added Ubc9. (A) Structures of Dex and DAC. (B) Organization of chimeric truncated GRs (GAL/GR525C). Residues 1–524 of wild type GR, including AF1 and DNA binding domains, are replaced by the GAL4 DNA binding domain (shaded box) while the GR LBD (diagonal striping) remains. (C) Variation in induction parameters of GAL/GR525C chimeric receptors in U2OS cells with elevated Ubc9. Data from triplicate transiently transfected cells (1 ng of GAL/GR525C with wild type or A625I GR LBD ± 150 ng Ubc9 plasmid) were generated, analyzed, and plotted along with predicted EC50 values as in Fig. 4 (error bars = S.E.M. from 7 independent experiments).P-values for mutant vs. wild type GR under the same conditions are * < 0.05, ** < 0.005, and *** < 0.0005. The predicted EC50 of Dex with each mutant GR was calculated as in Fig. 1. For DAC, the predicted EC50 is based upon the relative increase in Kd for DAC binding to the A625I mutant vs. wild type GR being 1.1 (9).
As previously reported (9), the A625I mutation in the context of the GAL/GR525C construct greatly decreases the Amax with Dex but not with DAC in U2OS cells (Fig. 5C). This disparity is maintained after the addition of excess Ubc9. The PAA of DM, relative to Dex, is also lower here for the A625I mutant than wild type GR in the GAL/GR525C construct. The addition of Ubc9 does not increase the PAA, as might be expected from Fig. 3, but rather lowers it even further. Finally, in the presence of Dex, the addition of Ubc9 has no effect on the EC50 of the mutant GR while that of the wild type GR is lowered. In contrast, with DAC, exogenous Ubc9 lowers the EC50 of both wild type and mutant GRs. Thus, the properties of the isolated AF2, ± Ubc9, are more severely diminished by the mutation with Dex as the bound steroid than with DAC. We conclude that the biological responses, and thus the governing biochemical interactions, of Ubc9 with DAC-bound mutant GR LBD are much more like those of the wild type GR LBD than Dex-bound mutant GR LBD.
Binding of Ubc9 to wild type and mutant GRs
The data with elevated concentrations of Ubc9 suggest that the defects of the mutant GRs are more evident in the absence of the AF1 domain (Fig. 5C), are sensitive to the structure of the bound steroid (Fig. 5C), and are not due to altered binding capacities of GR for Ubc9 (Figs. 3–4). In order to obtain more direct evidence for this last conclusion, we used a co-IP assay with GR and Flag-tagged Ubc9. Flag-tagged Ubc9 was used because the available antibodies to Ubc9 are not very sensitive (see Fig. 6A below) and because the biological activities of Flag-Ubc9 and Ubc9 are the same (data not shown and ref. 24).
Full length wild type and mutant receptors, along with Flag/Ubc9, were overexpressed in transiently transfected Cos-7 cells. The cells were then treated with different concentrations of Dex for 1 hr, followed by mild cross-linking with DTBP to freeze the weakly associated complexes (Tao and Simons, unpublished results). Cytosolic extracts were prepared and anti-Flag antibody was added to immunoprecipitate Flag/Ubc9 and any associated GR. Fig. 6A shows that the amount of GR that was co-immunoprecipitated (co-IP’d) with Flag/Ubc9 is the same for EtOH-treated mutant GRs as for wild type GRs. Likewise, the amount of wild type GR co-IP’d in the presence of 1 μM Dex is very similar to that of the mutant GRs treated with a higher amount of Dex (10 μM) to compensate for their lower steroid binding affinity. At the same time, the binding of Ubc9 to wild type and mutant (A625I) LBDs fused to the GAL DNA binding domain was examined (Fig. 6B). This shows that the GR LBD is sufficient for Ubc9 binding and that there is no significant difference between wild type and mutant GR binding of Ubc9. We conclude that the deficiencies in Ubc9 modulation of the Amax, PAA, and EC50 of the mutant GRs are not due to any alterations in the ability to bind Ubc9.
Induction of endogenous genes by wild type vs. GR A625I mutant bound by Dex vs. DAC
The above data suggest that the PAA and EC50 for gene induction by GR, with and without exogenous Ubc9, are much more sensitive than Amax to mutations in the GR LBD. These changes are influenced by steroid structure but dissociated from the GR binding affinity of steroid and the GR binding capacity of Ubc9. To determine whether these phenomena with an exogenous, synthetic reporter gene also occur under more physiologically relevant conditions, we examined the behavior of three endogenous genes with full length wild type and mutant GR (A625I) treated with Dex or DAC in the presence or absence of exogenous Ubc9. The three genes selected were IGFBP1, I6PK, and LAD1. IGFBP1 is a member of the family of structurally homologous proteins that circulates in the plasma and specifically binds and modulates the half-life of IGF-1 and IGF-2 and their interaction with cell surface receptors. Very little is known about I6PK except that it is glucocorticoid inducible (25). LAD1 (Ladinin) is a novel component of the basement membranes and may function in contributing to the stability of the association of the epithelial layers with the underlying mesenchyme (26).
In these studies, fold induction, as opposed to the often closely related Amax, was determined because the qRT-PCR determinations with SYBR Green cannot give quantitative measurements of cDNAs. In all cases with Dex, the fold induction is somewhat less with the mutant GR (Fig. 7). In contrast, the fold induction by mutant GR with DAC is always the same or more than that with wild type GR. Furthermore, added Ubc9 affords a greater increase in fold induction with DAC-bound mutant receptor. Large differences in the PAA of the antiglucocorticoid DM are seen between mutant and wild type receptors, both without and with added Ubc9. The PAA, whether without or with added Ubc9, is always between 11 and 21% of the value for wild type GRs. There is no significant difference between the Ubc9-induced changes in EC50 for Dex- vs. DAC-bound receptors. Interestingly, excess Ubc9 only exacerbates the difference between wild type and mutant GR EC50s, which are 4 to 30-fold higher than predicted in the absence of exogenous Ubc9. Thus, the consequences of the A625I mutation vary in severity with the parameter examined in a manner that is sensitive to steroid structure and endogenous gene. Similarly, the effect of elevated Ubc9 can be steroid- and gene-dependent. The PAA of the antiglucocorticoid DM is much lower with the mutant than with wild type GR, with or without exogenous Ubc9. The fold induction by mutant vs. wild type GR is always greater for DAC- than Dex-bound receptors; but, what happens with added Ubc9 is gene- and steroid-dependent. Finally, the EC50 of mutant receptors is always lower than predicted from the binding affinity of the inducing steroid and relatively insensitive to added Ubc9.
Fig. 7.

Variation in induction parameters of endogenous genes by mutant GRs in U2OS cells with more Ubc9. Data from triplicate transiently transfected cells (50 ng GR and 200 ng Ubc9) that had been treated with Dex, DM, or DAC were generated by qRT-PCR assay of mRNA preparations as described in Material and Methods. The data were then analyzed and plotted as in Fig. 4 (error bars = S.E.M. from 5–7 independent experiments with Dex and 4 with DAC).
Discussion
Selected point mutations in the LBD pocket of GR are shown to alter the three major parameters of gene induction (Amax, PAA, and EC50) by glucocorticoid receptors in a manner that depends upon numerous factors: the concentration of GR itself, the amount of the comodulator Ubc9, the structure of the bound steroid, and the induced gene. These changes can not be explained by mutation-induced differences in either steroid binding affinity of, or factor binding capacity with, GR. Several lines of evidence suggest that the altered transcriptional parameters derive from minor conformational changes in the GR LBD that modify the activities of bound cofactor without affecting cofactor binding to GR.
The point mutations examined here were among those predicted from x-ray studies (8) to have the greatest effect on the binding of two differently sized agonists, Dex and DAC (9). We previously found that these mutations do slightly reduce the affinity of each steroid for GR but that the greatest effect was on the gene induction parameters of GR with and without the added coactivator TIF2, which were affected much more than predicted from the changes in steroid binding affinity. The earlier studies with TIF2 led to the conclusion that the activity of GR-bound TIF2, but not the binding of TIF2 to GR, was altered in the mutant GRs in a manner that was much greater for Dex- than for DAC-bound receptors (9). The present work confirms and extends these conclusions to other GR-binding factors (i.e., GR and Ubc9) in two different cell lines and for selected endogenous genes. Added receptor decreases the EC50 of the mutant GRs much less than for the wild type GR in two cell lines (Figs. 1 and 2). This suggests that the mutant GRs are desensitized in a manner other than a reduced avidity for some factor, which would be expected to be counterbalanced by increasing the receptor concentration. With Ubc9 and the exogenous GREtkLUC reporter, the effects are somewhat masked by the presence of the amino-terminal AF1 domain. We have previously documented that the GR LBD in GAL/GR525C is sufficient to mediate the modulatory activity of Ubc9 (14). Therefore, the isolated GR LBD in GAL/GR525C should be more responsive to added Ubc9 than the full length GR. In fact, experiments with mutant GAL/GR525C chimera clearly show that none of the parameters (Amax, PAA, and EC50) with Dex-bound mutant receptor are appreciably modulated by added Ubc9 (Fig. 5C) even though Ubc9 binding is unchanged (Fig. 6). In contrast, the induction parameters of DAC-bound mutant GAL/GR525C remain responsive to Ubc9 (Fig. 5C).
The current experiments also support and expand our earlier observations with TIF2 siRNA, where the induction properties of endogenous genes in human peripheral blood mononuclear cells (PBMCs) were selectively affected in a gene-specific manner upon reducing the intracellular levels of TIF2 protein (27). In the current studies with three different endogenous genes in U2OS cells, the ability of added Ubc9 to modify the parameters of the mutant A625I receptor is again gene selective. Furthermore, in contrast to Dex-bound mutant receptor, the fold-induction of DAC-bound mutant receptor is greater than that of wt GR without or with added Ubc9 (Fig. 7). This selective modulation of individual induction parameters has frequently been documented for synthetic reporter genes with mutant receptors (estrogen, progesterone, and glucocorticoid) and with chemicals such as TSA and valproic acid (reviewed in 4). The present results with GR-binding proteins lead us to conclude that the ability to separate both factor binding to and factor activities with GRs appear to be a general feature of GR-regulated gene induction. This modular control of gene induction properties greatly expands the possible mechanisms for differential control of gene expression, particularly if it is found to be a property of steroid receptors in general.
Several alternative explanations could be eliminated as possible causes for the changes in the induction parameters. While the affinity of Dex, and to a lesser extent of DAC), was reduced by the mutations examined, steroid affinity differences per se cannot account for changes in Amax. The affinity of DAC for wild type GRs is about 20-fold higher than of Dex and yet the Amax is usually very similar (Figs. 5 and 7 and refs. 9, 23). For the same reason, the reduced affinity of the mutant GRs for steroid would not be expected to account for the greatly diminished PAA of the antiglucocorticoid DM. The EC50 is also increased in every instance much more than expected from the small reduction in steroid binding affinity. Nonetheless, the nuclear translocation of mutant and wild type receptors is about the same (Fig. 2B). Possible effects on GR dimerization are unlikely for two reasons. First, a decrease in GR avidity for dimer formation on DNA should be countered by an elevated GR concentration, which was not observed, especially when looking at EC50 values (Figs. 1&2). Second, the mutations are not in a region known to affect GR dimerization (18). Unequal effects on DNA binding can be ruled out by the observations with GAL/GR525C. None of the GR sequences in GAL/GR525C contact DNA. The only DNA-protein interactions are those of the GAL4 DBD. Nonetheless, the changes in Amax, PAA, and EC50 for mutant vs. wild type GAL/GR525C chimeras with a GAL-regulated reporter (Fig. 5C) are very similar to those seen with full length mutant vs. wild type GR that binds directly to a GRE-controlled reporter (Figs. 3 and 4). Thus, the GR LBD appears to be the dominant domain for mediating the consequences of the LBD mutations independent of whether GR sequences have any direct contact with DNA. Furthermore, the Amax value, which is indicative of the total DNA-bound GR, is relatively constant with the various LBD mutations compared to the larger changes in PAA or EC50 (Figs. 1–4 and 7). This separate modulation of transcription parameters has also been seen for three other endogenous genes in human PBMCs with reduced levels of TIF2 (27). Collectively, these data illustrate how increased mechanistic information is accessible when EC50 and PAA are determined in addition to the conventional Amax.
Two results discount the explanation that the LBD mutations reduce cofactor binding to the GR LBD. First, co-IP experiments did not reveal any lower binding of Ubc9 to full length or truncated (GAL/GR525C) receptors, especially when adjusted for the higher levels of Dex needed to achieve similar levels of GR-steroid complexes (Fig. 6). Second, excess exogenous Ubc9 or GR can not reverse the changes in Amax, PAA, or EC50 of the mutant GRs (Figs. 1–5 and 7). Simple mass-action considerations (16) indicate that increasing the concentration of one component of a complex in the entire steroid-regulated transcription scheme will increase the amount of that complex. Over-expression of GR or Ubc9 does have some effect in two cell lines and on exogenous and endogenous reporters. However, the minimal reversals of the consequences of each mutation with added factor argue that reduced binding of GR to itself, of GR to Ubc9, or of GR to any cofactor is not the explanation. These conclusions are identical to those reached previously with TIF2 (9).
The results of exogenous Ubc9 binding to GAL/GR525C are particularly instructive. Ubc9 binds to the GR LBD (14). The GR LBD with the AF2 domain is sufficient to mediate the Ubc9-induced changes in induction parameters displayed by the full length GR; and, complications due to the stronger AF1 domain are eliminated (Fig. 5 and ref. 14). Thus, GAL/GR525C is ideally constructed to reveal the maximal effects of Ubc9 binding. Fig. 5C shows that the mutant GAL/GR525C has more nearly wild type properties when bound by DAC than by Dex. The Amax of DAC-bound mutant GR, with or without added Ubc9, and the ability of exogenous Ubc9 to decrease the EC50 of DAC-bound mutant receptor is much closer to wild type GRs. Similar more nearly wild-type properties of DAC- vs. Dex-bound mutant receptors and greater sensitivity of GAL/GR525C vs. full length GRs induction parameters to added cofactor were seen in the activities of these mutant GRs with TIF2 (9).
It is difficult to quantitatively compare the effects of increased Ubc9 or GR to those of increased TIF2 because the mechanisms of action appear to be quite different (16) (Chow et al, submitted). The inability of Ubc9 and TIF2 to decrease the EC50, or increase the PAA, of the A625I mutation in the context of GAL/GR525C are comparable. In contrast, the changes in Amax are 30-fold with added TIF2 vs. 2-fold with exogenous Ubc9 (Fig. 5 and ref. 9). These differences further document the ability to separately modify selected induction parameters and emphasize the mechanistic benefits of examining more parameters for steroid receptor regulated gene transcription than just Amax. Because the site for TIF2 binding to GR appears different from those for Ubc9 association and GR dimerization (10), qualitative and quantitative difference might be expected. Conversely, these results argue for the generality of small changes in the tertiary structure of the receptor LBD having larger effects on the biological activity of the receptor-bound cofactor than on the binding of the factor. As these same mutations disproportionately modified the Amax, PAA, and EC50 of GR-repressed genes (9), we speculate that the dissociation of factor binding and of the three transcription parameters occurs both in GR-regulated induction and GR-mediated repression.
The ability of changes in factor activity in the presence of unaltered factor binding to a mutant receptor appears to involve action-at-a-distance effects. However, the mounting number of reports of changes that are not immediately recognizable as sequential modifications in contacting amino acids suggest that this may be a more prevalent phenomenon that hitherto considered (11, 28), particularly for DNA-bound proteins (29). The DNA binding of GRs is known to stabilize the GR AF1 domain (30). Reminiscent of what we report, DNA sequence can also act as an allosteric ligand of DNA-bound GRs, with mutant GRs having different fold induction patterns with assorted glucocorticoid response elements (GREs) that are unrelated to the DNA binding affinity of GR (31). Also, modification of ligand structure can cause decreased transactivation by GR with no change in the binding of TIF2 peptides (32). The Jun dimerization protein 2 (JDP2) binds to progesterone receptor LBD but affects activity of the progesterone receptor AF1 domain (33). Finally, an elegant compilation of 3D structures shows different binding sites on TIFIID of p53, Sp1, and c-Jun with action-at-a-distance changes in structure in the absence of any modifications in the intervening structures (12). Collectively, the above observations provide several plausible mechanisms for how the Amax, PAA, and EC50 could vary among endogenous genes with different receptor mutations and agonist steroid structures.
Acknowledgments
We thank Hans Luecke (NIDDK, NIH) and members of the Steroid Hormones Section for critical comments.
Footnotes
This research was supported by the Intramural Research Program of the NIDDK, NIH.
Abbreviations: Amax, maximal activity; AF1 and AF2, activation function 1 and 2 respectively; DAC, deacylcortivazol; Dex, dexamethasone; DM, Dex-21-mesylate; DBD, DNA binding domain; GR, glucocorticoid receptor; IGFBP1, Insulin-like growth factor-binding protein 1; I6PK, inositol hexaphosphate kinase 3; LAD1, ladinin1; LBD, ligand binding domain; and PAA, partial agonist activity.
Disclosure statement: The authors have nothing to disclose.
References
- 1.Haas DM, McCullough W, Olsen CH, Shiau DT, Richard J, Fry EA, McNamara MF. Neonatal outcomes with different betamethasone dosing regimens: a comparison. J Reprod Med. 2005;50:915–922. [PubMed] [Google Scholar]
- 2.Stahn C, Lowenberg M, Hommes DW, Buttgereit F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol Cell Endocrinol. 2007;275:71–78. doi: 10.1016/j.mce.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 3.Simons SS., Jr The importance of being varied in steroid receptor transactivation. TIPS. 2003;24:253–259. doi: 10.1016/S0165-6147(03)00101-9. [DOI] [PubMed] [Google Scholar]
- 4.Simons SS., Jr What goes on behind closed doors: physiological versus pharmacological steroid hormone actions. Bioessays. 2008;30:744–756. doi: 10.1002/bies.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes and Develop. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang D, Wang Q, Awasthi S, Simons SS., Jr Amino-terminal domain of TIF2 is involved in competing for corepressor binding to glucocorticoid and progesterone receptors. Biochemistry. 2007;48:8036–8049. doi: 10.1021/bi7004575. [DOI] [PubMed] [Google Scholar]
- 7.Chen S, Sarlis NJ, Simons SS., Jr Evidence for a common step in three different processes for modulating the kinetic properties of glucocorticoid receptor-induced gene transcription. J Biol Chem. 2000;275:30106–30117. doi: 10.1074/jbc.M005418200. [DOI] [PubMed] [Google Scholar]
- 8.Suino-Powell K, Xu Y, Zhang C, Tao YG, Tolbert WD, Simons SSJ, Xu HE. Doubling the size of the glucocorticoid receptor ligand binding pocket by deacylcortivazol. Mol Cell Biol. 2008;28:1915–1923. doi: 10.1128/MCB.01541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tao YG, Xu Y, Xu HE, Simons SS., Jr Mutations of glucocorticoid receptor differentially affect AF2 domain activity in a steroid-selective manner to alter the potency and efficacy of gene induction and repression. Biochemistry. 2008;47:7648–7662. doi: 10.1021/bi800472w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szapary D, Song LN, He Y, Simons SS., Jr Differential modulation of glucocorticoid and progesterone receptor transactivation. Mol Cell Endocrinol. 2008;283:114–126. doi: 10.1016/j.mce.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci U S A. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu WL, Coleman RA, Ma E, Grob P, Yang JL, Zhang Y, Dailey G, Nogales E, Tjian R. Structures of three distinct activator-TFIID complexes. Genes Dev. 2009;23:1510–1521. doi: 10.1101/gad.1790709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaul S, Blackford JA, Jr, Cho S, Simons SS., Jr Ubc9 is a novel modulator of the induction properties of glucocorticoid receptors. J Biol Chem. 2002;277:12541–12549. doi: 10.1074/jbc.M112330200. [DOI] [PubMed] [Google Scholar]
- 14.Cho S, Kagan BL, Blackford JA, Jr, Szapary D, Simons SS., Jr Glucocorticoid receptor ligand binding domain is sufficient for the modulation of glucocorticoid induction properties by homologous receptors, coactivator transcription intermediary factor 2, and Ubc9. Mol Endo. 2005;19:290–311. doi: 10.1210/me.2004-0134. [DOI] [PubMed] [Google Scholar]
- 15.Kim Y, Sun Y, Chow C, Pommier YG, Simons SS., Jr Effects of acetylation, polymerase phosphorylation, and DNA unwinding in glucocorticoid receptor transactivation. J Steroid Biochem Molec Biol. 2006;100:3–17. doi: 10.1016/j.jsbmb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Ong KM, Blackford JA, Jr, Kagan BL, Simons SS, Jr, Chow CC. A new theoretical framework for gene induction and experimental comparisons. Proc Natl Acad Sci U S A. 2010;107:7107–7112. doi: 10.1073/pnas.0911095107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szapary D, Xu M, Simons SS., Jr Induction properties of a transiently transfected glucocorticoid-responsive gene vary with glucocorticoid receptor concentration. J Biol Chem. 1996;271:30576–30582. doi: 10.1074/jbc.271.48.30576. [DOI] [PubMed] [Google Scholar]
- 18.Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. doi: 10.1016/s0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- 19.Simons SS, Jr, Pons M, Johnson DF. α-Keto mesylate: a reactive thiol-specific functional group. J Org Chem. 1980;45:3084–3088. [Google Scholar]
- 20.Wang Q, Blackford JA, Jr, Song LN, Huang Y, Simons SS., Jr Equilibrium interactions of corepressors and coactivators modulate the properties of agonist and antagonist complexes of glucocorticoid receptors. Mol Endocrinol. 2004;18:1376–1395. doi: 10.1210/me.2003-0421. [DOI] [PubMed] [Google Scholar]
- 21.He Y, Simons SS., Jr STAMP: a novel predicted factor assisting TIF2 actions in glucocorticoid receptor-mediated induction and repression. Mol Cell Biol. 2007;27:1467–1485. doi: 10.1128/MCB.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vicent GP, Ballare C, Nacht AS, Clausell J, Subtil-Rodriguez A, Quiles I, Jordan A, Beato M. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell. 2006;24:367–381. doi: 10.1016/j.molcel.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 23.Simons SS, Jr, Thompson EB, Johnson DF. Anti-inflammatory pyrazolo-steroids: potent glucocorticoids containing bulky A-ring substituents and no C3-carbonyl. Biochem Biophys Res Comm. 1979;86:793–800. doi: 10.1016/0006-291x(79)91782-0. [DOI] [PubMed] [Google Scholar]
- 24.Poukka H, Karvonen U, Janne OA, Palvimo JJ. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1) Proc Natl Acad Sci U S A. 2000;97:14145–14150. doi: 10.1073/pnas.97.26.14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, Darimont BD, Garabedian MJ, Yamamoto KR. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2003;100:13845–13850. doi: 10.1073/pnas.2336092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Motoki K, Megahed M, LaForgia S, Uitto J. Cloning and chromosomal mapping of mouse ladinin, a novel basement membrane zone component. Genomics. 1997;39:323–330. doi: 10.1006/geno.1996.4507. [DOI] [PubMed] [Google Scholar]
- 27.Luo M, Simons SS., Jr Modulation of glucocorticoid receptor induction properties by cofactors in peripheral blood mononuclear cells. Human Immunology. 2009;70:785–789. doi: 10.1016/j.humimm.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarkson MW, Gilmore SA, Edgell MH, Lee AL. Dynamic coupling and allosteric behavior in a nonallosteric protein. Biochemistry. 2006;45:7693–7699. doi: 10.1021/bi060652l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leatherwood J, Futcher B. King of the castle: competition between repressors and activators on the Mcm1 platform. Mol Cell. 2010;38:1–2. doi: 10.1016/j.molcel.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 30.Kumar R, Thompson EB. Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol Endocrinol. 2003;17:1–10. doi: 10.1210/me.2002-0258. [DOI] [PubMed] [Google Scholar]
- 31.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biggadike K, Bledsoe RK, Coe DM, Cooper TW, House D, Iannone MA, Macdonald SJ, Madauss KP, McLay IM, Shipley TJ, Taylor SJ, Tran TB, Uings IJ, Weller V, Williams SP. Design and x-ray crystal structures of high-potency nonsteroidal glucocorticoid agonists exploiting a novel binding site on the receptor. Proc Natl Acad Sci U S A. 2009;106:18114–18119. doi: 10.1073/pnas.0909125106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill KK, Roemer SC, Jones DN, Churchill ME, Edwards DP. A progesterone receptor co-activator (JDP2) mediates activity through interaction with residues in the carboxyl-terminal extension of the DNA binding domain. J Biol Chem. 2009;284:24415–24424. doi: 10.1074/jbc.M109.003244. [DOI] [PMC free article] [PubMed] [Google Scholar]